

Abstract
The “3-color, 46-bead” model of a folding polypeptide is the vehicle for adapting to proteins a mode of analysis used heretofore for atomic clusters, to relate the topography of the potential surface to the dynamics that lead to formation of selected structures. The analysis is based on sequences of stationary points—successive minima, joined by saddles—that rise monotonically in energy from basin bottoms. Like structure-seeking clusters, the potential surface of the model studied here is staircase-like, rather than sawtooth-like, with highly collective motions required for passage from one minimum to the next. The surface has several deep basins whose minima correspond to very similar structures, but which are separated by high energy barriers.
Keywords: folding pathways, stable structures, β-barrel
A challenge to chemical theory has been finding a way to infer from attainable data at the atomic level why some systems readily form glasses and others fall into very selective structures. It is possible to do this now for atomic clusters by examining sequences of linked stationary points on the potential surface, particularly sequences whose minima rise monotonically from the bottoms of basins on the surface (1–4). The signature of a glass-former seems to be a complex potential with a sawtooth topography—a potential whose successive minima differ little in energy, relative to the heights of the energy barriers that separate them. In contrast, the signature of a “structure-seeker” appears to be a complex potential with a staircase topography—a potential some of whose adjacent minima differ considerably in energy, relative to the energy barrier between them. The mechanistic difference between the two, at the atomic level, is that only a very few particles move when a glass-former passes from one local minimum to the next, whereas in a structure-seeker, many particles move in the well-to-well passages, and in some of these, the internal potential energy changes considerably. Examples of glass-formers are such rare-gas clusters as Ar19 and Ar55 (2, 3, 5); an example of a good structure-seeker is (KCl)32, which, when quenched (in molecular dynamics simulations) from liquid, finds one of its few hundred rocksalt structures rather than one of its (roughly) 1013 amorphous structures (6). Only if the cluster is quenched at a rate above 1013 K/s—i.e., if the energy is removed during only 5–10 vibrational periods—can this system be trapped in an amorphous structure (6, 7).
Even as the results emerged for clusters, it was apparent that the same characteristics might govern the structure-seeking propensities of proteins and other biopolymers. The interatomic forces in proteins differ very much indeed from those between the atoms of clusters, but if the characterization is correct, that difference is irrelevant to the generic issue of why some species are good structure-seekers and others are good glass-formers. The crucial issue is the topography of the potential surface, not the microscopic origin of the forces that give rise to that topography. We therefore undertook an analysis of the topography of the potential surface of a simple prototype, a continuously movable, “3-color, 46-bead” model that had been developed and examined previously (8–11), which in turn is related to a lattice model (12, 13). The present study has pursued the following questions. (i) To what extent is the topography of the potential surface of this system sawtooth-like or staircase-like, and to what extent is this answer consistent with the inference from clusters that the former is associated with few-particle motions in well-to-well passage and the latter, with highly collective motions? (ii) What is the large-scale basin structure of the potential landscape of this model system and to what structures do the basin minima correspond? (iii) How are the kinetics and mechanism of relaxation, folding, and interbasin passage related to the topography of the surface?
A fourth question looms ahead, but is not addressed here, except in speculations arising from the answers, such as they are now, to the first three: how is this model related to real proteins?
The force field and units we have used are almost the same as those of Thirumalai et al. (8–11). We did replace rigid bonds with stiff but harmonic, spring-like bonds. The model consists of hydrophilic beads (L), hydrophobic beads (B), and neutral beads (N). The sequence, in terms of these beads, is B9N3(LB)4N3B9N3(LB)5L, so there are four strands, two consisting of only hydrophobic beads and two of alternating hydrophobic and hydrophilic beads. The strands are joined by short segments of neutral beads that accommodate the folding of the strands together because of their very low torsional forces. This object has, as its lowest-energy structure, a β-barrel in which the four strands are parallel (or antiparallel). The effects of an aqueous solvent are subsumed in the parameters of the model that put the hydrophilic beads on the outside of the outer chains in the low-energy structures. Temperature is meaningful only in terms of the units of ɛ, the well-depth parameter of the pair interactions, which has a value 0.01 eV or 121 K, in this model.
The terminology of the discussion is this paper is as in refs. 2 and 14: all local minima are merely called “minima;” stationary points on the barriers separating local minima are saddles. The lowest points in sequences of minima monotonic in energy are called “basin bottoms,” and the sets of such monotonic sequences that lead to the same basin bottom define basins on the potential surface. The saddles that link monotonic sequences in adjacent basins are called “divides.” The basin containing the global minimum is called the “primary basin;” a basin separated by one divide from the primary basin is a “secondary basin,” and so forth. Within the variety of basins we consider, some might be wide at high energies and either wide or very constricted at their bottoms. Hence we prefer to impose no prejudgment on the form of the topographies of the surfaces, and therefore, at least at this stage of the analysis, use the more general term “basin” rather than “funnel,” which carries a vivid pictorial implication (see refs. 15 and 16).
We located minima and saddles largely by a variant of the conjugate peak refinement method (17) and the ridge method (18) supplemented with eigenvector-following (19–21) as implemented in the program optim (22), and with the slowest-slides method (23). We also used molecular dynamics simulations, both to help us find interwell saddles and interbasin divides and to study the dynamics of relaxation and folding in this system. The dynamics calculations used the velocity Verlet algorithm. The density of locally stable states was computed by the binning method applied previously to the (KCl)32 cluster (6). The logarithm, ln[G(E)], of this density rises linearly with energy from zero at the global minimum up to a rounded peak at 38, at a value of approximately 35 ɛ, where ɛ is the energy parameter in the model (8–11). At higher energies, the density of stable states drops; the states in the high-energy range are not simply the highly extended states. There are many contorted but locally stable forms, some with energies well above the energies of the most extended structures.
The collectivity index developed by Wales et al. (22) from the normed quartic moment of Stillinger and Weber (24) shows the effective number of particles moving when the system passes from one minimum to the next in a sequence. Several monotonic sequences connected to the global minimum structure are shown in Fig. 1a; Fig. 1b shows sequences to another minimum. Collectivity indices are given for each step of one of the sequences in each panel. This figure makes it evident that the topography of this surface is indeed staircase-like, and that the motions are highly collective. In short, this simplified protein model fits unambiguously into the pattern associated with structure-seekers, consistent with the strong folding tendency the model was constructed to have (8–10). This is the first point we wish to make, and is the one with the most general implications regarding structure-seeking species: that they appear to be characterized by staircase topographies and highly collective motions in their well-to-well passages.
Figure 1.

Typical sequences of minima, with their linking saddles, rising monotonically from basin bottoms. (a) Sequences from the global minimum (primary basin). (b) Sequences from the third structure shown in the following figure (secondary basin). The numbers shown for one example in each panel are collectivity indices, the effective number of particles that move as the system passes from one local minimum to the next, along the sequences shown as solid lines.
Next, we turn to the second question. It was known that this model has at least eight somewhat similar barrel structures (8–11), in addition to and different from those that involve only small rearrangements of the flexible regions of the global minimum structure. We have explored the topography associated with these structures in some detail to determine energy barriers between the lower-lying basin bottoms. Fig. 2 shows the global minimum structure and two other similar structures with their energies, and typical sequences of minima that link them. At first glance, these may appear extremely like one another, as their energies might suggest. However close examination reveals certain important differences. In the global minimum structure as displayed, the first bead, no. 1, is in the same horizontal plane as beads 21, 23, and 45. In the structure with energy −0.5344 eV, bead 1 is coplanar with beads 21–24 and 45. That is, these two structures differ by a half-cycle, in the relative positions of the two purely hydrophobic chains containing beads 1 and 24. The structure with energy −0.5350 eV has beads 1, 20, 24, and 44 coplanar, so that its mixed hydrophobe-hydrophil chains are shifted a half-cycle relative to the first two structures. Likewise, another deep minimum and basis bottom is the structure not shown here, in which beads 1 and 25 are matched, corresponding to a full cycle shift from the global minimum structure.
Figure 2.

Structures and energies of the global minimum geometry and two other low-lying, basis-bottom structures of this model, and of the saddles that link them; beneath each “side” view is an “end-on” view, showing how passage between one basin bottom and another occurs by screw-like rotation and translation of one of the strands.
Fig. 2 also shows structures and energies at divides that separate the basins of these structures. We can only say that these are the lowest-energy divides that we have found and hence are upper limits to the activation energies for basin crossing. On surfaces of the complexity of these, it would be a considerable task to determine with certainty the pathway of lowest energy between basin bottoms. Even finding the minimum-energy path between adjacent minima is nontrivial in spaces of high dimensionality (132 independent variables in this instance), and the number of possible paths must be very high indeed. Nonetheless, to find alternatives to the saddle structures shown in Fig. 2, we have tried starting the system along several possible mechanistic paths, such as introducing a kink that might propagate along a chain. No such alternative yielded any divide with an energy as low as those shown. Unless there are divides much lower than any we have found by any of the methods we have used, we can say that the energies of the interbasin divides separating the low-lying structures are very high, much higher than typical saddles between adjacent wells on this surface. A model protein must surmount a rather rough, high hill, climbing several steep faces along the way, to pass from one of those structures to another.
The “mechanisms”—i.e., lowest-energy pathways—we have found for the interbasin pathways have an elegant simplicity. (We reiterate that these need not be the lowest-energy pathways for the system; they are only the lowest we have found.) In the examples shown in Fig. 2, one strand of hydrophobes rotates, screw-like, through approximately 180° as it moves a half-cycle along the other chain of hydrophobes. The “side” views of the structures in Fig. 2 give an indication of how this happens. It is probably easier to infer the precise pathway from the “end-on” views of Fig. 2, where the hydrophobic chains are clearly approximately aligned with their long axes nearly parallel but with a relative azimuthal angle of about 90° in the saddle structures but a relative azimuth of 0° in the low-energy structures. Animations of these motions are available on the World Wide Web at the URL <http://rainbow.uchicago.edu/∼nelmaci>. One can think of these processes as collective edge-crossing processes.
The topographical analyses derived by locating sequences of minima and saddles suggest, and isothermal Molecular Dynamics simulations of the dynamics of relaxation confirm, that the energy scale of configurations of this polymer model may be divided roughly into four regions. The structures corresponding to deep local minima (with E < −0.50 eV), are generated mainly by two types of distortions of the global minimum structure (E = −0.536 eV). The first, associated with low activation energy, involves deformations of the bending (neutral) regions of the chain. These lie deep in the primary basin of the global minimum structure. The structures of the second type are those barrels described previously that, relative to the global minimum, require translational shifts of different strands of the folded polymer. These structures, separated from the global minimum by high potential barriers, are linked dynamically with the global minimum and with each other by rotation-translation rearrangements of the kind mentioned above. We have been unable to find any evidence in the Molecular Dynamics simulations for propagation of a “kink,” or for any simple sliding or reptation of one part of the chain along another. Next in energy (−0.50 < E < −0.25 eV) are configurations generated by changes of dihedral angles within either a hydrophobic or an alternating hydrophobe–hydrophil chain, that bends one of the chains of the folded polymer into a short hairpin or into a partially “disordered” (“glassy” or “melted”) kernel. Configurations at still higher energy (−0.25 < E < −0.10 eV) exhibit partial unfolding; typically the unfolding starts by separation, almost tearing off, the “38–46” mixed hydrophobe–hydrophil tail from the rest of the barrel. Almost totally unfolded configurations have energies above −0.1 eV. In contrast to the situation with clusters, the density of locally stable states of polymers (barring dissociation) has at least two local minima: one, as in clusters, for the global minimum, and at least one other at quite high energy, where the structures are mostly extended. (The most stretched structure of this model is not the locally stable structure of highest energy.) For that reason it is improbable to find the polymer in highly extended configurations even at rather high temperatures.
Now we turn to the third question, the dynamics and relaxation. We have simulated the isothermal structural relaxation of vibrationally cold polymers (with mean kinetic energies corresponding to temperatures < 70 K for bead masses of 40 atomic units) that have, initially, configurationally hot, unfolded structures (with typical configuration energies 0.0–0.3 eV). During the relaxation the polymer collapses very rapidly (t < 10−10 sec or < 20,000 time steps) to one of the partly folded configurations with a distorted hydrophobic kernel (E ≈ −0.2 to −0.3 eV). After this first stage, if the vibrational temperature is low (<30 K), the system may stay trapped at this configuration (Fig. 3a, T = 15). As this figure shows, at higher temperatures (30 < T < 40 K), the polymer continues, but much slower, equilibrating thermally as it goes, along its way downhill to more stable and more ordered structures whose energies are approximately −0.40 to −0.47 eV; some of these have one misfolded chain, and others, a deformed kernel. So long as the temperature is below about 50 K, the system, starting from a high-energy structure, is very unlikely to find the global minimum or any other of the deep structures with energies below about −0.51 eV. The thermal equilibration suggests that linear, Markovian master equations with rate coefficients based on standard, thermalized kinetic models are likely to be adequate to describe the kinetics of folding (2, 4, 14, 25, 26). Temperatures between about 50 and 55 K are the most favorable for relaxing to the global minimum (Fig. 3b shows a trajectory for 55 K); even so, most examples visit many structures at higher energies, within the interval of 1–2 × 106 time steps interval of these simulations. At slightly higher temperatures (>55 K) the entropy contribution to the free energy changes the state occupancies in favor of metastable configurations with E ≈ −0.45eV; at these temperatures, the polymer occasionally visits but never stays at or near the global minimum. The history for 62 K in Fig. 3b illustrates this behavior. Thus for this type of isothermal relaxation there appears to be only a rather narrow “temperature window” for optimal isothermal relaxation to the global minimum.
Figure 3.

Time histories of quenched energies along typical molecular dynamics pathways taken in the folding process, showing (a) relaxation and descent when the vibrational temperature is relatively low; (b) two isothermal histories showing descent but followed by failure to remain in a single well, if the temperature is relatively high, and a contrasting example of an annealing history from approximately the same initial condition; and (c) three typical annealing histories, each leading to a different deep basin bottom. The time scale is of course scaled; if the masses of all the particles are set at m = 40 da, one time step corresponds to 1 × 10−14 s; the scale increases as 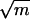 .
.
A much easier, more forgiving way to induce the system to relax to the global minimum is to apply a gradual decrease of temperature—i.e., to carry out an annealing process—so that the temperature remains high enough in the middle of relaxation to prevent trapping at intermediate configurations, and then low enough at the end to make the global minimum heavily populated. However, even this is not sufficient to bring all the systems of an ensemble based on this 46-bead model to the global minimum and remain there. We have simulated annealing from a high temperature of ≈55 K to low final temperatures between 12 and 25 K, thus far only with cooling schedules linear over 500,000 time-step intervals. The system seems almost always to find its way to one or another of the deep, β-barrel basin bottoms, but only about as often to the global minimum as to one or another of the other deep minima. This is consistent with the notion that a system that has started its way down a staircase potential into a deep basin is very likely to progress all the way to the bottom of that basin. One such history for 53 to 18 K is included in Fig. 3b; three more that reach the bottoms of different basins are shown in Fig. 3c. Many other possibilities occur. In one annealing run from a very high initial energy, the system ended in a basin with energy of approximately −0.46 eV with only 3.5 “properly” folded chains and one hydrophobe-hydrophil chain bent 180° at its midpoint.
These observations bring us to a set of inferences, I-i–I-v, and to a set of questions, Q-i–Q-v. The inferences are as follows. (I-i) The readiness of this model to fold into its β-barrel arises from the same considerations that make some clusters efficient structure-seekers—i.e., that the topography of the potential surface is staircase-like. (I-ii) That, again like clusters, the microscopic origin of the staircase topography is the highly collective character of the well-to-well steps. (I-iii) The topography of the potential surface of this system is analogous to a high, rolling plane with at least eight large craters that drop precipitously from that plane, each with a few stable local minima in its bottom. (I-iv) any system that finds its way over the rim of a crater drops into that crater and makes its way to the structures in the vicinity of the basin bottom, unless the temperature is high enough to allow the system eventually to climb back out. (I-v) Corresponding to the large perimeters of the crater rims, the polymers may start their way down a crater in many different ways—i.e., by nucleating in many different ways, again analogous to the behavior of structure-seeking clusters.
Some of the questions raised by these results are the following. (Q-i) Can one estimate the hyper-lengths of the perimeter rims and infer from them the likelihood that the system will fold into each particular structure as a result of its relaxation? Can we replace the unrealistic odds of the Levinthal paradox with numbers that reflect the likelihood of a system finding a jumping-off point for entering a particular crater and, thereby, almost inevitably reaching the structure corresponding to the bottom of that crater? (Q-ii) Do any real polypeptides behave at all like this model, in the sense of having several similar, low-lying structures separated by high barriers, and, if so, may two or more of these play similar active functional roles in vivo? (Q-iii) Are real proteins similar to this model with respect to the narrowness of the temperature window in which the system can find its way to a native structure? (Q-iv) If their potential surfaces do have such multiple basins, do real polypeptides pass from one basin to another by the screw-like motion exhibited by this model? Or do side chains complicate the potential surfaces so much that the simplicity of this motion is lost—or even that the multiplicity of similar deep minima is lost? Because the bonds to the side chains in β-sheets lie out of and roughly perpendicular to the planes of the sheets, the side chains may well not affect the stable structures very much, but may greatly change the reaction paths linking different basins (27). (Q-v) Can master equations based on statistical samples of sequences of linked minima and saddles give reliable representations of the kinetics of the folding process, analogous to the way such equations can represent the kinetics of (at least small) clusters (4)?
Answering these questions will, we hope, help provide a clearer understanding of how biopolymers and other complex systems find their ways to their function-specific structures.
Acknowledgments
N.E. would like to thank the Scientific and Technical Research Council of Turkey for the fellowship that enabled her to participate in this work. We would all like to acknowledge the support of the National Science Foundation for this research.
References
- 1.Ball K D, Berry R S, Proykova A, Kunz R E, Wales D J. Science. 1996;271:963–966. [Google Scholar]
- 2.Kunz R E, Berry R S. J Chem Phys. 1995;103:1904–1912. [Google Scholar]
- 3.Kunz R E, Berry R S, Astakhova T. Surf Rev Lett. 1996;3:307–312. [Google Scholar]
- 4.Kunz, R. E., Blaudeck, P., Hoffmann, K. H. & Berry, R. S. (1997), in press.
- 5.Vekhter B, Ball K D, Rose J, Berry R S. J Chem Phys. 1997;106:4644–4650. [Google Scholar]
- 6.Rose J P, Berry R S. J Chem Phys. 1993;98:3262–3274. [Google Scholar]
- 7.Cheng H-P, Landman U. Science. 1993;260:1304–1307. doi: 10.1126/science.260.5112.1304. [DOI] [PubMed] [Google Scholar]
- 8.Honeycutt J D, Thirumalai D. Proc Natl Acad Sci USA. 1990;87:3526–3529. doi: 10.1073/pnas.87.9.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Honeycutt J D, Thirumalai D. Biopolymers. 1992;32:695–709. doi: 10.1002/bip.360320610. [DOI] [PubMed] [Google Scholar]
- 10.Guo Z, Thirumalai D, Honeycutt J D. J Chem Phys. 1992;97:525–535. [Google Scholar]
- 11.Guo Z, Thirumalai D. In: Energy Landscape and Folding Mechanisms in Proteins. Bohr H, Brunak S, editors. Cleveland: CRC; 1995. pp. 233–239. [Google Scholar]
- 12.Skolnick J, Kolinski A, Yaris R. Proc Natl Acad Sci USA. 1989;85:5057–5061. doi: 10.1073/pnas.85.14.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skolnick J, Kolinski A, Yaris R. Biopolymers. 1989;28:1059–1095. doi: 10.1002/bip.360280604. [DOI] [PubMed] [Google Scholar]
- 14.Berry R S, Breitengraser-Kunz R E. Phys Rev Lett. 1995;74:3951–3954. doi: 10.1103/PhysRevLett.74.3951. [DOI] [PubMed] [Google Scholar]
- 15.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onuchic J N, Socci N D, Luthey-Schulten Z, Wolynes P G. Folding Design. 1996;1:441–450. doi: 10.1016/S1359-0278(96)00060-0. [DOI] [PubMed] [Google Scholar]
- 17.Fischer S, Karplus M. Chem Phys Lett. 1992;194:252–261. [Google Scholar]
- 18.Ionova I V, Carter E A. J Chem Phys. 1993;98:6377–6386. [Google Scholar]
- 19.Pancik J. Coll Czech Chem Commun. 1975;40:1112–1118. [Google Scholar]
- 20.Hilderbrandt R L. Comput Chem. 1977;1:179–186. [Google Scholar]
- 21.Cerjan C J, Miller W H. J Chem Phys. 1981;75:2800–2806. [Google Scholar]
- 22.Wales D J, Popelier P L A, Stone A J. J Chem Phys. 1995;102:5551–5565. [Google Scholar]
- 23.Davis H L, Wales D J, Berry R S. J Chem Phys. 1990;92:4473–82. [Google Scholar]
- 24.Stillinger F H, Weber T A. Phys Rev A. 1983;28:2408–2416. [Google Scholar]
- 25.Zwanzig R. Proc Natl Acad Sci USA. 1995;92:9801–9804. doi: 10.1073/pnas.92.21.9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker O, Karplus M. J Chem Phys. 1997;106:1495–1517. [Google Scholar]
- 27.Smith C K, Regan L. Science. 1997;30:153–161. [Google Scholar]