

Abstract
The outer kinetochore binds microtubules to control chromosome movement. Outer kinetochore assembly is restricted to mitosis, whereas the inner kinetochore remains tethered to centromeres throughout the cell cycle. The cues that regulate this transient assembly are unknown. We find that inhibition of Aurora B kinase significantly reduces outer kinetochore assembly in Xenopus laevis and human tissue culture cells, frog egg extracts, and budding yeast. In X. leavis M phase extracts, preassembled kinetochores disassemble after inhibiting Aurora B activity with either drugs or antibodies. Kinetochore disassembly, induced by Aurora B inhibition, is rescued by restraining protein phosphatase 1 (PP1) activity. PP1 is necessary for kinetochores to disassemble at the exit from M phase, and purified enzyme is sufficient to cause disassembly on isolated mitotic nuclei. These data demonstrate that Aurora B activity is required for kinetochore maintenance and that PP1 is necessary and sufficient to disassemble kinetochores. We suggest that Aurora B and PP1 coordinate cell cycle–dependent changes in kinetochore assembly though phosphorylation of kinetochore substrates.
Introduction
Kinetochores assemble onto centromeres during mitosis, where they coordinate the chromosome movements that segregate DNA replication products into two daughter cells. They also resolve improper microtubule attachments and generate spindle checkpoint signals. The inner kinetochore, which organizes the underlying chromatin, is generated from several proteins including centromere protein C (Cenp-C) and a modified nucleosome containing the histone H3 variant Cenp-A (Cse4 in yeast). The inner kinetochore proteins are constitutively associated with the centromere throughout the cell cycle. The outer kinetochore, which directly contacts and regulates microtubules, is assembled onto the inner kinetochore during mitosis. In vertebrates, outer kinetochore proteins begin to assemble during prophase and kinetochores are fully assembled by early prometaphase.
The outer kinetochore proteins that assemble in mitosis are numerous. They include the KMN network that is believed to constitute the outer kinetochores core microtubule binding activity and contains the Knl1/Blinkin (Knl1) protein together with the hetero-tetrameric subcomplexes Mis12 (Mis12, Nsl1, Nnf1, and Dsn1) and Ndc80 (Ndc80, Nuf2, Spc24, and Spc25; Wigge and Kilmartin, 2001; Deluca et al., 2002, 2006; McCleland et al., 2003, 2004; Cheeseman et al., 2004, 2006; Obuse et al., 2004; Kline et al., 2006; Kiyomitsu et al., 2007). There are several microtubule motors and microtubule-binding proteins that play critical roles in chromosome alignment and segregation, including dynein/dynactin, Cenp-E, and Cep57 (Yen et al., 1991; Sharp et al., 2000; Emanuele and Stukenberg, 2007). Proteins required for spindle checkpoint signaling (Bub1, BubR1, Bub3, Mad1, Mad2, MPS1, Rod, Zw10, and Zwilch) also assemble onto kinetochores in prophase/prometaphase (for review see Musacchio and Salmon, 2007). The interdependencies of these proteins for assembly has been studied (for review see Maiato et al., 2004).
The effectors that trigger outer kinetochore assembly and disassembly at the beginning and end of mitosis remain unclear. Kinetochores begin to assemble in prophase when cyclin A/Cdk1 accumulates in the nucleus. Also in prophase, the Aurora B kinase, a member of a four-protein complex called the chromosome passenger complex, is activated and relocalizes from mitotic chromatin to the inner centromere. Aurora B has been implicated in kinetochore assembly. Overexpression of a kinase-dead Aurora B mutant displaces Cenp-E and dynein from kinetochores (Murata-Hori and Wang, 2002a). Treatment of HeLa cells with the small molecule inhibitor of Aurora B, ZM447439, displaces the spindle checkpoint proteins BubR1 and Mad2 and the kinesin motor Cenp-E (Ditchfield et al., 2003). Surprisingly, treatment with a second compound, hesperadin, or siRNA knockdown of Aurora B only affects BubR1 localization (Hauf et al., 2003; Liu et al., 2006). Yeast cells deficient for the Aurora B orthologue Ipl1 cannot localize the Dam1/DASH complex and Mad2 to centromeres but most other proteins have not been examined for dependence on Ipl1 activity (Cheeseman et al., 2002; Gillett et al., 2004).
Aurora B has been implicated in many kinetochore functions and it is unclear if the phenotypes are caused by direct regulation of assembled kinetochores or if they are the indirect consequence of improper kinetochore assembly. Ipl1-deficient yeast cells cluster centromeres close to spindle poles and cannot biorient chromosomes or signal the spindle checkpoint in response to a lack of tension (Biggins and Murray, 2001; Tanaka et al., 2002). In vertebrates, Aurora B kinase is required for checkpoint signaling, chromosome alignment, and release of syntelic and merotelic attachments (Kallio et al., 2002; Ditchfield et al., 2003; Hauf et al., 2003; Cimini et al., 2006; Knowlton et al., 2006). Aurora B phosphorylates a serine (S10 in yeast, frogs, and humans) on the N-terminal tail of histone H3 (Hsu et al., 2000). Phosphorylation of S10 on histone H3 (pH3S10) marks mitotic cells under normal growth conditions and provides an indicator of Aurora B activity.
Aurora B/Ipl1 kinase is essential for viability in budding yeast, and the viability of ipl1 mutant cells is rescued by specific mutant alleles of the protein phosphatase 1 (PP1) orthologue Glc7 (Francisco et al., 1994). The phosphatase activity of PP1/Glc7 is repressed potently and specifically by Glc8 (Inhibitor-2 [I-2] in vertebrates) in vitro, and ipl1 mutants can also be rescued by overexpression of Glc8 (Tung et al., 1995). In vertebrates, Aurora B activity and I-2 levels peak in mitosis and subsequently decrease in telophase (Brautigan et al., 1990).
Immunodepletion of Aurora B from frog egg extracts blocked the accumulation of Mis12, Ndc80, Zwint, and dynein/dynactin at kinetochores (Emanuele et al., 2005). This effect was more severe than that observed in human cells when Aurora B was inhibited with drugs. In this study, we have further explored the role of Aurora B kinase in kinetochore assembly. We show that inhibition of Ipl1 in budding yeast, using temperature-sensitive mutants in both Ipl1 and Sli15 (an inner centromere protein [INCENP] homologue), disrupts accumulation of Ndc80 and Mtw1 (a Mis12 homologue) at centromeres. Inhibition of Aurora B using small molecule inhibitors also effected the accumulation of Ndc80 at kinetochores in human and Xenopus laevis tissue culture cells. In addition, we show a novel role for Aurora B kinase activity in maintaining outer but not inner kinetochore proteins at the centromere. In frog egg extracts, Aurora B inhibition causes rapid disassembly of the outer kinetochore. Inhibition of PP1 simultaneously with Aurora B rescues this effect, demonstrating that these enzymes work in opposition to one another with respect to kinetochore assembly. This is not caused by a reactivation of Aurora B after repression of phosphatases. Furthermore, extracts exiting M phase undergo PP1-dependent kinetochore disassembly as they enter interphase. Finally, purified PP1 alone is sufficient to disassemble kinetochores on nuclei isolated from egg extracts. We conclude that Aurora B and PP1 play an important role in the cell cycle–dependent assembly and disassembly of the kinetochore.
Results
Kinetochore assembly depends on Aurora B/Ipl1
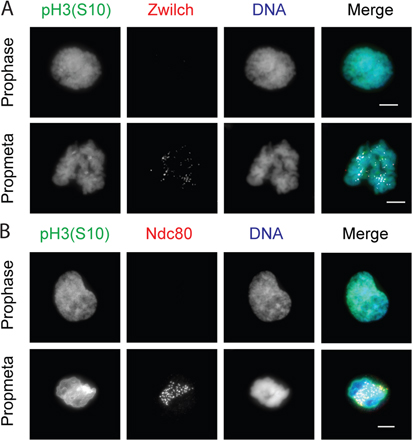
To examine the role of Aurora B kinase in kinetochore assembly, X. laevis–derived S3 tissue culture cells were treated with the Aurora kinase inhibitor hesperadin and prepared for immunofluorescence. Because many proteins are removed from kinetochores after microtubule attachment, we added nocodazole 15 min before fixing cells. Hesperadin treatment depleted pH3S10 staining on chromatin, indicating the effectiveness of the drug treatment (Fig. 1 A). In DMSO controls, 90% of mitotic cells that had progressed beyond prophase had Ndc80 kinetochore staining on chromatin. After treatment with 250 nM hesperadin, 49% of cells had visibly undetectable levels of Ndc80 and 47% showed strongly reduced Ndc80 staining. Quantification of immunofluorescent intensity at centromeres shows that Ndc80 kinetochore staining was reduced 76% in cells treated with 250 nM hesperadin (Fig. 1 A, right). Immunostaining with antibodies to Zwilch and the dynactin subunit p150Glued produced similar results (unpublished data). These experiments also revealed that Zwilch and Ndc80 appear at kinetochores after Aurora B has been activated, which is indicated by costaining for pH3S10 (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200710019/DC1). This suggests that Aurora B kinase activity is required for the localization of Ndc80, Zwilch, and dynactin at kinetochores in X. laevis tissue culture cells.
Figure 1.

Aurora B inhibition compromises kinetochore assembly in X. laevis and human tissue culture cells and in budding yeast. (A) Asynchronous X. laevis S3 cells were treated with hesperadin and fixed for immunofluorescence. Cells were immunostained with antibodies to Ndc80 and phosphorylated serine 10 on histone H3 (pH3S10). The graph shows levels of Ndc80 staining at kinetochores in control and hesperadin-treated cells (n = 60 kinetochores in three different cells per condition). (B) A549 human lung epithelial cells and primary human foreskin fibroblasts were treated with either DMSO or hesperadin and ZM447439 (Hesp + ZM) together. Cells were fixed and processed for immunofluorescence with ACA and Ndc80 antibodies. The amount of Ndc80 staining at kinetochores relative to ACA staining on the same kinetochore was determined and is graphed (n = 60 kinetochores in three cells per condition). Bars, = 5 μm. (C) Wild type or sli15-3 yeast cells were grown at a restrictive temperature of 37° for 3 h and kinetochore assembly was assessed by ChIP for Ndc80, Mtw1, Kip1, Bim1, Ipl1, Dam1, Cse4, and Ctf19. The outer kinetochore proteins are displaced from kinetochores in sli15-3 cells. IN and + indicate PCR performed on either total input or immunoprecipitated DNA, respectively. Centromeres on chromosome 16 (Cen16) were amplified. Graphs show mean plus standard deviation.
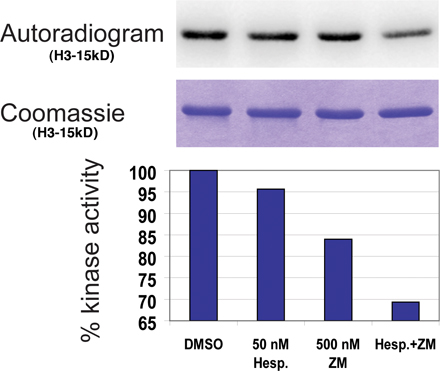
Treatment of human tissue culture cells with hesperadin or ZM447439 alone did not effect Ndc80 recruitment to kinetochores, as has been found previously (unpublished data: Ditchfield et al., 2003; Hauf et al., 2003; Lampson et al., 2004). However, these treatments do not completely inhibit Aurora kinase activity. To more fully inhibit Aurora B enzymatic activity but avoid amplifying off-target effects caused by treatment with high drug concentrations, human tissue culture cells were treated simultaneously with both hesperadin and ZM447439. Double drug treatment additively inhibits the kinase activity of the purified Aurora B complex relative to using either drug alone (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200710019/DC1).
Treatment of human A549 lung epithelial cells with 250 nM hesperadin and 500 nM ZM447439 together caused a 70% reduction in the amount Ndc80 localized at kinetochores compared with control treated cells (Fig. 1 B). Ndc80 levels were quantified relative to the amount of anti-centromere antigen (ACA) staining in the same region, as described previously (Ditchfield et al., 2003). An identical drug treatment of human primary foreskin fibroblasts caused a 38% reduction in Ndc80 staining at kinetochores (Fig. 1 B, right). Further, hesperadin- and ZM447439-treated A549 cells were unable to maintain a nocodazole-induced mitotic arrest. The mitotic index of nocodazole-treated A549 cells was reduced from 18.3% (control) to 5.3% after double drug treatment (n > 500 cells). These data suggest that the kinase activity of Aurora B is required for outer kinetochore assembly in human cultured cells. Interestingly, HeLa cells treated simultaneously with both drugs showed no effect on Ndc80 recruitment to kinetochores (unpublished data).
Aurora B/Ipl1 activity in yeast can be compromised with temperature-sensitive mutants in either Ipl1 or its activator Sli15. Asynchronous cultures of sli15-3 or wild-type yeast were shifted to the restrictive temperature (37°) for 3 h and their ability to recruit and/or maintain kinetochore proteins to the centromere was measured by chromatin immunoprecipitation (ChIP; Fig. 1 C). When grown at the restrictive temperature, the ability of Ndc80, Mtw1 (a Mis12 orthologue), Kip1 (a kinesin motor), Bim1 (an EB-1 orthologue), Ipl1, and Dam1 to be cross-linked to centromeres is either totally abolished or greatly reduced in sli15-3 cells, thus indicating that the abundance of these proteins at centromeres is reduced in the absence of Ipl1 activity. The inner kinetochore protein Cse4 (Cenp-A orthologue) and the central kinetochore protein Ctf19 (COMA complex) localized normally (Fig. 1 C). Each of these proteins is present at centromeres in wild-type cells at the restrictive temperature. We tested a subset of the proteins (Ndc80, Bim1, and Dam1) for centromere accumulation in ipl1-2 cells and found similar results (unpublished data; Cheeseman et al., 2002). This demonstrates that Ipl1 and Sli15 are required to recruit outer kinetochore proteins to centromeres in budding yeast.
Aurora B activity drives kinetochore assembly in frog egg extracts
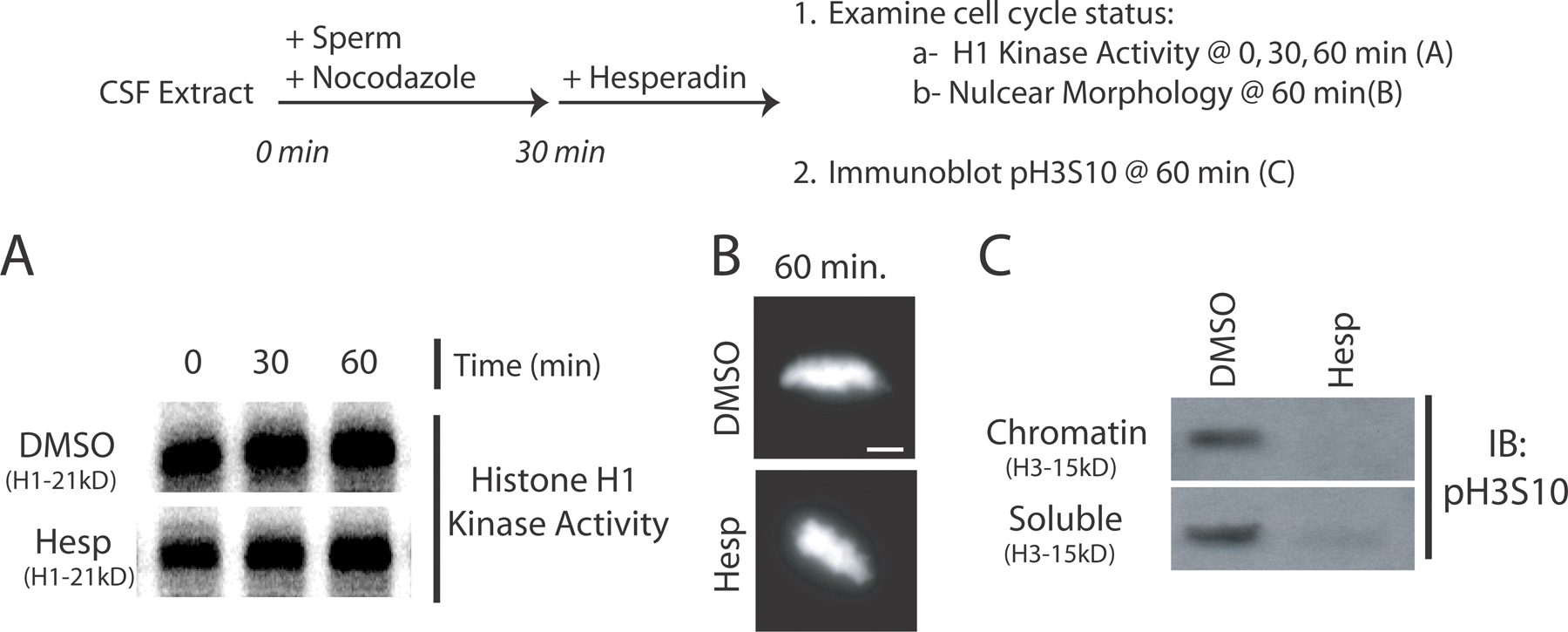
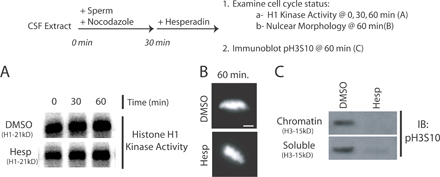
To further dissect the role of Aurora B kinase in kinetochore assembly, we used X. laevis egg extracts that assemble functional kinetochores onto centromeres of sperm chromatids. Hesperadin was titrated into cytostatic factor (CSF)-arrested M phase extracts. Sperm nuclei were added for 45 min (Fig. 2 A, top) and then reactions were fixed and examined by immunofluorescence. Aurora kinase was inhibited by 2 μM hesperadin as determined by pH3S10 staining (Fig. 2 A; Zhang et al., 2007). Staining for pH3S10 was quantified as a ratio to a nuclear stain (Hoechst 33342) in the presence of increasing concentrations of hesperadin (Fig. 2 A, right). In a functional assay for Aurora B activity, 2 μM hesperadin inhibited spindle checkpoint signaling in nocodazole-treated extracts (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200710019/DC1). Aurora inhibition does not drive the extracts out of M phase. Histone H1 kinase activity, a readout of Cdk1 activity, remained high, and nuclear morphology appeared condensed for at least 60 min after hesperadin addition, demonstrating that it does not affect cell cycle state in egg extracts (Fig. S4).
Figure 2.

Aurora B activity is required for kinetochore assembly in X. laevis CSF extracts. (A and B) Hesperadin was titrated into X. laevis CSF egg extracts. Extracts were supplemented with nocodazole and sperm nuclei and, after 45 min, fixed and processed for immunofluorescence (A, top). (A) Nuclei stained with antibodies to pH3S10 and the nuclear stain Hoechst33342. The intensity of pH3S10 staining relative to the nuclear stain is graphed (right). Graphs show mean plus standard deviation. (B) Nuclei immunostained for the kinetochore protein Zwilch. Kinetochore assembly is inhibited at 2 μM hesperadin. (C) Kinetochores were assembled in CSF extracts preincubated with either anti–Aurora A or B antibodies. Nuclei were stained with antibodies to dynactin subunit p150Glued. Bars, 5 μm.
We confirmed that Aurora B activity was required for kinetochore assembly in X. laevis extracts. Hesperadin was titrated into CSF extracts, and sperm nuclei and nocodazole were added to initiate kinetochore assembly. X. laevis sperm have 18 chromosomes, and the assembly of proteins onto kinetochores can be detected by immunostaining, in which they appear as discrete spots on the chromatin mass. Hesperadin displaced the kinetochore/spindle checkpoint protein Zwilch from chromatin at the same concentration required to abolish pH3S10 (2 μM; Fig. 2 B). After hesperadin treatment, <3% of nuclei have any discernable Zwilch kinetochore staining. In DMSO controls, >95% of nuclei stain positive for Zwilch (n > 100 nuclei per condition). These data suggest a role for Aurora kinase activity in kinetochore assembly. Importantly, this system provided us with a rapid, robust, and biochemically amenable assay for dissecting the contributions of Aurora B to kinetochore assembly.
Because hesperadin can inhibit both Aurora A and B kinases, we confirmed that the effect of hesperadin on kinetochore assembly is caused by inhibition of Aurora B. Kinetochores were assembled in CSF extracts preincubated with antibodies to either kinase. We examined kinetochore assembly with a mouse monoclonal antibody to the p150Glued subunit of dynactin to avoid cross-reaction of our secondary antibody with the Aurora B antibodies. Treatment with the Aurora B antibody blocked accumulation of p150Glued at kinetochores (Fig. 2 C), whereas adding Aurora A antibodies had no effect. We used a concentration of Aurora A antibody that disrupts microtubule aster formation (unpublished data). We conclude that the affect of hesperadin on kinetochore assembly is caused by inhibition of Aurora B and not Aurora A. These data are further supported by our previous demonstration that extracts immunodepleted of Aurora B are unable to assemble outer kinetochore proteins (Emanuele et al., 2005).
Aurora B activity maintains kinetochore assembly status
We next tested if continuous Aurora B activity is required to maintain assembly of the kinetochore. In vitro egg extracts are well suited for this because they allow us to temporally control Aurora B activity. We preassembled kinetochores on sperm chromatin in extracts for 30 min and then added hesperadin (Fig. 3 A, top). Reactions were fixed at 5-min intervals and immunostained with Ndc80 antibodies (Fig. 3 A). The ratio of Ndc80 to Cenp-A staining on the same centromeres was calculated as described previously (Ditchfield et al., 2003). Ndc80 staining was reduced >90% after 5 min and >95% after 10 min (Fig. 3 A). Identical effects were seen after adding anti–Aurora B (Fig. 3 B) or anti-INCENP (not depicted) antibodies instead of hesperadin. Therefore, Aurora B is required to both establish and maintain the Ndc80 protein at centromeres.
Figure 3.

Aurora B activity is required for kinetochore maintenance in CSF extracts. (A) Kinetochore maintenance was assessed after hesperadin treatment in extracts containing nuclei with preassembled kinetochores. The relative intensity of Ndc80 to Cenp-A immunostaining on the same kinetochore was determined and graphed (bottom; n = 60 individual kinetochores on four nuclei per condition). Graphs show mean plus standard deviation. (B) The addition of anti–Aurora B antibodies caused preassembled kinetochores to disassemble in an identical manner. (C) A panel of antibodies was used to assess the sensitivity of outer kinetochore proteins to hesperadin treatment in extracts with preassembled kinetochores. The outer kinetochore proteins Dsn1, Knl1, Ndc80, and Zwint and the checkpoint proteins BubR1, Mad1, and Mad2 are displaced after hesperadin treatment. The inner kinetochore proteins Cenp-A and Cenp-C are unaffected. Bars, 5 μm.
We used a panel of antibodies to identify the proteins that require Aurora B activity to be maintained at kinetochores. All outer kinetochore proteins tested, including Knl1, Ndc80, Zwint, Cep57, Dsn1 (Mis12 complex), CLIP-170, p150Glued (dynactin complex), and the spindle checkpoint components Mad1, Mad2, and BubR1 (Fig. 3 C and Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200710019/DC1) were displaced from kinetochores within 15 min of hesperadin treatment. In contrast, the inner kinetochore components Cenp-A and Cenp-C are retained at centromeres (Fig. 3 C). We conclude that in CSF extracts, Aurora B is critical for the maintenance of the outer kinetochore.
To confirm that Aurora B was also required to maintain kinetochore assembly on replicated chromosomes, CSF extracts containing sperm were cycled through S phase and rearrested in the subsequent mitosis with fresh CSF extract. Knl1, Mis12 (Fig. 4, A and B), Ndc80, Zwilch, and p150Glued (not depicted) were displaced after hesperadin treatment. Together, our data demonstrate that kinetochores require persistent Aurora B kinase activity.
Figure 4.

Aurora B activity is required for kinetochore maintenance in cycled extracts. (A and B) Cycled X. laevis extracts containing replicated chromatids and assembled kinetochores were tested for their sensitivity to hesperadin (or DMSO). After 15 min in hesperadin, Knl1 (A) and Mis12 (B) were displaced from kinetochores. (C) Maintenance reactions were repeated in CSF and cycled egg extracts. Aurora B complex proteins were displaced from chromatin by the addition of hesperadin after assembly. X. laevis S3 cells also lost Aurora B staining on chromatin after treatment with hesperadin. Bars, 5 μm.
Treatment of CSF extracts with hesperadin, after kinetochore assembly, resulted in a loss of Aurora B staining on chromatin (Fig. 4 C, left). Identical results were obtained in cycled extracts immunostained for Dasra A and INCENP (Fig. 4 C, middle; and not depicted) and in X. laevis tissue culture cells (Fig. 4 C, right). These data suggest that Aurora B kinase activity is required for its association with the centromere.
PP1 opposes Aurora B in kinetochore assembly
If phosphatases act directly on the Aurora B substrates that are critical for kinetochore assembly, then phosphatase inhibition should rescue the kinetochore disassembly defects caused by hesperadin. To test this, kinetochores were preassembled on nuclei in extracts (Fig. 5 A, top). Hesperadin and the phosphatase inhibitor okadaic acid were added alone or in combination. After 15 min, kinetochore status was assayed by immunostaining for Zwilch (Fig. 5 A). Because the RZZ (Rod, Zw10, and Zwilch) complex requires Ndc80 for assembly and is itself essential for dynein/dynactin recruitment, it is an excellent indicator of kinetochore assembly status (Starr et al., 1998; Emanuele et al., 2005). We found that simultaneous addition of okadaic acid rescued the kinetochore disassembly caused by hesperadin (Fig. 5 A).
Figure 5.

Phosphatases oppose Aurora B in maintaining an assembled kinetochore. (A) Hesperadin and okadaic acid were added alone or together and Zwilch maintenance at kinetochores was examined after 15 min. The addition of 1 μM okadaic acid rescued the effect of hesperadin on kinetochore maintenance. The percentage of nuclei staining positive for the kinetochore marker Zwilch is graphed (n > 100 nuclei per condition). (B) Kinetochores were assembled on sperm nuclei for 30 min in extracts. Hesperadin was added to turn off Aurora B and, after 15 min, 1 μM okadaic acid was also added to restrain phosphatase activity. Neither Zwilch nor pH3S10 staining returned to chromatin after 15 min in okadaic acid. Bars, 5 μm.
We tested if okadaic acid treatment was reactivating Aurora B despite the presence of its inhibitor (hesperadin). In an order-of-addition experiment, kinetochores were first preassembled onto sperm nuclei. Hesperadin was added, disassembling kinetochores, and then phosphatases were inhibited by adding okadaic acid. No discernable pH3S10 or kinetochore staining was observed after treatment with both drugs (Fig. 5 B), which suggests that Aurora B was not reactivated by phosphatase inhibition.
The concentration of okadaic acid required (1.0 μM; Fig. 5 A) suggested that PP1 and not PP2A was modulating kinetochore assembly. To test this hypothesis, we added hesperadin and the PP1-specific inhibitory protein I-2 to extracts containing preassembled kinetochores (Fig. 6). Zwilch, Ndc80, Knl1, Dsn1, and p150Glued were retained at kinetochores after simultaneous treatment with hesperadin and I-2 (Fig. 6, A–C). After the addition of hesperadin and I-2, Aurora B remains dissociated from centromeres, whereas the kinetochore remains intact (Fig. 6 C). Thus, Aurora B localization at centromeres is not essential for maintaining kinetochore assembly in the absence of PP1 activity. This also suggests that Aurora B does not play a structural role at the centromere for the recruitment of kinetochore proteins. We conclude that in the kinetochore assembly pathway, Aurora B is opposed by PP1.
Figure 6.

PP1 opposes Aurora B in the assembly pathway of the kinetochore. Hesperadin and the PP1-specific inhibitor I-2 were added alone or in combination in a kinetochore maintenance assay. (A) Zwilch is retained at kinetochores after coaddition of hesperadin and I-2. (B) The percentage of nuclei staining positive for the kinetochore proteins Ndc80, Mis12, and Knl1 after treatment with DMSO, hesperadin, or hesperadin and I-2 together is graphed (n > 100 nuclei per condition). Graphs show mean plus standard deviation. (C) Nuclei from the same experiment were stained with antibodies to p150Glued and Aurora B. Aurora B is not retained at kinetochores after treatment with hesperadin and I-2 but kinetochores are still intact, as assessed by p150 Glued staining. Bar, 5 μm.
PP1 and kinetochore disassembly
We asked whether PP1 is important for kinetochore disassembly after mitotic exit. Kinetochores were assembled onto sperm nuclei in CSF extracts containing nocodazole. Extracts were driven out of M phase by the addition of calcium in the presence or absence of I-2. Histone H1 kinase activity was lost 10 min after calcium addition in BSA control and I-2–treated extracts (Fig. 7 A). In control extracts, immunostaining for Ndc80 was reduced after 10 min and was undetectable after 20 min (Fig. 7 B). In I-2–treated extracts, where PP1 is inactivated, the levels of Ndc80 staining on chromatin remain unchanged for 30 min after triggering M phase exit (Fig. 7 B). Both BSA- and I-2–treated extracts displayed interphase chromatin morphology by 45 min (unpublished data). We conclude that PP1 activity is necessary for kinetochore disassembly after exit from M phase.
Figure 7.

PP1 is both necessary and sufficient to disassemble kinetochores on nuclei in extracts. (A and B) Kinetochores were assembled for 30 min in CSF extracts. I-2 or BSA was added to extracts and, 15 min later, the extract was driven out of M phase. (A) Both BSA- and I-2–treated extracts exited mitosis biochemically after the addition of calcium, as judged by histone H1 kinase activity. (B) In I-2–treated extracts, Ndc80 was retained at the kinetochore for 30 min after M phase exit. In control BSA-treated extracts, Ndc80 staining was reduced by 10 min and was undetectable by 20 min. (C) Nuclei with assembled kinetochores were isolated, washed, and mixed with purified PP1 enzyme. The addition of PP1 caused the dephosphorylation of H3S10 and the loss of kinetochore staining on the nuclei compared with BSA-treated nuclei. In control BSA-treated samples, 92% of nuclei stained positive for Ndc80. In PP1-treated samples, 39% of nuclei stained positive for Ndc80 (n > 100 nuclei per condition). Graphs show mean plus standard deviation. Bar, 5 μm.
To determine whether PP1 is sufficient to disassemble the kinetochore, nuclei with assembled kinetochores were isolated through a glycerol cushion, washed thoroughly, and mixed with purified PP1. Nuclei incubated with buffer and BSA alone retained Ndc80 at kinetochores and stained positive for pH3S10. Treatment with purified PP1 enzyme for 15 min caused dephosphorylation of H3S10 and displacement of the Ndc80 protein. We conclude that PP1 dephosphorylates either a chromatin- or kinetochore-bound substrate to induce kinetochore disassembly.
Discussion
Cell cycle regulation of kinetochore assembly
The outer kinetochore proteins assemble in prophase/prometaphase and are disassembled in late anaphase. We have shown that these kinetochore assembly events are regulated by Aurora B kinase activity. Initial observations suggested that Aurora B was important for kinetochore assembly after immunodepletion from frog egg extracts (Emanuele et al., 2005). In this study, we demonstrate that Aurora B activity is required for assembly and maintenance of the outer kinetochore from yeast to humans. Aurora B inhibition, using multiple methods, leads to rapid disassembly of the kinetochore. Every outer kinetochore protein we examined was sensitive to Aurora B inhibition. In contrast, the inner kinetochore proteins, which include Cenp-A (Cse4) and Cenp-C, assemble independently of Aurora B activity. Importantly, the recruitment of the KMN network (Knl1, Mis12, and Ndc80 complexes), which is part of the kinetochores' core microtubule binding machinery, is compromised after Aurora inhibition. These data suggest that Aurora B is a cell cycle regulator of outer kinetochore assembly.
In budding yeast, Ipl1 mutants are rescued by specific mutant alleles of Glc7/PP1, demonstrating that their activities oppose each other. We have shown here that Aurora B's role in kinetochore assembly is opposed by PP1 in vertebrates. Restraining PP1 activity using the small inhibitory protein I-2 protected the kinetochore from disassembly after Aurora B inhibition. PP1 activity was required for kinetochore disassembly at mitotic exit and the purified enzyme was sufficient to disassemble kinetochores on isolated nuclei. PP1 and Aurora B affect assembly of an identical subset of kinetochore proteins. Cenp-A and Cenp-C are insensitive to changes in Aurora B and PP1 activity, as they remain centromere associated throughout the cell cycle. Inhibition of Aurora B and PP1 in extracts before the addition of sperm nuclei does not rescue kinetochore assembly, demonstrating that Aurora B activity is essential for initial assembly of the outer kinetochore even in the absence of PP1 activity (unpublished data). These data suggest that PP1 is a regulator of kinetochore disassembly at mitotic exit. We conclude that Aurora B and PP1 act in opposition to each other as cell cycle–dependent regulators of outer kinetochore assembly.
A model depicting the activities of Aurora B and PP1 during the somatic cell cycle relative to the times of kinetochore assembly and disassembly is depicted in Fig. 8. The various steps of the mitotic cell cycle, starting in late G2 and proceeding through telophase, are shown relative to Aurora B and PP1 activity. In prophase, Aurora B becomes activated and, by prometaphase, Aurora B is localized to inner centromere. At this time, the outer kinetochore proteins are recruited and assembled onto centromeres. At the metaphase-to-anaphase transition, Aurora B is released from the inner centromere but remains catalytically active in a gradient of activity concentrated on the spindle midzone and diffuses throughout the cell (unpublished data). We hypothesize that one role for the gradient of Aurora B activity is to keep kinetochores assembled in anaphase A. By anaphase B, kinetochores pass beyond the periphery of Aurora B's zone of activity, and outer kinetochore proteins are displaced from the centromere. Recent work has demonstrated that Ncd80 levels are reduced 5–10-fold at kinetochores in anaphase B (McAinsh et al., 2006). In late anaphase B and telophase, I-2 levels, which remain high throughout mitosis, begin to decrease, which is concomitant with the complete loss of outer kinetochore proteins at centromeres, a decrease in Aurora B activity, and, finally, mitotic exit.
Figure 8.

Model of Aurora B and PP1 activity relative to kinetochore assembly in the mitosis. G2 and the mitotic phases (prophase, prometaphase, metaphase, anaphases A and B, and telophase) are depicted. Microtubules are displayed in green, chromosomes in blue, kinetochores in yellow, and active Aurora B kinase in red. The times at which Aurora B (red lines) and PP1 (green lines) are activated are shown relative to the morphological changes of mitosis. In prophase, Aurora B is activated and begins to move from chromosome arms to centromeres. By prometaphase, activated Aurora B is fully localized to the inner centromere and outer kinetochore proteins are assembled. Complete chromosome alignment occurs in metaphase, at which point the spindle checkpoint signal is extinguished and sister chromatids separate and begin poleward anaphase movements. Aurora B dissociates from the inner centromere in anaphase and relocalizes to overlapping spindle midzone microtubules, where it remains active in a gradient of kinase activity that spreads from out from the spindle midzone (fading red). Kinetochores disassemble as they move outside of the zone of Aurora B activity in anaphase B. PP1 is initially activated in anaphase, and its small protein inhibitor I-2 is further degraded in anaphase B and telophase. Active PP1, in the absence of Aurora B, leads to outer kinetochores disassembly in telophase before the ensuing G1.
We propose a model wherein two mechanisms are essential for outer kinetochore assembly in vertebrates. The first mechanism spatially directs kinetochore assembly to a specific site, the centromere. This is accomplished by Cenp-A loading at centromeres. However, from yeast to vertebrates, Cenp-A localization is insufficient to induce kinetochore assembly. In yeast cells, Cse4, but not the outer kinetochore proteins, is loaded at centromeres in Ipl1 mutants. Similarly, in frog extracts and cells, Cenp-A is properly localized but outer kinetochore proteins fail to assemble in the absence of Aurora B activity. Thus, the second mechanism driving outer kinetochore assembly is the activation of Aurora B, which temporally constrains outer kinetochore assembly to mitosis. Aurora B localizes to the centromere in late prophase, when both Cdk1 and Aurora B are activated. In addition, I-2 levels peak in mitosis, keeping PP1 inactive and preventing disassembly. Because Aurora B is necessary for assembly and is spatially localized to the kinetochore assembly site, we predict that its role will be direct. A large number of kinetochore proteins contain Aurora B phospho-consensus motifs, including several conserved sites on proteins in the Ndc80 and Mis12 complexes. Identification and characterization of kinetochore substrates of Aurora B is an important future direction.
Because inhibition of Aurora B reduced Ndc80 levels at kinetochores in yeast cells, X. laevis cells and extracts, and in most human cells tested, we conclude that the role of Aurora B kinase activity in outer kinetochore assembly is evolutionarily conserved. In HeLa cells, we were unable to affect Ndc80 recruitment to centromeres even after prolonged treatment (24 h) with high concentrations of the Aurora inhibitors hesperadin, ZM447439, and VE-645 (a VX-680 derivative, Aurora B selective, small molecule inhibitor) used alone and in combination (unpublished data). These results are consistent with previously published findings (Hauf et al., 2003; Lampson et al., 2004; Meraldi and Sorger, 2005). It is disconcerting that our results could not be confirmed in HeLa cells. The differences between our results in A549 and PFF cells and those found in HeLa cells by us and others could be attributed to changes that have occurred in the phospho-signaling cascades in this immortalized cell line. Importantly, treatment of human cells with either ZM447439 or hesperadin alone can represses pH3S10 staining but not outer kinetochore assembly, which suggests that only a small amount of Aurora B kinase activity is required for outer kinetochore assembly in human cultured cells.
Are the phenotypes resulting from reduced Aurora B/Ipl1 activity caused by improper kinetochore assembly?
It is important to reexamine the phenotypes of Aurora B/Ipl1 in light of its critical role for assembling kinetochores. Centromeres in ipl1 mutants cluster close to spindle poles and do not oscillate or breathe on the metaphase plate (He et al., 2001; Tanaka et al., 2002). These experiments suggested that Ipl1 was required for kinetochore biorientation because of a role in releasing improperly attached (syntelic) microtubules. The ipl1 phenotype is clearly different than kinetochore null mutants (ndc10-1) where chromosomes randomly float throughout the nucleus (He et al., 2001; Tanaka et al., 2002). Our data suggest that centromeres remain associated with spindle poles in ipl1 mutant cells with little or no outer kinetochore protein at centromeres. Thus, the kinetochore–microtubule attachments in Ipl1 mutants are mediated through some inner kinetochore protein or complex whose centromere association is refractory to Ipl1 activity. Therefore, biorientation may be the role of the outer kinetochore proteins that are recruited by Ipl1 rather than Ipl1 regulating outer kinetochore proteins to generate biorientation.
Interestingly, there is a discrepancy in the literature with respect to the localization of kinetochore proteins using ChIP versus fluorescent imaging. In yeast harboring ipl1 mutants, Ndc80 and Mtw1 appear properly localized at centromeres at the restrictive temperature when assayed by fluorescence (Pinsky et al., 2006; Maure et al., 2007). However, we have shown that these proteins are mislocalized under similar conditions when assayed by ChIP (Fig. 1). Furthermore, the same laboratory has reported that in ipl1 mutants, Dam1 localizes to centromeres by fluorescence imaging but is mislocalized by ChIP (Kang et al., 2001; Cheeseman et al., 2002). These differing results could be a result of many factors, including different mutants, the amount of time cells spend at the restrictive temperature, differing growth conditions, or some inherent, unexplained difference between the sensitivity of ChIP and fluorescent imaging of kinetochore proteins. Moving forward, it will be critical to analyze the kinetochores' assembly status by multiple methods under the conditions used to draw these conclusions. Importantly, a role for Ipl1 in kinetochore assembly and microtubule release is not mutually exclusive.
Poor outer kinetochore assembly can explain the phenotypes of chromosome passenger complex knockdown in tissue culture cells. Overexpression of kinase-dead Aurora B generated prometaphase spindles lacking any sign of K-fiber formation or chromosome congression (Murata-Hori and Wang, 2002b). RNAi knockdown of either Aurora B or INCENP in Drosophila melanogaster cells “abolished the ability of cells to achieve a metaphase chromosome alignment,” generating “anaphase-like spindles with chromosomes distributed along their length” (Adams et al., 2001). Injection of Aurora B antibodies into X. laevis XTC cells generated severe chromosome misalignment defects and the absence of K-fibers, as judged by cold-calcium lysis (Kallio et al., 2002). Aurora B RNAi in human tissue culture cells caused “the absence of kinetochore–microtubule interactions” (Ditchfield et al., 2003). These phenotypes are easily explained by the lack of proper outer kinetochore formation. In fact, it has already been shown that Aurora is required for the affiliation of BubR1, CENP-E, and dynein with centromeres in HeLa cells, where the requirement for Aurora in assembly is diminished. Our results support the hypothesis espoused by Johnson et al. (2004), that Aurora B may provide a priming phosphorylation on specific substrates to both establish and maintain proteins on kinetochores. Corroborating the data presented here, a recent study has also shown that recruitment of the RZZ complex to kinetochores that are not under tension is dependent on Aurora B activity in human cells (Famulski and Chan, 2007). We conclude that the kinase activity of Aurora B is required for proper assembly of outer kinetochore proteins.
Is it reasonable to conclude that Aurora B is required to release improper microtubule attachments in vertebrate cells? Syntelic attachments require Aurora B activity to be resolved in HeLa cells, where we do not see a dramatic effect on kinetochore assembly (Hauf et al., 2003; Lampson et al., 2004). In X. laevis S3 cells, Aurora B is enriched at merotelic kinetochore attachment sites and is required to either prevent or resolve them (Knowlton et al., 2006). These experiments were performed in the same X. laevis S3 cells used in this study but with short 30-min incubations in hesperadin. This treatment generates merotelic microtubule attachments but does not affect Ndc80 levels at kinetochores. We conclude that Aurora B has a role in both building kinetochores and resolving microtubule attachments in vertebrates. However, it is not clear if the role of Aurora B in resolving improper attachments is direct or through the recruitment of other known or unknown outer kinetochore proteins other than Ndc80 and the KMN. In support of the latter hypothesis, treatment of human cells with ZM447439 alone causes to the dissociation of BubR1 and Cenp-E from kinetochores, both of which are important factors in generating productive kinetochore–microtubule attachments. There are likely to be specific substrates that are phosphorylated by Aurora B in response to its multiple roles in mitosis. The identification of the specific kinetochore substrates that control these activities is an important area of future study.
Materials and methods
X. laevis extracts
X. laevis CSF extracts, sperm nuclei, and extract buffers (XB and CSF-XB) were prepared as described previously (Murray, 1991). Kinetochore assembly reactions were initiated by the addition of nocodazole (5 μg/ml final concentration, diluted first in extract; Sigma-Aldrich) and sperm nuclei (∼1,000/μl) to extract prewarmed at room temperature (∼22°C). The PP1 inhibitor I-2 was purified and used as described previously (Satinover et al., 2004). XB + 5 mM CaCl2 was added at a 1:10 dilution to the extract to relieve CSF arrest. (Murray, 1991).
Kinetochore assembly and immunofluorescence
In CSF extracts, kinetochore assembly reactions were incubated at room temperature and processed for immunofluorescence as described previously (Emanuele and Stukenberg, 2007). X. laevis S3 tissue culture cells were incubated with hesperadin or DMSO for 8 h. Wild-type yeast cells and those containing the ipl-1 or sli15-3 temperature-sensitive alleles were grown in synchronous culture at 26°C. Cultures were shifted to the restrictive temperature of 37°C for 3 h, after which ChIPs were preformed as described previously (Kang et al., 2001). In X. laevis S3 cells, hesperadin was added for 8 h and cells were fixed and processed for immunofluorescence.
To immunostain M phase nuclei, extracts were fixed for 10 min by ∼20-fold dilution in BRB80 (80 mM Pipes + 1 mM MgCl2 + 1 mM EGTA, pH 6.8) + 20% glycerol + 0.5% Triton X-100 + 3.7% formaldehyde (Thermo Fisher Scientific) at room temperature. Fixed reactions were layered onto a cushion of BRB80 + 40% glycerol overlaying a poly-l-lysine–coated coverslips (No. 1). The coverslips and cushions were set up in 12- or 24-well cell culture dishes. Nuclei were pelleted onto coverslips on plate holders at 3,000 rpm for 25 min at 18°C in a centrifuge (CR412; Jouan). Cushions were washed with BRB80 and coverslips were postfixed in ice-cold methanol for 5 min, blocked with TBS + 0.1% Tween20 + 5% BSA, and processed using standard immunostaining techniques. The xCenp-A and xCenp-C antibodies were provided by K. Milks and A. Straight (Stanford University, Paolo Alto, CA).
X. laevis tissue culture cells were grown in 66% Leibovitz L-15 media containing 10% fetal calf serum, 1 mM sodium pyruvate, and antibiotics at ∼20°C. Cells were fixed with PHEM buffer containing 0.5% Triton X-100 and 2% paraformaldehyde (Electron Microscopy Sciences) and were immunostained with standard techniques. Primary foreskin fibroblasts were maintained in Iscove's Modified Dulbecco's Medium + 10% fetal bovine serum and the human lung epithelial A549 line was maintained in DME + 10% fetal bovine serum. Cells were treated with hesperadin and/or ZM447439 for 8 h. X. laevis S3 cells were treated with nocodazole for no longer than 15 min before fixation because it can cause detachment of cells. Hesperadin was a gift of N. Kraut (Boehringer Ingelheim, Ingelheim, Germany), ZM447439 was a gift of C. Crafter (AstraZeneca, London, UK), and VE-645 was a gift of J. Pollard (Vertex, Prescot, UK) and C. Buser (Merck, Whitehouse Station, NJ).
Secondary antibodies (goat anti–rabbit, –mouse, or –chicken) coupled to Alexa 488 or 555 were used for detection (Invitrogen). DNA was stained with Hoechst 33342 (Sigma-Aldrich). Primary antibodies are described in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200710019/DC1).
Fluorescence imaging
Fluorescent images were collected with a camera (Cool Snap ES; Photometrics) mounted on a microscope (E600 Eclipse; Nikon) with 60× NA 1.40 or 100× NA 1.40 Plan-Apo oil immersion objectives (Nikon) at room temperature using MetaVue software (MDS Analytical Technologies). Image processing was performed with MetaMorph (MDS Analytical Technologies). Z stacks were collected through specimens and sum projections were analyzed. Comparison of pH3S10 staining was performed by creating a mask around all Hoechst-stained material. The integrated intensity of pH3S10 staining in the masked region was than quantified relative to nuclear stain in the same region. Kinetochore staining was quantified with respect to Cenp-A stain on the same kinetochore, as described previously (Ditchfield et al., 2003). The percentage of nuclei staining positive for kinetochore markers was quantified visually, counting >100 nuclei per experimental condition.
PP1 treatment of nuclei
Kinetochores were assembled on sperm nuclei in extracts containing nocodazole. Extract was diluted 100-fold in CSF-XB + 1 μM okadaic acid, and nuclei were spun onto coverslips through a cushion of BRB80 + 40% glycerol + 0.1 μM okadaic acid. Cushions were washed with CSF-XB and coverslips were blocked in CSF-XB + 5% BSA + 1 μM okadaic acid for 45 min at room temperature. Coverslips were washed once in CSF-XB and turned upside-down in a solution of PP1 enzyme (10 U/ml) or buffer alone (purified and provided by D. Brautigan, University of Virginia, Charlottesville, VA). Coverslips were returned to culture dishes, washed once in CSF-XB, and fixed with BRB80 + 3.7% formaldehyde for 15 min. Reactions were postfixed in ice-cold methanol and processed for immunofluorescence. PP1 enzyme was purified and used as described previously (Brautigan et al., 1985).
Online supplemental material
Fig. S1 demonstrates that Aurora B is activated in the nucleus, before kinetochore assembly, during prophase. Fig. S2 shows that ZM447439 and hesperadin additively inhibit the kinase activity of Aurora B in vitro. Fig. S3 shows that Aurora B inhibition, using hesperadin, blocks spindle checkpoint signaling in X. laevis egg extracts. Fig. S4 shows that the addition of hesperadin to egg extracts does not affect the CSF-controlled cell cycle state of the extract. Fig. S5 demonstrates that the kinetochore proteins CLIP-170, Cep57, and p150/Glued require Aurora B kinase activity for their maintenance at preassembled kinetochores. Table S1 describes all antibodies used in this study. Table S2 describes all yeast strains generated for and used in yeast ChIP experiments. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710019/DC1.
Supplemental Material
Acknowledgments
We thank Courtney Kestner and John Tooley for generating and characterizing antibody reagents; David Brautigan for providing PP1 enzyme; Norbert Kraut for generously providing hesperadin; Claire Crafter for ZM447439; John Pollard and Carolyn Buser for VE-465; Dan Burke for critical manuscript revisions; Iain Cheeseman for helpful conversations; and the Burke and Stukenberg laboratory members for helpful criticisms during the course of this study. Special thanks to Aaron Straight and Kristin Milks for generously sharing with us X. laevis–specific Cenp-A and Cenp-C antibodies.
This work was supported by grants from the American Cancer Society (RSG-04-021-01-CCG) and the National Institutes of Health (5R01GM063045-06).
Abbreviations used in this paper: ACA, anti-centromere antigen; Cenp, centromere protein; ChIP, chromatin immunoprecipitation; CSF, cytostatic factor; I-2, Inhibitor-2; INCENP, inner centromere protein; PP1, protein phosphatase 1.
References
- Adams, R.R., H. Maiato, W.C. Earnshaw, and M. Carmena. 2001. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J. Cell Biol. 153:865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins, S., and A.W. Murray. 2001. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 15:3118–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigan, D.L., C.L. Shriner, and P.A. Gruppuso. 1985. Phosphorylase phosphatase catalytic subunit. Evidence that the Mr = 33,000 enzyme fragment is derived from a native protein of Mr = 70,000. J. Biol. Chem. 260:4295–4302. [PubMed] [Google Scholar]
- Brautigan, D.L., J. Sunwoo, J.C. Labbe, A. Fernandez, and N.J. Lamb. 1990. Cell cycle oscillation of phosphatase inhibitor-2 in rat fibroblasts coincident with p34cdc2 restriction. Nature. 344:74–78. [DOI] [PubMed] [Google Scholar]
- Cheeseman, I.M., S. Anderson, M. Jwa, E.M. Green, J. Kang, J.R. Yates III, C.S. Chan, D.G. Drubin, and G. Barnes. 2002. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell. 111:163–172. [DOI] [PubMed] [Google Scholar]
- Cheeseman, I.M., S. Niessen, S. Anderson, F. Hyndman, J.R. Yates III, K. Oegema, and A. Desai. 2004. A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev. 18:2255–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman, I.M., J.S. Chappie, E.M. Wilson-Kubalek, and A. Desai. 2006. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 127:983–997. [DOI] [PubMed] [Google Scholar]
- Cimini, D., X. Wan, C.B. Hirel, and E.D. Salmon. 2006. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr. Biol. 16:1711–1718. [DOI] [PubMed] [Google Scholar]
- Deluca, J.G., B. Moree, J.M. Hickey, J.V. Kilmartin, and E.D. Salmon. 2002. hNuf2 inhibition blocks stable kinetochore-microtubule attachment and induces mitotic cell death in HeLa cells. J. Cell Biol. 159:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluca, J.G., W.E. Gall, C. Ciferri, D. Cimini, A. Musacchio, and E.D. Salmon. 2006. Kinetochore microtubule dynamics and attachment stability are regulated by hec1. Cell. 127:969–982. [DOI] [PubMed] [Google Scholar]
- Ditchfield, C., V.L. Johnson, A. Tighe, R. Ellston, C. Haworth, T. Johnson, A. Mortlock, N. Keen, and S.S. Taylor. 2003. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 161:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele, M.J., and P.T. Stukenberg. 2007. Xenopus cep57 is a novel kinetochore component involved in microtubule attachment. Cell. 130:893–905. [DOI] [PubMed] [Google Scholar]
- Emanuele, M.J., M.L. McCleland, D.L. Satinover, and P.T. Stukenberg. 2005. Measuring the stoichiometry and physical interactions between components elucidates the architecture of the vertebrate kinetochore. Mol. Biol. Cell. 16:4882–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulski, J.K., and G.K. Chan. 2007. Aurora B kinase-dependent recruitment of hZW10 and hROD to tensionless kinetochores. Curr. Biol. 17:2143–2149. [DOI] [PubMed] [Google Scholar]
- Francisco, L., W. Wang, and C.S. Chan. 1994. Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol. Cell. Biol. 14:4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillett, E.S., C.W. Espelin, and P.K. Sorger. 2004. Spindle checkpoint proteins and chromosome-microtubule attachment in budding yeast. J. Cell Biol. 164:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf, S., R.W. Cole, S. LaTerra, C. Zimmer, G. Schnapp, R. Walter, A. Heckel, J. van Meel, C.L. Rieder, and J.M. Peters. 2003. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X., D.R. Rines, C.W. Espelin, and P.K. Sorger. 2001. Molecular analysis of kinetochore-microtubule attachment in budding yeast. Cell. 106:195–206. [DOI] [PubMed] [Google Scholar]
- Hsu, J.Y., Z.W. Sun, X. Li, M. Reuben, K. Tatchell, D.K. Bishop, J.M. Grushcow, C.J. Brame, J.A. Caldwell, D.F. Hunt, et al. 2000. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 102:279–291. [DOI] [PubMed] [Google Scholar]
- Johnson, V.L., M.I. Scott, S.V. Holt, D. Hussein, and S.S. Taylor. 2004. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 117:1577–1589. [DOI] [PubMed] [Google Scholar]
- Kallio, M.J., M.L. McCleland, P.T. Stukenberg, and G.J. Gorbsky. 2002. Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr. Biol. 12:900–905. [DOI] [PubMed] [Google Scholar]
- Kang, J., I.M. Cheeseman, G. Kallstrom, S. Velmurugan, G. Barnes, and C.S. Chan. 2001. Functional cooperation of Dam1, Ipl1, and the inner centromere protein (INCENP)-related protein Sli15 during chromosome segregation. J. Cell Biol. 155:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyomitsu, T., C. Obuse, and M. Yanagida. 2007. Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev. Cell. 13:663–676. [DOI] [PubMed] [Google Scholar]
- Kline, S.L., I.M. Cheeseman, T. Hori, T. Fukagawa, and A. Desai. 2006. The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J. Cell Biol. 173:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton, A.L., W. Lan, and P.T. Stukenberg. 2006. Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr. Biol. 16:1705–1710. [DOI] [PubMed] [Google Scholar]
- Lampson, M.A., K. Renduchitala, A. Khodjakov, and T.M. Kapoor. 2004. Correcting improper chromosome-spindle attachments during cell division. Nat. Cell Biol. 6:232–237. [DOI] [PubMed] [Google Scholar]
- Liu, S.T., J.B. Rattner, S.A. Jablonski, and T.J. Yen. 2006. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J. Cell Biol. 175:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiato, H., J. DeLuca, E.D. Salmon, and W.C. Earnshaw. 2004. The dynamic kinetochore-microtubule interface. J. Cell Sci. 117:5461–5477. [DOI] [PubMed] [Google Scholar]
- Maure, J.F., E. Kitamura, and T.U. Tanaka. 2007. Mps1 kinase promotes sister-kinetochore bi-orientation by a tension-dependent mechanism. Curr. Biol. 17:2175–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAinsh, A.D., P. Meraldi, V.M. Draviam, A. Toso, and P.K. Sorger. 2006. The human kinetochore proteins Nnf1R and Mcm21R are required for accurate chromosome segregation. EMBO J. 25:4033–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland, M.L., R.D. Gardner, M.J. Kallio, J.R. Daum, G.J. Gorbsky, D.J. Burke, and P.T. Stukenberg. 2003. The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev. 17:101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland, M.L., M.J. Kallio, G.A. Barrett-Wilt, C.A. Kestner, J. Shabanowitz, D.F. Hunt, G.J. Gorbsky, and P.T. Stukenberg. 2004. The vertebrate Ndc80 complex contains Spc24 and Spc25 homologs, which are required to establish and maintain kinetochore-microtubule attachment. Curr. Biol. 14:131–137. [DOI] [PubMed] [Google Scholar]
- Meraldi, P., and P.K. Sorger. 2005. A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J. 24:1621–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata-Hori, M., and Y.L. Wang. 2002. a. The kinase activity of aurora B is required for kinetochore-microtubule interactions during mitosis. Curr. Biol. 12:894–899. [DOI] [PubMed] [Google Scholar]
- Murata-Hori, M., and Y.L. Wang. 2002. b. The kinase activity of aurora B is required for kinetochore-microtubule interactions during mitosis. Curr. Biol. 12:894–899. [DOI] [PubMed] [Google Scholar]
- Murray, A.W. 1991. Cell cycle extracts. Methods Cell Biol. 36:581–605. [PubMed] [Google Scholar]
- Musacchio, A., and E.D. Salmon. 2007. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8:379–393. [DOI] [PubMed] [Google Scholar]
- Obuse, C., O. Iwasaki, T. Kiyomitsu, G. Goshima, Y. Toyoda, and M. Yanagida. 2004. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat. Cell Biol. 6:1135–1141. [DOI] [PubMed] [Google Scholar]
- Pinsky, B.A., C. Kung, K.M. Shokat, and S. Biggins. 2006. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat. Cell Biol. 8:78–83. [DOI] [PubMed] [Google Scholar]
- Satinover, D.L., C.A. Leach, P.T. Stukenberg, and D.L. Brautigan. 2004. Activation of Aurora-A kinase by protein phosphatase inhibitor-2, a bifunctional signaling protein. Proc. Natl. Acad. Sci. USA. 101:8625–8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, D.J., G.C. Rogers, and J.M. Scholey. 2000. Cytoplasmic dynein is required for poleward chromosome movement during mitosis in Drosophila embryos. Nat. Cell Biol. 2:922–930. [DOI] [PubMed] [Google Scholar]
- Starr, D.A., B.C. Williams, T.S. Hays, and M.L. Goldberg. 1998. ZW10 helps recruit dynactin and dynein to the kinetochore. J. Cell Biol. 142:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, T.U., N. Rachidi, C. Janke, G. Pereira, M. Galova, E. Schiebel, M.J. Stark, and K. Nasmyth. 2002. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 108:317–329. [DOI] [PubMed] [Google Scholar]
- Tung, H.Y., W. Wang, and C.S. Chan. 1995. Regulation of chromosome segregation by Glc8p, a structural homolog of mammalian inhibitor 2 that functions as both an activator and an inhibitor of yeast protein phosphatase 1. Mol. Cell. Biol. 15:6064–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge, P.A., and J.V. Kilmartin. 2001. The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J. Cell Biol. 152:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen, T.J., D.A. Compton, D. Wise, R.P. Zinkowski, B.R. Brinkley, W.C. Earnshaw, and D.W. Cleveland. 1991. CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J. 10:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., W. Lan, S.C. Ems-McClung, P.T. Stukenberg, and C.E. Walczak. 2007. Aurora B phosphorylates multiple sites on MCAK to spatially and temporally regulate its function. Mol. Biol. Cell. 18:3264–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}