

Abstract
To date, the glutamate-glutamine cycle has been the dominant paradigm for understanding the coordinated, compartmentalized activities of phosphate-activated glutaminase (PAG) and glutamine synthetase (GS) in support of functional glutamate trafficking in vivo However, studies in cell cultures have repeatedly challenged the notion that functional glutamate trafficking is accomplished via the glutamate-glutamine cycle alone. The present study introduces and elaborates alternative cycles for functional glutamate trafficking that integrate glucose metabolism, glutamate anabolism, transport, and catabolism, and trafficking of TCA cycle intermediates from astrocytes to presynaptic neurons. Detailed stoichiometry for each of these alternative cycles is established by strict application of the principle of conservation of atomic species to cytosolic and mitochondrial compartments in both presynaptic neurons and astrocytes. In contrast to the glutamate-glutamine cycle, which requires ATP, but not necessarily oxidative metabolism, to function, cycles for functional glutamate trafficking based on intercellular transport of TCA cycle intermediates require oxidative processes to function. These proposed alternative cycles are energetically more efficient than, and incorporate an inherent mechanism for transporting nitrogen from presynaptic neurons to astrocytes in support of the coordinated activities of PAG and GS that is absent in, the glutamate-glutamine cycle. In light of these newly elaborated alternative cycles, it is premature to presuppose that functional glutamate trafficking in synaptic neurotransmission in vivo is sustained by the glutamate-glutamine cycle alone.
Keywords: Brain metabolism, glutamate-glutamine cycle, glutamate trafficking, synaptic function
Introduction
Functional glutamate trafficking in synaptic neurotransmission is fundamental to human brain function. The vast majority of excitatory synapses in the human brain employ glutamate as an excitatory neurotransmitter (Nicholls, 1993). Researchers today generally accept that the glutamate-glutamine cycle (Van den Berg and Garfinkel, 1971; Benjamin and Quastel, 1975), i.e., conversion of glutamine to glutamate in presynaptic neurons, trafficking of neurotransmitter glutamate from presynaptic neurons to astrocytes, conversion of glutamate to glutamine in astrocytes, and trafficking of non-neuroactive glutamine from astrocytes to presynaptic neurons, plays a major role in functional glutamate trafficking in vivo. Consistent with, and in support of, this view, nuclear magnetic resonance studies of brain metabolism and functional glutamate trafficking in vivo suggest that the glutamate-glutamine cycle is a major metabolic pathway in the cerebral cortex (Gruetter et al., 1998; Sibson et al., 1998; Shen et al., 1999; Gruetter et al., 2001; Lebon et al. 2002) and that its rate increases in proportion to increases in neuronal glucose oxidation (Sibson et al., 1998; Patel et al., 2004; Oz et al. 2004).
Despite its widespread acceptance, the glutamate-glutamine cycle may not be sufficient to explain functional glutamate trafficking in vivo in its entirety. The basic premise of the glutamate-glutamine cycle is that astrocytes recycle neurotransmitter glutamate in the form of glutamine. Consistent with this basic premise, astrocytes convert exogenous glutamate to endogenous glutamine which is released giving rise to exogenous glutamine in culture (Waniewski and Martin, 1986) and clear neurotransmitter glutamate from synaptic clefts in vivo (Rothstein et al., 1996). However, the glutamate-glutamine cycle depends on a major, yet presently unexplained, intercellular flux of ammonium from neurons to astrocytes associated with the deamidation of glutamine to form glutamate in presynaptic terminals and the amidation of glutamate to form glutamine in astrocytes (Benjamin and Quastel, 1975; Yudkoff et al., 1996; Hutson et al., 1998; Waagepetersen et al., 2000a; Marcaggi and Coles, 2001). Therefore, the extent to which the glutamate-glutamine cycle sustains functional glutamate trafficking in vivo is constrained by intercellular transport of ammonium from neurons to astrocytes. Furthermore, studies in cell culture also suggest that astrocytes degrade glutamate by means of oxidative processes (Yu et al., 1982; Sonnewald et al., 1993; McKenna et al., 1996a; Peng et al., 2001; Waagepetersen et al., 2002; Hertz and Hertz, 2003). If astrocytes degrade neurotransmitter glutamate by oxidative processes in vivo, then this would deplete the pool of neurotransmitter glutamate available to be recycled by means of the glutamate-glutamine cycle, and thereby undermine its ability to sustain functional glutamate trafficking in vivo. Are there alternative cycles for functional glutamate trafficking in vivo that are consistent not only with the premise that astrocytes recycle neurotransmitter glutamate in the form of glutamine but also with the possibility that astrocytes degrade neurotransmitter glutamate by means of oxidative processes? If so, do these alternative cycles explain how presynaptic terminals effectively transport ammonium to astrocytes in support of neurotransmitter glutamate recycling in the form of glutamine?
Neurons and astrocytes differ with respect to the expression of glutamine synthetase (GS), and likely differ with respect to the function of phosphate-activated glutaminase (PAG), enzymes that mediate conversions between glutamate and glutamine. GS and PAG catalyze the following irreversible reactions:
GS, which catalyzes the conversion of glutamate to glutamine, is expressed in astrocytes and not in neurons (Martinez-Hernandez et al., 1977). This compartmentalization of GS in astrocytes is consistent with the function of astrocytes, as consumers of glutamate, in functional glutamate trafficking (Patel et al., 1982). The absence of GS in neurons is also consistent with the function of neurons as producers, and not consumers, of glutamate under normal conditions. PAG, which catalyzes the conversion of glutamine to glutamate, is expressed in both neurons and astrocytes (Aoki et al., 1991; Wurdig and Kugler, 1991), but is primarily active in neurons (Kvamme et al., 1982; Hogstad et al., 1988) which is also supported by experimental data in vivo (Shen et al., 1999; Lebon et al., 2002). Neuronal PAG activity is consistent with the function of neurons as producers of glutamate in functional glutamate trafficking.
PAG produces, and GS consumes, ammonium in presynaptic neurons and astrocytes, respectively. Therefore, in order to operate at steady state, the coordinated activities of PAG and GS in functional glutamate trafficking in vivo, the glutamate-glutamine “cycle,” must be supported necessarily by processes that effectively transport ammonium from presynaptic neurons to astrocytes at the same rate that PAG and GS operate. Direct transport of ammonium from presynaptic neurons to astrocytes appears to be unlikely. Although there is evidence that astrocytes take up ammonium directly (Nagaraja and Brookes, 1998), there is no evidence that presynaptic neurons release ammonium, or that ammonium uptake by astrocytes is coupled to synaptic function. Three mechanisms for the effective transfer of ammonium from presynaptic neurons to astrocytes, presumably coupled to the glutamate-glutamine “cycle,” have been proposed previously. These are ammonia diffusion (Benjamin and Quastel, 1975), a branchedchain amino acid-branched chain keto acid (BCAA-BCKA) nitrogen shuttle (Yudkoff et al., 1996; Yudkoff, 1997; Hutson et al., 1998), and an alanine-lactate nitrogen shuttle (Waagepetersen et al., 2000a). The extent to which each of these previously proposed mechanisms for ammonium transfer supports the coordinated activities of PAG and GS in functional glutamate trafficking in vivo has yet to be established experimentally (Zwingmann and Leibfritz, 2003).
Glutamine synthesis in astrocytes, processes that package glutamate into synaptic vesicles within presynaptic neurons, and processes that maintain ionic gradients that drive glutamate transport from synaptic clefts to astrocytes and glutamine transport to presynaptic neurons all consume ATP. Consequently, the coordinated activities of PAG and GS in vivo must be supported necessarily by processes in presynaptic neurons and astrocytes that produce ATP at the rates that these processes consume ATP. Consistent with this necessity, differences in rates of the glutamate-glutamine cycle are proportional to differences in rates of the neuronal TCA cycle in vivo (Sibson et al., 1998), which, under normal physiological conditions, is sustained by brain glucose catabolism (Siesjo, 1978; Dienel and Hertz, 2001). However, neither the compartmentalization of glucose catabolism, nor the mechanism by which its rate is matched to that of functional glutamate trafficking, in support of functional glutamate trafficking in vivo, has, to date, been established experimentally.
Glutamate dehydrogenase (GDH), an enzyme central to glutamate metabolism, supports both the oxidative degradation of glutamate in astrocytes (Yu et al., 1982; Sonnewald et al., 1993; McKenna et al., 1996a; Peng et al., 2001; Waagepetersen et al., 2002; Hertz and Hertz, 2003) and the de novo synthesis of glutamate in either astrocytes (Hertz et al., 1999) or neurons (Hassel and Brathe, 2000). Consequently, GDH may contribute directly to sustaining functional glutamate trafficking in vivo. GDH is active in synaptic terminals, expressed in both neurons and astrocytes, and likely performs a different function in these differing cell types (McKenna et al., 2000; Zaganas et al., 2001). GDH catalyses the following reaction:
In the direction of glutamate synthesis, presumably favored in neurons (Faff-Michalak and Albrecht, 1993; Zaganas et al., 2001), GDH consumes ammonium and oxoglutarate (OG) and oxidizes nicotinamide adenine dinucleotide (NAD). In the direction of glutamate degradation, presumably favored in astrocytes (Zaganas et al., 2001; Yu et al., 1982; Westergaard et al., 1996; McKenna et al., 1996b), GDH produces ammonium and OG and reduces NAD. This presumed differential role of GDH in neurons and astrocytes is consistent with the function of each of these cell types in, and may have important implications for the understanding of, functional glutamate trafficking in synaptic neurotransmission. More specifically, a composite process consisting of GDH-mediated glutamate synthesis in presynaptic neurons, glutamate release by presynaptic neurons into synaptic clefts, glutamate uptake by astrocytes, and GDH-mediated degradation of glutamate in astrocytes, represents, in theory, a second mechanism (in addition to the one implicit in the coordinated activities of PAG and GS) for functional glutamate trafficking.
In order to operate at steady state, this hypothesized mechanism for functional glutamate trafficking in vivo, based on the ompartmentalized, coordinated, differential role of GDH in presynaptic neurons and astrocytes (Zaganas et al., 2001) must necessarily be supported by other processes. In glutamate synthesis GDH consumes, and in glutamate degradation GDH produces, ammonium. Therefore, the differential activities of GDH in presynaptic neurons and astrocytes in functional glutamate trafficking in vivo must be supported necessarily by a process that effectively transports ammonium from astrocytes to presynaptic neurons at the same rate that GDH operates differentially in these cell types. Furthermore, in glutamate synthesis GDH consumes, and in glutamate degradation GDH produces, OG. Therefore, GDH-mediated glutamate synthesis in presynaptic neurons and GDH-mediated glutamate degradation in astrocytes, must necessarily be supported by processes that, respectively and at the same rate, supply and demand OG. GDH-mediated glutamate synthesis in neurons may be supported by import of OG, or of some TCA cycle intermediate from which OG can be derived, from astrocytes (Shank and Campbell, 1982, 1984a; Westergaard et al., 1994a; Westergaard et al., 1994b; Schousboe et al., 1997). GDH-mediated glutamate degradation in astrocytes may be supported by either OG oxidation via pyruvate recycling (Sonnewald et al., 1996; Waagepetersen et al., 2002) or export of OG, or of some TCA cycle intermediate derived from OG, from astrocytes. Finally, GDH-mediated glutamate synthesis and degradation, respectively, oxidize and reduce NAD. Therefore, the hypothesized compartmentalized, coordinated, differential activities GDH in presynaptic neurons and astrocytes in functional glutamate trafficking in vivo must necessarily be supported by processes that reduce and oxidize, respectively and at the same rates, NAD in presynaptic neurons and astrocytes.
The present study is a theoretical investigation designed to specify, from a systems perspective and in detail, theoretically plausible cycles for functional glutamate trafficking in synaptic neurotransmission. It introduces and elaborates hypothetical cycles for functional glutamate trafficking, including cycles consistent with evidence that astrocytes degrade neurotransmitter glutamate by means of oxidative processes (Huang et al., 1993), that have the potential to provide a more complete understanding of the biochemical and transport processes necessary to sustain the well-documented, coordinated activities of PAG and GS in vivo. The elaboration of these cycles is intended to provide theoretical insight into potential mechanisms by which ammonium is effectively transported from presynaptic neurons to astrocytes in support of the coordinated activities of PAG and GS and by which ATP is available for glutamine synthesis in astrocytes, glutamine transport to presynaptic neurons, and processes that transport neurotransmitter glutamate between presynaptic neurons and astrocytes. The present study also introduces and elaborates a hypothetical cycle for functional glutamate trafficking that explicitly incorporates the carboxylation of pyruvate in astrocytes as an essential step in the de novo synthesis of neurotransmitter glutamate in presynaptic neurons.
Methods
The present investigation adopts an analytic approach grounded in the principle of conservation of matter that ensures that each of the proposed cycles for functional glutamate trafficking between presynaptic neurons and astrocytes that result from this investigation provides a complete, compartmentalized account of the origin and fate of all atomic and molecular species implicated in the process of functional glutamate trafficking in synaptic neurotransmission. The overall system for analysis is conceptualized as the composition of regions in space occupied by functional components of neuronal-glial glutamate trafficking, that is, regions in space occupied by presynaptic neurons, cooperating astrocytes, and the extracellular space between these presynaptic neurons and astrocytes through which these cell types exchange molecular species. Two subsystems, regions occupied by presynaptic neuons and astrocytes, respectively, are each divided into two sub-component regions, a cytosolic compartment, bounded by plasma and organelle membranes, and a mitochondrial compartment,bounded by mitochondrial membranes. A third subsystem, the extracellular space (including synaptic clefts and vesicles produced by presynaptic neurons), mediates net exchanges of compounds between presynaptic neurons and astrocytes. Although there is evidence that cellular or even subcellular compartmentalization may be considerably more intricate than that adopted in the present analyses (e.g., see Bakken et al., 1997; Waagepetersen et al., 2000b; McKenna et al., 2000; Waagepetersen et al., 2001; Dienel and Cruz, 2003; Zwingmann and Leibfritz, 2003; Hertz, 2004; Sonnewald et al., 2004), the simple compartmentalization adopted here suffices for the purposes of the present study. This is not a limitation of the present methodology per se, which in principle can be applied to systems that contain any number of compartments. For each cycle, the overall system, each subsystem, and the individual sub-compartments of each subsystem, are each contrained to operate in a stationary “steady” state in which there is no net production of any atomic or molecular species. Furthermore, the overall system is constrained to interact with the blood stream only, with which it exchanges CO2 and H2O for glucose and O2.
In the analysis, each molecular species within each compartment is constrained to satisfy a general “bookkeeping” relationship in which the net change in the number of molecules of species i within compartment j, Δmij, is equal to the combination of the net transport of that molecular species to the compartment and the net production of that molecular species by means of chemical reactions within that compartment, as follows:
In this equation, Rtm represents the net rate of transport mechanism m, Tij,m represents the number of molecules of species i transported into compartment j per transport mechanism m, Rxn represents the rate of chemical reaction n, and Pij,n represents the number of molecules of species i produced in compartment j per chemical reaction n. Individual terms in each summation, Rtm Ti,j,m and Rxn Pij,n, that assume negative values correspond to a net rate of transport of molecular species i out of compartment j by transport mechanism m and a net rate of consumption of molecular species i within compartment j by chemical reaction n, respectively. These “bookkeeping” relationships for each molecular species were used to construct systems of linear algebraic equations, representing linear combinations of compartmentalized biochemical reactions and localized transport processes. These systems of equations were used to develop cycles for functional glutamate trafficking that may operate between presynaptic neurons and astrocytes and that explicitly account for the origin and fate of each molecular species within each compartment.
In this analysis, chemical reactions were restricted to cellular compartments, plasma and mitochondria compartments of presynaptic neurons and astrocytes. Plasma compartments of presynaptic neurons contain a composite process representing glycolysis (G), lactate dehydrogenase (LDH), a branched-chain aminotransferase (BCAT), alanine aminotransferase (ALAT), and phosphate-activated glutaminase (PAG). Plasma compartments of astrocytes contain glycolysis (G), LDH, GDH, a BCAT, ALAT, glutamine synthetase (GS) and malic enzyme (ME). Although ME is generally considered to be an NADP-dependent enzyme, human brain cytosolic malic enzyme (cME) displays both NAD- and NADP-linked activity (Kam et al., 1988; Bukato et al., 1995b). Below pH ≈ 6.9, NAD-linked brain cME activity is greater than NADP-linked cME activity (Bukato et al., 1995b). Given that synaptic clefts are acidified by exocytosed protons to pH ≈ 6.9 (Palmer et al., 2003) and that astrocytes display intracellular pH less than extracellular pH (Mellergard et al., 1994), astrocytic cME is treated as an NADdependent enzyme in the present analyses. The mitochondria compartments of presynaptic neurons and astrocytes each contain pyruvate dehydrogenase (PDH), TCA cycle enzymes [citrate synthetase (CS), aconitate hydratase (AH), isocitrate dehydrogenase (IDH), oxoglutarate dehydrogenase (OGDH), succinyl-CoA synthetase (SCS), succinate dehydrogenase (SDH), fumarate hydratase (FH), and malate dehydrogenase (MDH)], glutamate dehydrogenase (GDH), and a nucleoside diphosphate kinase (mNDK) to convert GTP produced by SCS to ATP. Enzymes bound to the inner mitochondrial membrane were placed in either the mitochondrial compartment (e.g., SDH) or the plasma compartment (e.g., PAG; Kvamme et al. 2000) of a given cell type, depending on whether, in each case, the functional domain of the enzyme resides, respectively, inside or outside the inner mitochondrial membrane.
Material transport processes, representing the transfer of molecular species from one compartment to another, facilitated by specific membrane proteins, were localized to membranes that contain those specific transport proteins. In this analysis, plasma membranes of presynaptic neurons contain a glucose transporter (e.g., GLUT3), monocarboxylate transporters (MCTs) to transport lactate (e.g, MCT2) and BCKAs, sodium-coupled neutral amino acid transporters (SNAT1s) to transport glutamine and alanine, a sodium-coupled BCAA transporter (SBAT1), and a set of sodium-coupled citrate transporters (NaCTs) to transport TCA cycle intermediates citrate, oxoglutarate, and malate (Halestrap and Price, 1999; Broer and Brookes, 2001; Inoue et al., 2004; Mackenzie and Erickson, 2004; Takanaga et al., 2005; Pellerin et al., 2005; Pierre and Pellerin, 2005). Plasma membranes of astrocytes contain a glucose transporter (e.g., GLUT1), MCTs to transport lactate (e.g., MCT1) and BCKAs, a sodium-coupled glutamine transporter (SNAT3), a SNAT1 to transport alanine, SBAT1 to transport BCAAs, a set of NaCTs to transport citrate, OG and malate, and a specific glutamate transporter (GLAST). Plasma membranes for both presynaptic neurons and astrocytes contain a sodium-hydrogen exchanger (NHE) and a potassium leak channel (KCNK) to support the maintenance of cellular ionic balances (Orlowski and Grinstein, 1997; Talley et al., 2003). Mitochondrial membranes, in both presynaptic neurons and astrocytes, contain an MCT to transport pyruvate, a glutamate carrier (GC), an oxoglutarate carrier (OGC), a citrate carrier (CIC), a dicarboxylate carrier (DIC). a phosphate carrier (PiC), and an ADP/ATP carrier (AAC) (Palmieri, 2004).
Several additional composite processes that couple specific chemical reactions to specific transport process were included in the development of cycles for functional glutamate trafficking. Some of these composite processes reflect the effective transport or oxidation of nicotinamide adenine dinucleotide (NAD), or the oxidation of flavin adenine dinucleotide (FAD), in support of compartmentalized cellular chemical reactions that consume NAD+ and FAD+, respectively. These processes were a malate-aspartate shuttle (MAS) and two effective electron transport (ET) mechanisms associated with the oxidation of NADH (ET1) and FADH2 (ET2) at the inner mitochondrial membrane. The MAS was composed of mitochondrial and cytosolic MDH, mitochondrial and cytosolic aspartate aminotransferase (AAT), mitochondrial membrane OGC and mitochondrial membrane aspartate/glutamate carrier (AGC). Use of the individual component processes of the MAS, i.e., cMDH, mMDH, cAAT, mAAT, mmAGC and mmOGC, rather than the MAS composite process in the analyses produces equivalent results for the hypothetical cycles elaborated in the present study. That is, the net effect of the individual components of the MAS is identical to that of the composite MAS for these cycles. ET1 was composed of NADH-coenzyme Q reductase (ETC: Complex I), coenzyme Q-cytochrome c reductase (ETC: Complex III) and cytochrome c oxidase (ETC: Complex IV). ET2 was composed of succinate-coenzyme Q reductase (ETC: Complex II), coenzyme Q-cytochrome c reductase and cytochrome c oxidase. Other composite processes reflect the production or consumption of ATP coupled to specific transport processes. These were a plasma membrane Na-K ATPase and a mitochondrial membrane ATP synthase (ETC: Complex V) in bothpresynaptic neurons and astrocytes, and an ATPase representing the packaging of glutamate into vesicles (transport of glutamate across a plasma membrane) in presynaptic neurons. Plasma compartments of presynaptic neurons and astrocytes each included a process representing a generic ATPase to account for deficits or surpluses of ATP associated with each cycle.
For each cycle, relative localized material transport and compartmentalized chemical reaction rates were determined by solving systems of “bookkeeping” equations (described above) representing material balances in astrocytes and presynaptic neurons, respectively. Table 1 provides a complete list of process equations used to compose these systems of equations for the analyses presented herein. The present analyses were designed specifically to elaborate previously proposed and alternative cycles for functional (neurotransmitter) glutamate trafficking between presynaptic neurons and astrocytes, and not other metabolic functions of neurons and astrocytes (e.g., glucose consumption dedicated solely to the production of ATP or to biosynthetic processes unrelated to neurotransmitter glutamate synthesis). Furthermore, although individual enzymes may participate in other pathways, the present analyses focus solely on their potential participation in cycles for functional glutamate trafficking as determined by the stoichiometries of those cycles. The present analyses represent cycles for functional glutamate trafficking for which all of the glutamate released by presynaptic neurons into synaptic clefts is taken up by astrocytes. In these analyses, astrocytes import glutamate, BCAAs and alanine from, and export glutamine, BCKAs, lactate, and TCA cycle intermediates (oxoglutarate, malate and citrate) to, the extracellular space. Presynaptic neurons export glutamate, BCAAs and alanine to, and import glutamine, BCKAs, lactate, and TCA cycle intermediates (oxoglutarate, malate and citrate) from, the extracellular space. Both astrocytes and presynaptic neurons import glucose from the blood stream. For each cycle, rates of material exchanges between astrocytes and the extracellular space, and between presynaptic neurons and the extracellular space, were matched to ensure no net rates of change of any molecular species within the extracellular space. For any given cycle, cellular localization of glucose uptake by either presynaptic neurons or astrocytes was determined by localized demand for either NADH or acetyl-CoA that could be met by either glycolysis or a combination of glycolysis and PDH, respectively, within each cell type. In principle, glucose uptake and glycolysis in either presynaptic neurons or astrocytes could support variants of each of the proposed cycles elaborated in the present analyses due to the presence of LDH and monocarboxylate transporters in each of these cell types.
Table 1.
Process Equations
| Chemical Reaction | Equation |
|---|---|
| cGS | 1 cATP + 1 cGlu + 1 cNH4+ → 1 cGln + 1 cH+ + 1 cADP + 1 cPi |
| cPAG* | 1 cGln + 1 dH2O → 1 cGlu + 1 cNH4+ |
| cN | 1 cNH4+ → 1 cH+ + 1 dNH3 |
| cLDH | 1 cLac + 1 cNAD+ → 1 cH+ + 1 cPyrv + 1 cNADH |
| cME | 1 cMalate + 1 cNAD+ → 1 cPyrv + 1 dCO2 + 1 cNADH |
| cBCAT | 1 cGlu + 1 cBCKA → 1 cBCAA + 1 cOG |
| cALAT | 1 cGlu + 1 cPyrv → 1 cAla + 1 cOG |
| mGDH | 1 mGlu + 1 mNAD+ + 1 dH2O → 1 mH+ + 1 mOG + 1 mNADH + 1 mNH4+ |
| mN | 1 mNH4+ → 1 mH+ + 1 dNH3 |
| mPDH | 1 CoA + 1 mNAD+ + 1 mPyrv → 1 dCO2 + 1 mActCoA + 1 mNADH |
| mCS | 1 mActCoA + 1 mOAA + 1 dH2O → 1 mCit + 1 mH+ + 1 CoA |
| mAH | 1 mCit → 1 mIsocit |
| mIDH | 1 mIsocit + 1 mNAD+ → 1 dCO2 + 1 mNADH + 1 mOG |
| mOGDH | 1 mOG + 1 CoA + 1 mNAD+ → 1 dCO2 + 1 mNADH + 1 mSucCoA |
| mSCS | 1 mPi + 1 mSucCoA + 1 mGDP → 1 mGTP + 1 mSuc + 1 CoA |
| mSDH | 1 mSuc + 1 FAD+ → 1 FADH2 + 1 mFum |
| mFH | 1 mFum + 1 dH2O → 1 mMalate |
| mMDH | 1 mMalate + 1 mNAD+ → 1 mH+ + 1 mNADH + 1 mOAA |
| mNDK | 1 mGTP + 1 mADP → 1 mATP + 1 mGDP |
| Transport Mechanism | Equation |
| pmGLAST(Glu) | 1 cK+ + 1 xH+ + 1 xGlu + 3 xNa+ → 1 cGlu + 1 cH+ + 3 cNa+ + 1 xK+ |
| pmSNAT3 (Gln) | 1 cGln + 1 cNa+ + 1 xH+ → 1 cH+ + 1 xGln + 1 xNa+ |
| pmSNAT 1(Gln) | 1 cGIn + 1 cNa+ → 1 xGln + 1 xNa+ |
| pmGLUT(Gluc) | 1 xGluc → 1 cGluc |
| pmSBAT1(BCAA) | 1 cBCAA + 1 cNa+ → 1 xBCAA + 1 xNa+ |
| pmMCT(BCKA) | 1 cH+ + 1 cBCKA → 1 xH+ + 1 xBCKA |
| pmSNAT1(Ala) | 1 cAla + 1 cNa+ → 1 xAla + 1 xNa+ |
| pmMCT(Lac) | 1 cH+ + 1 cLac → 1 xH+ + 1 xLac |
| pmNaCT(OG) | 4 cNa+ + 1 cOG → 1 xOG + 4 xNa+ |
| pmNaCT(Mal) | 1 cMalate + 4 cNa+ → 1 xMalate + 4 xNa+ |
| pmNaCT(Cit) | 1 cCit + 4 cNa+ → 1 xCit + 4 xNa+ |
| pmNHE | 1 cH+ 1 xNa+ → 1 cNa+ + 1 xH+ |
| pmKCNK | 1 cK+ → 1 xK+ |
| mmGC | 1 cH+ + 1 cGlu → 1 mH+ + 1 mGlu |
| mmMCT(Pyr) | 1 cH+ + 1 cPyrv → 1 mH+ + 1 mPyrv |
| mmPiC | 1 cH+ + 1 cPi → 1 MH+ + 1 mPi |
| mmAAC | 1 mATP + 1 cADP → 1 cATP + 1 mADP |
| mmOGC | 1 cMalate + 1 mOG → 1 cOG + 1 mMalate |
| mmCIC | 1 cMalate + 1 mCit + 1 mH+ → 1 cCit + 1 cH+ + 1 mMalate |
| mmDIC | 1 cMalate + 1 mPi → 1 mMalate + 1 cPi |
| Composite Process | Equation |
| cG | 1 cGluc + 2 cADP + 2 cNAD+ + 2 cPi → 2 cATP + 2 cH+ + 2 cPyrv + 2 cNADH + 2 dH2O |
| MAS | 2 cH+ + 1 cNADH + 1 mNAD+ → 2 mH+ + 1 mNADH + 1 cNAD+ |
| ET1 | 1 dO2 + 11 mH+ + 1 mNADH → 10 cH+ + 1 dH2O + 1 mNAD+ |
| ET2 | 1 dO2 + 1 FADH2 + 6 mH+ → 6 cH+ + 1 dH2O + 1 FAD+ |
| mmATPase | 4 cH+ + 1 mPi + 1 mADP → 1 mATP + 3 mH+ + 1 dH2O |
| pmNaKATPase | 1 cATP + 3 cNa+ + 1 dH2O + 2 xK+ → 1 cH+ + 2 cK+ + 1 cADP + 1 cPi + 3 xNa+ |
| pmExoATPase | 2 cATP + 1 cGlu + 2 dH2O → 1 cH+ + 1 xH+ + 2 cADP + 2 cPi + 1 xGlu |
| cATPase | 1 cATP + 1 dH2O → 1 cH+ + 1 cADP + 1 cPi |
Prefixes: cytosolic (c), mitochondrial (m), extracellular (x), diffusible (d) plasma membrane (pm), and mitochondrial membrane (mm).
PAG, bound to the outer side of the inner mitochondrial membrane, is treated here as a cytosolic enzyme because it converts glutamine to glutamate outside of the mitochondria.
Results
The methods described were used to elaborate functional glutamate trafficking in synaptic neurotransmission in terms of three variants of the glutamate-glutamine cycle each based on a previously proposed mechanism of ammonium transport from presynaptic neurons to astrocytes (i.e., ammonia diffusion, a BCAA-BCKA nitrogen shuttle, or an alanine-lactate nitrogen shuttle) and in terms of three newly proposed alternative cycles each based on the transport of a representative TCA cycle intermediate (i.e., OG, malate or citrate) from astrocytes to presynaptic neurons. In principle, any TCA cycle intermediate could have been chosen for these latter analyses. The solution to the system of “bookkeeping” equations for each cycle is represented in terms of a diagram for net carbon and nitrogen pathways and in terms of tables for relative process rate and relative molecular species production rate stoichiometries.
Figure 1 diagrammatically represents the glutamate-glutamine cycle based on three previously proposed mechanisms for the transport of ammonium from presynaptic neurons to astrocytes, i.e., ammonia diffusion (Benjamin and Quastel, 1975), a BCAA-BCKA shuttle (Yudkoff et al., 1996; Yudkoff, 1997; Hutson et al., 1998) and an alanine-lactate shuttle (Waagepetersen et al., 2000a), derived from the present analysis. As anticipated, in each of these variants of the glutamate-glutamine cycle, presynaptic neurons exchange glutamate for glutamine with astrocytes in a ratio of 1:1. In the ammonia diffusion model, Figure 1a, ammonia diffuses, and protons (not shown) are effectively transported (via the combination of protonlinked neuronal glutamate exocytosis and proton-linked astrocytic glutamate uptake), from presynaptic neurons to astrocytes. In the BCAA-BCKA shuttle model, Figure 1b, ammonium is effectively transported from presynaptic neurons to astrocytes as presynaptic neurons exchange a BCAA for a BCKA with astrocytes. In this model, glycolysis in presynaptic neurons serves as a source of NADH. The reducing equivalents from this cytosolic NADH are transferred via the neuronal MAS to mitochondrial NADH, which enters the BCAA-BCKA shuttle in the neuronal reductive amination of OG to glutamate via mitochondrial GDH. Mitochondrial NADH exits the BCAA-BCKA shuttle in the oxidative deamination of glutamate to OG via mitochondrial GDH in astrocytes and is oxidized via ET1 in astrocytes. In the alanine-lactate shuttle model, Figure 1c, ammonium is effectively transported from presynaptic neurons to astrocytes as presynaptic neurons exchange alanine for lactate with astrocytes. Here, glycolysis in astrocytes serves as a source of NADH, which is effectively transported to neuronal GDH via a combination of astrocytic LDH, lactate transport from astrocytes to presynaptic neurons, neuronal LDH, and the neuronal MAS. NADH exits the alanine-lactate shuttle via mitochondrial GDH in astrocytes and is oxidized via ET1 in astrocytes.
Figure 1.

Proposed Variants of the Glutamate-Glutamine Cycle
Table 2 displays the relative process rate stoichiometries for the three variants of the glutamate-glutamine cycle displayed in Figure 1. For each cycle, neuronal PAG and astrocytic GS operate in a ratio of 1:1. The BCAA-BCKA shuttle and alanine-lactate shuttle models, for which glycolysis serves as a source of NADH, include glycolysis and TCA cycle activity. The ammonia diffusion model consumes a net 4 1/3 ATP, with ATP deficits in both presynaptic neurons and astrocytes. The BCAA-BCKA shuttle and alanine-lactate shuttle models each result in an overall net surplus of 11 5/12 mole ATP based on the catabolism of 1/2 mole glucose. The BCAA-BCKA shuttle model produces a surplus of ATP in presynaptic neurons and deficit of ATP in astrocytes, and the alanine-lactate shuttle model produces a deficit of ATP in presynaptic neurons and a surplus of ATP in astrocytes. Table 3 displays the relative molecular species production rate stoichiometries for the variants of the glutamate-glutamine cycle displayed in Figure 1.
Table 2.
Relative Process Rates for Proposed Variants of the Glutamate-Glutamine Cycle
| Process | Mechanism for Ammonium Transport | |||||
|---|---|---|---|---|---|---|
| Ammonium Diffusion | BCAA-BCKA Shuttle | Alanine-Lactate Shuttle | ||||
| PN | A | PN | A | PN | A | |
| cGS | N/A | 1 | N/A | 1 | N/A | 1 |
| cPAG | 1 | N/A | 1 | N/A | 1 | N/A |
| cN | 1 | −1 | 1 | −1 | 1 | −1 |
| cLDH | 0 | 0 | 0 | 0 | 1 | −1 |
| cBCAT | 0 | 0 | 1 | −1 | 0 | 0 |
| cALAT | 0 | 0 | 0 | 0 | 1 | −1 |
| mGDH | 0 | 0 | −1 | 1 | −1 | 1 |
| mN | 0 | 0 | −1 | 1 | −1 | 1 |
| mTCA | 0 | 0 | 1 | 0 | 0 | 1 |
| mNDK | 0 | 0 | 1 | 0 | 0 | 1 |
| pmGLAST(Glu) | N/A | 1 | N/A | 1 | N/A | 1 |
| pmSNAT3(Gln) | N/A | 1 | N/A | 1 | N/A | 1 |
| pmSNAT1(Gln) | −1 | N/A | −1 | N/A | −1 | N/A |
| pmGLUT(Gluc) | 0 | 0 | 1/2 | 0 | 0 | 1/2 |
| pmSBAT1(BCAA) | 0 | 0 | 1 | −1 | 0 | 0 |
| pmMCT(BCKA) | 0 | 0 | −1 | 1 | 0 | 0 |
| pmSNAT1(Ala) | 0 | 0 | 0 | 0 | 1 | −1 |
| pmMCT(Lac) | 0 | 0 | 0 | 0 | −1 | 1 |
| pmNHE | 0 | 1 | 0 | 1 | 0 | 1 |
| pmKCNK | 2/3 | 1 | 0 | 1 2/3 | 0 | 1 2/3 |
| mmGC | 0 | 0 | −1 | 1 | −1 | 1 |
| mmMCT(Pyr) | 0 | 0 | 1 | 0 | 0 | 1 |
| mmPiC | 0 | 0 | 13 1/2 | 1 1/4 | 1 | 13 3/4 |
| mmAAC | 0 | 0 | 12 1/2 | 2 1/4 | 0 | 14 3/4 |
| mmOGC | 0 | 0 | −1 | 1 | −1 | 1 |
| mmDIC | 0 | 0 | 1 | −1 | 1 | −1 |
| cG | 0 | 0 | 1/2 | 0 | 0 | 1/2 |
| MAS | 0 | 0 | 1 | 0 | 1 | 0 |
| ET1 | 0 | 0 | 4 | 1 | 0 | 5 |
| ET2 | 0 | 0 | 1 | 0 | 0 | 1 |
| mmATPase | 0 | 0 | 11 1/2 | 2 1/4 | 0 | 13 3/4 |
| pmNaKATPase | 1/3 | 1 | 0 | 1 1/3 | 0 | 1 1/3 |
| pmExoATPase | 1 | N/A | 1 | N/A | 1 | N/A |
| cATPase | −2 1/3 | −2 | 11 1/2 | −1/12 | −2 | 13 5/12 |
Prefixes: cytosolic (c), mitochondrial (m), plasma membrane (pm), and mitochondrial membrane (mm).
mTCA is an abbreviation for a composite process consisting of mPDH and TCA cycle enzymes mCS mAH, mIDH, mOGDH, mSCS, mSDH, mFH, and mMDH.
Table 3.
Relative Molecular Species Production Rates for Proposed Variants of the Glutamate-Glutamine Cycle
| Molecule | Mechanism for Ammonium Transport | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ammonium Diffusion | BCAA-BCKA Shuttle | Alanine-Lactate Shuttle | |||||||
| PN | A | Net | PN | A | Net | PN | A | Net | |
| xGlucose | 0 | 0 | 0 | 0 | −1/2 | −1/2 | −1/2 | 0 | −1/2 |
| dO2 | 0 | 0 | 0 | 0 | −3 | −3 | −2 1/2 | −1/2 | −3 |
| dCO2 | 0 | 0 | 0 | 0 | 3 | 3 | 3 | 0 | 3 |
| dH2O | −1 | 1 | 0 | 0 | 3 | 3 | 2 | 1 | 3 |
| xGlutamate | 1 | −1 | 0 | 1 | −1 | 0 | 1 | −1 | 0 |
| xGlutamine | −1 | 1 | 0 | −1 | 1 | 0 | −1 | 1 | 0 |
| dNH3 | 1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| xBCAA | 0 | 0 | 0 | 0 | 0 | 0 | 1 | −1 | 0 |
| xBCKA | 0 | 0 | 0 | 0 | 0 | 0 | −1 | 1 | 0 |
| xAlanine | 0 | 0 | 0 | 1 | −1 | 0 | 0 | 0 | |
| xLactate | 0 | 0 | 0 | −1 | 1 | 0 | 0 | 0 | 0 |
| xH+ | 1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Prefixes: extracellular (x) and diffusible (d).
Figure 2 diagrammatically represents three alternative cycles for functional glutamate trafficking derived from the present analysis. Each of these cycles is based on the transport of a TCA cycle intermediate from astrocytes to presynaptic neurons (Shank and Campbell, 1982, 1984a; Westergaard et al., 1994a; Westergaard et al., 1994b; Schousboe et al., 1997) and integrates glucose catabolism with functional glutamate trafficking. In each of these cycles, presynaptic neurons exchange glutamate for glutamine and a TCA cycle intermediate. Overall glucose catabolism associated with glutamate trafficking is distributed between presynaptic neurons and astrocytes. In the Glutamate-Glutamine/Oxoglutarate Cycle (GGOC), Figure 2a, the only direct tie between glucose catabolism and glutamate trafficking is that glycolysis effectively supplies NADH to GDH in presynaptic neurons. This NADH is effectively transported to GDH in astrocytes via neurotransmitter glutamate and ultimately oxidized by ET1 in astrocytes. In the Glutamate-Glutamine/Malate Cycle (GGMC) and Glutamate-Glutamine/Citrate Cycle (GGCC), Figure 2b and Figure 2c, respectively, carbon entering the overall system in glucose is ultimately used in the synthesis of glutamate in presynaptic neurons via GDH. Carbon from glutamate consumed in astrocytes via GDH enters the astrocytic TCA cycle as OG and leaves in another TCA cycle intermediate, malate or citrate, transported from astrocytes to presynaptic neurons. In the GGMC, citrate synthetase activity associated with glutamate trafficking occurs in presynaptic neurons in support of de novo synthesis of neurotransmitter glutamate. In the GGCC, citrate synthetase activity directly associated with glutamate trafficking occurs in astrocytes.
Figure 2.

Proposed Alternative Cycles for Functional Glutamate Trafficking
Table 4 displays the relative process rate stoichiometries for the newly proposed alternative cycles for functional glutamate trafficking based on the transport of TCA cycle intermediates between astrocytes and presynaptic neurons displayed in Figure 2. As anticipated, for each of these cycles, neuronal PAG and astrocytic GS operate in a ratio of 1:1 and GDH operates in opposite directions in presynaptic neurons and astrocytes. The GGOC, GGMC, and GGCC each result in an overall net surplus of 4 5/24 mole ATP based on the catabolism of 1/4 mole glucose. The GGOC produces a surplus of ATP in presynaptic neurons and deficit of ATP in astrocytes, the GGMC produces surpluses of ATP in both presynaptic neurons and astrocytes, and the GGOC produces a deficit of ATP in presynaptic neurons and a surplus of ATP in astrocytes.
Table 4.
Relative Process Rates for Proposed Alternative Cycles for Glutamate Trafficking
| Process | Cycle for Functional Glutamate Trafficking | |||||
|---|---|---|---|---|---|---|
| GGOC | GGMC | GGCC | ||||
| PN | A | PN | A | PN | A | |
| cGS | N/A | 1/2 | N/A | 1/2 | N/A | 1/2 |
| cPAG | 1/2 | N/A | 1/2 | N/A | 1/2 | N/A |
| cN | 1/2 | −1/2 | 1/2 | −1/2 | 1/2 | −1/2 |
| cBCAT | 0 | 0 | 0 | 0 | 0 | 0 |
| cALAT | 0 | 0 | 0 | 0 | 0 | 0 |
| mGDH | −1/2 | 1/2 | −1/2 | 1/2 | −1/2 | 1/2 |
| mN | −1/2 | 1/2 | −1/2 | 1/2 | −1/2 | 1/2 |
| mPDH | 1/2 | 0 | 1/2 | 0 | 0 | 1/2 |
| mCS | 1/2 | 0 | 1/2 | 0 | 0 | 1/2 |
| mAH | 1/2 | 0 | 1/2 | 0 | 1/2 | 0 |
| mIDH | 1/2 | 0 | 1/2 | 0 | 1/2 | 0 |
| mOGDH | 1/2 | 0 | 0 | 1/2 | 0 | 1/2 |
| mSCS | 1/2 | 0 | 0 | 1/2 | 0 | 1/2 |
| mSDH | 1/2 | 0 | 0 | 1/2 | 0 | 1/2 |
| mFH | 1/2 | 0 | 0 | 1/2 | 0 | 1/2 |
| mMDH | 1/2 | 0 | 1/2 | 0 | 0 | 1/2 |
| mNDK | 1/2 | 0 | 0 | 1/2 | 0 | 1/2 |
| pmGLAST(Glu) | N/A | 1 | N/A | 1 | N/A | 1 |
| pmSNAT3(Gln) | N/A | 1/2 | N/A | 1/2 | N/A | 1/2 |
| pmSNAT1(Gln) | −1/2 | N/A | −1/2 | N/A | −1/2 | N/A |
| pmGLUT(Gluc) | 1/4 | 0 | 1/4 | 0 | 0 | 1/4 |
| pmNaCT(OG) | −1/2 | 1/2 | 0 | 0 | 0 | 0 |
| pmNaCT(Mal) | 0 | 0 | −1/2 | 1/2 | 0 | 0 |
| pmNaCT(Cit) | 0 | 0 | 0 | 0 | −1/2 | 1/2 |
| pmNHE | −1 | 1 1/2 | −1 | 1 1/2 | −1 1/2 | 2 |
| pmKCNK | 1 | 1/3 | 1 | 1/3 | 2/3 | 2/3 |
| mmGC | −1/2 | 1/2 | −1/2 | 1/2 | −1/2 | 1/2 |
| mmMCT(Pyr) | 1/2 | 0 | 1/2 | 0 | 0 | 1/2 |
| mmPiC | 6 3/4 | 5/8 | 4 1/4 | 3 1/8 | 5/8 | 6 3/4 |
| mmAAC | 6 1/4 | 1 1/8 | 3 3/4 | 3 5/8 | 1/8 | 7 1/4 |
| mmOGC | −1/2 | 1/2 | 0 | 0 | 0 | 0 |
| mmCIC | 0 | 0 | 0 | 0 | −1/2 | 1/2 |
| mmDIC | 1/2 | −1/2 | 1/2 | −1/2 | 1/2 | −1/2 |
| cG | 1/4 | 0 | 1/4 | 0 | 0 | 1/4 |
| MAS | 1/2 | 0 | 1/2 | 0 | 0 | 1/2 |
| ET1 | 2 | 1/2 | 1 1/2 | 1 | 0 | 2 1/2 |
| ET2 | 1/2 | 0 | 0 | 1/2 | 0 | 1/2 |
| mmATPase | 5 3/4 | 1 1/8 | 3 3/4 | 3 1/8 | 1/8 | 6 3/4 |
| pmNaKATPase | 1/2 | 2/3 | 1/2 | 2/3 | 1/3 | 5/6 |
| pmExoATPase | 1 | N/A | 1 | N/A | 1 | N/A |
| cATPase | 4 1/4 | −1/24 | 1 3/4 | 2 11/24 | −2 5/24 | 6 5/12 |
Prefixes: cytosolic (c), mitochondrial (m), plasma membrane (pm), and mitochondrial membrane (mm).
Table 5 displays the relative molecular species production rate stoichiometries for the alternative cycles for functional glutamate trafficking displayed in Figure 2. The net material effect of all three alternative cycles for functional glutamate trafficking based on the transport of TCA cycle intermediates from astrocytes to presynaptic neurons is to consume glucose and O2 while producing CO2 and H2O in a ratio of 1:6:6:6. In all three of these newly proposed cycles, presynaptic neurons exchange glutamate for glutamine and a TCA cycle intermediate with astrocytes in a ratio of 2:1:1.
Table 5.
Relative Molecular Species Production Rates for Proposed Alternative Cycles for Glutamate Trafficking
| Molecule | Cycle for Functional Glutamate Trafficking | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GGOC | GGMC | GGCC | |||||||
| PN | A | Net | PN | A | Net | PN | A | Net | |
| xGlucose | −1/4 | 0 | −1/4 | −1/4 | 0 | −1/4 | 0 | −1/4 | −1/4 |
| dO2 | −1 1/4 | −1/4 | −1 1/2 | −3/4 | −3/4 | −1 1/2 | 0 | −1 1/2 | −1 1/2 |
| dCO2 | 1 1/2 | 0 | 1 1/2 | 1 | 1/2 | 1 1/2 | 1/2 | 1 | 1 1/2 |
| dH2O | 1 | 1/2 | 1 1/2 | 1 | 1/2 | 1 1/2 | 0 | 1 1/2 | 1 1/2 |
| xGlutamate | 1 | −1 | 0 | 1 | −1 | 0 | 1 | −1 | 0 |
| xGlutamine | −1/2 | 1/2 | 0 | −1/2 | 1/2 | 0 | −1/2 | 1/2 | 0 |
| xOG | −1/2 | 1/2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| xMalate | 0 | 0 | 0 | −1/2 | 1/2 | 0 | 0 | 0 | 0 |
| xCitrate | 0 | 0 | 0 | 0 | 0 | 0 | −1/2 | 1/2 | 0 |
| xH+ | 0 | 0 | 0 | 0 | 0 | 0 | −1/2 | 1/2 | 0 |
Prefixes: extracellular (x) and diffusible (d)
One of the alternative cycles, the GGMC, was investigated further to explore the possibility that a particular anaplerotic enzyme restricted to astrocytes, cME (Kurz et al., 1993), mediates functional glutamate trafficking in synaptic neurotransmission. The GGMC, as elaborated in Figure 2b and Table 4 and Table 5, requires no net anaplerotic reaction. However, because cME displays bi-directional activity (Bukato et al., 1995a), it is possible an anaplerotic process that occurs in astrocytes during one phase of the GGMC is reversed during another phase of the GGMC. Here we considered the GGMC to be, hypothetically, the net effect of a two phase process, one phase committed to the production of glutamate in presynaptic neurons, and a second phase committed to the consumption of glutamate in astrocytes.
Figure 3 displays the results of this investigation diagrammatically. In the hypothesized glutamate production phase of the proposed GGMC (Figure 3a), astrocytes and presynaptic neurons cooperate in a composite process that consumes glucose, lactate and glutamine and produces neurotransmitter glutamate in a ratio of 1/2:1:1:2. During this phase of the GGMC, astrocytes consume lactate to produce malate via LDH and cME, and transport equal amounts of malate and glutamine to presynaptic neurons as materials for the production of neurotransmitter glutamate. Presynaptic neurons, through oxidative processes and TCA cycle enzymes, combine pyruvate derived from glycolysis and malate imported from astrocytes to produce OG, which they use to produce 1/2 of the neurotransmitter glutamate via GDH. Presynaptic neurons produce the other 1/2 of the neurotransmitter glutamate from glutamine via PAG. In the hypothesized glutamate consumption phase of the GGMC (Figure 3b), astrocytes consume neurotransmitter glutamate and produce glutamine and lactate, in a ratio of 2:1:1. During this phase of the GGMC, astrocytes consume 1/2 of the neurotransmitter glutamate in the production of glutamine via GS, and the other 1/2 of the neurotransmitter glutamate in the production of lactate via cGDH, a series of TCA enzymes (mOGDH, mSCS, mSDH, mFH), cME, and cLDH. Over the complete GGMC (Figure 2b) there is no net flux through cME. In astrocytes, the formation of malate frompyruvate during the glutamate production phase of the cycle is exactly matched to the formation of pyruvate from malate during the glutamate consumption phase of the cycle. Table 6 displays the relative process rate stoichiometry for two hypothetical phases of the proposed GGMC, a glutamate production phase with enzymatic activity largely confined to presynaptic neurons, and a glutamate consumption phase with enzymatic activity entirely restricted to astrocytes. Table 7 displays the relative molecular species production rate stoichiometry associated with each of the hypothesized phases of the proposed GGMC.
Figure 3.
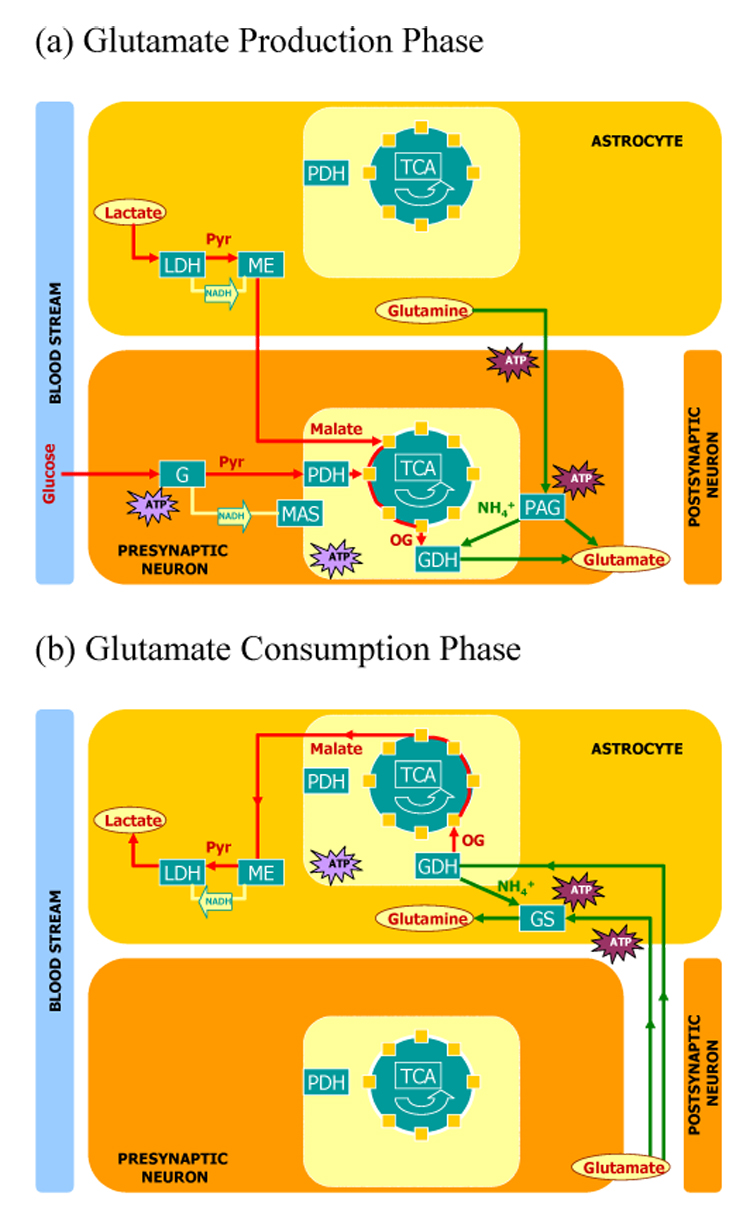
Hypothesized Phases of the Proposed Glutamate-Glutamine/Malate Cycle
Table 6.
Relative Process Rates for Hypothesized Phases of the Proposed GGMC Cycle
| Process | Glutamate-Glutamine/Malate Cycle | |||
|---|---|---|---|---|
| Glutamate Production Phase | Glutamate Consumption Phase | |||
| PN | A | PN | A | |
| cGS | N/A | 0 | N/A | 1/2 |
| cPAG | 1/2 | N/A | 0 | N/A |
| cN | 1/2 | 0 | 0 | −1/2 |
| cLDH | 0 | 1/2 | 0 | −1/2 |
| cME | N/A | −1/2 | N/A | 1/2 |
| mGDH | −1/2 | 0 | 0 | 1/2 |
| mN | −1/2 | 0 | 0 | 1/2 |
| mPDH | 1/2 | 0 | 0 | 0 |
| mCS | 1/2 | 0 | 0 | 0 |
| mAH | 1/2 | 0 | 0 | 0 |
| mIDH | 1/2 | 0 | 0 | 0 |
| mOGDH | 0 | 0 | 0 | 1/2 |
| mSCS | 0 | 0 | 0 | 1/2 |
| mSDH | 0 | 0 | 0 | 1/2 |
| mFH | 0 | 0 | 0 | 1/2 |
| mMDH | 1/2 | 0 | 0 | 0 |
| mNDK | 0 | 0 | 0 | 1/2 |
| pmGLAST(Glu) | N/A | 0 | N/A | 1 |
| pmSNAT3(Gln) | N/A | 1/2 | N/A | 0 |
| pmSNAT1(Gln) | −1/2 | N/A | 0 | N/A |
| pmGLUT(Gluc) | 1/4 | 0 | 0 | 0 |
| pmNaCT(Mal) | −1/2 | 1/2 | 0 | 0 |
| pmNaCT(Cit) | 0 | 0 | 0 | 0 |
| pmNHE | −1 | 1 | 0 | 1/2 |
| pmKCNK | 1 | 0 | 0 | 1/3 |
| mmGC | −1/2 | 0 | 0 | 1/2 |
| mmMCT(Pyr) | 1/2 | 0 | 0 | 0 |
| mmPiC | 4 1/4 | 0 | 0 | 3 1/8 |
| mmAAC | 3 3/4 | 0 | 0 | 3 5/8 |
| mmDIC | 1/2 | 0 | 0 | −1/2 |
| cG | 1/4 | 0 | 0 | 0 |
| MAS | 1/2 | 0 | 0 | 0 |
| ET1 | 1 1/2 | 0 | 0 | 1 |
| ET2 | 0 | 0 | 0 | 1/2 |
| mmATPase | 3 3/4 | 0 | 0 | 3 1/8 |
| pmNaKATPase | 1/2 | 0 | 0 | 2/3 |
| pmExoATPase | 1 | N/A | 0 | N/A |
| cATPase | 1 3/4 | 0 | 0 | 2 11/24 |
Prefixes: cytosolic (c), mitochondrial (m), plasma membrane (pm), and mitochondrial membrane (mm).
Table 7.
Relative Molecular Species Production Rates for Hypothesized Phases of the Proposed GGMC Cycle
| Molecule | Glutamate-Glutamine/Malate Cycle | |||||
|---|---|---|---|---|---|---|
| Glutamate Production Phase | Glutamate Consumption Phase | |||||
| PN | A | Net | PN | A | Net | |
| xGlucose | −1/4 | 0 | −1/4 | 0 | 0 | 0 |
| dO2 | −3/4 | 0 | −3/4 | 0 | −3/4 | −3/4 |
| dCO2 | 1 | −1/2 | 1/2 | 0 | 1 | 1 |
| dH2O | 1 | 0 | 1 | 0 | 1/2 | 1/2 |
| xGlutamate | 1 | 0 | 1 | 0 | −1 | −1 |
| xGlutamine | −1/2 | 1/2 | 0 | 0 | 0 | 0 |
| xMalate | −1/2 | 1/2 | 0 | 0 | 0 | 0 |
| xH+ | 0 | 1/2 | 1/2 | 0 | −1/2 | −1/2 |
| xNa+ | 0 | 1 1/2 | 1 1/2 | 0 | −1 1/2 | −1 1/2 |
| cGlutamine | 0 | −1/2 | −1/2 | 0 | 1/2 | 1/2 |
| cLactate | 0 | −1/2 | −1/2 | 0 | 1/2 | 1/2 |
| cNa+ | 0 | −1 1/2 | −1 1/2 | 0 | 1 1/2 | 1 1/2 |
Prefixes: extracellular (x), diffusible (d) and cytosolic (c).
Discussion
Each proposed cycle elaborated in the current analyses represents a coherent, unique explanation of functional glutamate trafficking in synaptic neurotransmission. Each cycle is a consequence of viewing compartmentalized enzymes and localized transporters as components of a larger system of molecular mechanisms designed to execute a specific, specialized function, i.e., to sustain a net unidirectional flux of glutamate from presynaptic neurons to astrocytes, through a set of interdependent processes distributed within and between these cell types. The presence and localization of the structural elements of each cycle, i.e., component enzymes and transporters, are well documented for synaptic terminals. The functional capabilities of these structural elements are also well established and consistent with the functions they perform within the context of each cycle. The methods of analysis ensure that, within each cycle, each compartment satisfies the basic principle of conservation of atomic species.
Although all of the proposed cycles for functional glutamate trafficking share some common elements, each cycle is distinct. Each cycle has unique implications for intracellular and intercellular transport of metabolites and compartmentalization of metabolism, including specialization of mitochondrial function compatible with reports of cellular mitochondrial heterogeneity (Sonnewald et al., 1998; Waagepetersen et al., 2000b; Waagepetersen et al., 2006). The proposed cycles differ with respect to mechanisms of ammonium transport from presynaptic neurons to astrocytes in support of the coordinated activities of PAG and GS, the participation of TCA cycle enzymes and metabolites, and the potential participation of a carbon fixing enzyme, in synaptic neurotransmission. These differences inform expectations about the potential significance of each particular cycle in functional glutamate trafficking.
Mechanisms of Ammonium Transport
In the ammonia diffusion variant of the glutamate-glutamine cycle, presynaptic neurons effectively transport ammonium to astrocytes by means of a combination of distinct transport mechanisms, i.e., molecular diffusion for the transport of ammonia and a combination of protonlinked substrate transport mechanisms (neuronal glutamate exocytosis, astrocytic glutamate uptake) for the transport of protons. Transport of ammonia by molecular diffusion would need to be driven by local concentration gradients of ammonia. Assuming that ammonia is produced in presynaptic neurons near sites of PAG activity and consumed in astrocytes near sites of GS activity, one might expect a local ammonia concentration gradient to support a net flux of ammonia from presynaptic neurons to astrocytes. However, one would not expect that all of the ammonia produced as a consequence of PAG activity at a single synapse to be transported by diffusion to sites of GS activity at that same synapse: Ammonia produced by PAG activity would be expected to diffuse in all directions, not entirely in a single direction toward a local site of GS activity. Transport of protons by proton-linked substrate transport mechanisms would be expected to be influenced by local, compartmentalized differences in proton concentration between presynaptic neurons and astrocytes. There is evidence that intracellular pH decreases in neurons and increases in astrocytes in an activity-dependent manner (Chesler and Kraig, 1987, 1989). Therefore, one might expect a local, compartmentalized difference in proton concentration to support a net flux of protons from presynaptic neurons to astrocytes. Indeed, it is possible that activity-dependent shifts in intracellular pH in neurons and astrocytes in opposite directions are due to proton production and consumption associated with the coordinated, compartmentalized activities of PAG and GS, respectively. In any case, the ammonia diffusion variant of the glutamate-glutamine cycle would be expected to be operative only to the extent that the fluxes of glutamate, ammonia, and protons from presynaptic neurons to astrocytes are matched in a ratio of 1:1:1.
In the BCAA-BCKA shuttle and alanine-lactate shuttle variants of the glutamateglutamine cycle, presynaptic neurons effectively transport ammonium to astrocytes by means an exchange of a non-neuroactive amino acid, a BCAA or alanine, respectively, for a carboxylic acid, a BCKA or lactate, respectively, in support of the coordinated activities of PAG and GS in synaptic neurotransmission. In these models, ammonium from PAG enters the shuttle via GDH in presynaptic neurons and ammonium to GS exits the shuttle via GDH in astrocytes. Experimental evidence in support of a BCAA-BCKA nitrogen shuttle, as suggested by Yudkoff and as elaborated by Hutson and her colleagues, has been limited to studies in cell cultures (Yudkoff et al., 1996; Yudkoff, 1997; Hutson et al., 1998; Lieth et al., 2001). Bak et al. (2005) report no direct coupling between an alanine-lactate nitrogen shuttle and the glutamate-glutamine cycle in neuronal-astrocytic cocultures, and that only the glutamate-glutamine cycle appears to be activity dependent. Therefore, it would appear that an alanine-lactate shuttle does not play any essential role in functional glutamate trafficking in glutamatergic synapses.
In the alternative cycles for functional glutamate trafficking based on the transport of TCA cycle intermediates from astrocytes to presynaptic neurons, proposed for the first time in this study, ammonium is effectively transported from presynaptic neurons to astrocytes in the form of glutamate, facilitated by neurotransmitter glutamate trafficking and the differential role of GDH in presynaptic neurons and astrocytes. In these cycles, presynaptic neurons exchange glutamate for glutamine with astrocytes in a ratio of 2:1. In presynaptic neurons, ammonium produced by PAG in the conversion of one molecule of glutamine to one molecule of glutamate is used in the de novo synthesis of a second molecule of glutamate produced by GDH. In astrocytes, one molecule of glutamate serves as a precursor to glutamine, and a second molecule of glutamate serves as a source of ammonium produced by GDH, per molecule of glutamine produced in GS. Because ammonium is effectively carried from presynaptic neurons to astrocytes by neurotransmitter glutamate in these cycles, ammonium transport by this mechanism is targeted from sites of GDH activity in presynaptic neurons to sites of GDH activity in astrocytes. In these cycles, mechanisms that transport neurotransmitter glutamate from presynaptic neurons to astrocytes effectively transport ammonium as well. That is, the transport of ammonium from presynaptic neurons to astrocytes is inherent in neurotransmitter glutamate transport.
In each of the proposed cycles for neurotransmitter glutamate trafficking resulting from the present analysis, with the sole exception of the ammonia diffusion variant of the glutamateglutamine cycle, GDH mediates the transport of ammonium from presynaptic neurons to astrocytes. In the BCAA-BCKA shuttle and alanine-lactate shuttle variants of the glutamateglutamine cycle and in each of the alternative cycles for functional glutamate trafficking based on the transport of a TCA cycle intermediate from astrocytes to presynaptic neurons, GDH in presynaptic neurons consumes ammonium produced by PAG and GDH in astrocytes produces ammonium consumed by GS. Within the context of these cycles, the compartmentalized activities of GDH are directly linked to the compartmentalized activities of PAG and GS.
Participation of TCA cycle enzymes and metabolites
Each of the proposed cycles for functional glutamate trafficking requires ATP to function. However, the ammonia diffusion variant of the glutamate-glutamine cycle differs from the other cycles with respect to direct, obligatory participation of TCA cycle enzymes and metabolites. The ammonia diffusion variant of the glutamate-glutamine cycle presupposes, but does not explicitly incorporate, processes that produce ATP in presynaptic neurons and astrocytes. This cycle can proceed, in principle, independently of other specific metabolic processes that produce ATP. The extent to which the ammonia diffusion variant of the glutamate-glutamine cycle operates on ATP derived from TCA cycle activity as opposed to ATP derived from other sources is an empirical question. The availability of ATP for the function the ammonia diffusion variant of the glutamate-glutamine cycle must be explained by mechanisms outside that cycle.
The BCAA-BCKA shuttle and alanine-lactate shuttle variants of the glutamate-glutamine cycle and the GGOC each require a supplementary source of reducing equivalents in the form of NADH in the mitochondrial compartment of presynaptic neurons in support of GDH in the reductive amidation of OG to glutamate. In the BCAA-BCKA shuttle variant of the glutamateglutamine cycle and the GGOC, glycolysis in presynaptic neurons produces these reducing equivalents that are transported via the neuronal MAS to the mitochondrial compartment in support of glutamate production via GDH. In these two cycles, glycolysis in presynaptic neurons also produces pyruvate that is oxidized in the neuronal TCA cycle to provide ATP to presynaptic neurons. In the alanine-lactate shuttle variant of the glutamate-glutamine cycle, glycolysis in astrocytes produces the requisite reducing equivalents that are effectively transported via astrocytic LDH, neuronal LDH, and the neuronal MAS to the mitochondrial compartment of presynaptic neurons in support of glutamate production via GDH. In this cycle, glycolysis in astrocytes also produces pyruvate that is oxidized in the astrocytic TCA cycle to provide ATP to astrocytes. In all three of these cycles, the oxidative deamidation of glutamate via GDH in astrocytes produces reducing equivalents in the form of NADH that are oxidized via the electron transport chain to provide ATP to astrocytes.
In the GGMC and GGCC, TCA cycle reactions play an integral part of larger, composite cycles that produce neurotransmitter glutamate in presynaptic neurons (Hamberger et al., 1979), transport glutamate from presynaptic neurons to astrocytes (Rothstein et al., 1996), and consume glutamate in astrocytes (Waniewski and Martin, 1986; Yu et al., 1982; Sonnewald et al., 1993; McKenna et al., 1996a; Peng et al., 2001; Waagepetersen et al., 2002; Hertz and Hertz, 2003). Within these composite cycles, trafficking of neurotransmitter glutamate from presynaptic neurons to astrocytes and TCA cycle intermediates from astrocytes to presynaptic neurons (Shank and Campbell, 1982, 1984a; Westergaard et al., 1994a; Westergaard et al., 1994b; Schousboe et al., 1997) mediates the complete oxidative metabolism of glucose, with a portion of the TCA cycle executed in presynaptic neurons and the remainder of the TCA cycle executed in astrocytes. A consequence of this integration of functional glutamate trafficking with oxidative metabolism of glucose, and this partitioning of oxidative processes between presynaptic neurons and astrocytes, is that these composite cycles produce ATP in both presynaptic neurons and astrocytes at rates directly proportional to the corresponding rates of neurotransmitter glutamate trafficking. Therefore, ATP produced in these cycles is available to support ATP consuming processes associated with functional glutamate trafficking.
In the current analyses, OG, malate and citrate were chosen to illustrate alternative cycles for neurotransmitter glutamate trafficking based on the transport of TCA cycle intermediate metabolites from astrocytes to presynaptic neurons because studies have demonstrated that these metabolites are readily released from astrocytes in cell culture (Westergaard et al., 1994a; Westergaard et al., 1994b). Although small in comparison to reported rates of neuronal glutamate release in cell culture (Schousboe et al., 1988), reported rates of OG, malate and citrate release from astrocytes in cell culture (Westergaard et al., 1994a; Westergaard et al., 1994b) are approximately 20%, 10%, and 100%, respectively, of rates of neurotransmitter glutamate trafficking in vivo (e.g., see Rothman et al., 2003). Transport of OG and malate from astrocytes to presynaptic neurons may be particularly relevant to neurotransmitter glutamate trafficking: Glutamate stimulates the release of OG and malate from astrocytes in cell culture (Westergaard et al., 1994b), OG and malate are taken up and metabolized by synaptosomes (Shank and Campbell, 1984a,b), and OG (Peng et al., 1991) and perhaps malate (Westergaard et al., 1995) serve as precursors for glutamate synthesis in neurons. Although studies in cell culture have failed to demonstrate that citrate in combination with alanine serves as a precursor for neuronal synthesis of neurotransmitter glutamate (Westergaard et al. 1994b), this may not have a direct bearing the potential efficacy of the proposed GGCC in vivo, for which, hypothetically, neurons produce glutamate from citrate and glutamine in a ratio of 2:1:1.
Glucose, oxygen and ATP consumption
The BCAA-BCKA shuttle and alanine-lactate shuttle variants of the glutamate-glutamine cycle and the newly proposed alternative cycles for functional glutamate trafficking each based on transport of a TCA cycle intermediate consume glucose and oxygen in support of glutamate synthesis and degradation in presynaptic neurons and astrocytes, respectively, in fixed, stoichiometric proportions to the rate of functional glutamate trafficking. For the BCAA-BCKA shuttle and alanine-lactate shuttle variants of the glutamate-glutamine cycle, presynaptic neurons and astrocytes, combined, consume 1/2 mole glucose and 3 mole oxygen per mole of neurotransmitter glutamate trafficked. For the GGOC, GGMC, and GGCC, presynaptic neurons and astrocytes, combined, consume 1/4 mole glucose and 1 1/2 mole oxygen per mole of neurotransmitter glutamate trafficked. In each of the cycles based on transport of a TCA cycle intermediate, neurotransmitter glutamate undergoes oxidative catabolism, to different degrees, in astrocytes. For the GGOC, this oxidative catabolism of neurotransmitter glutamate in astrocytes is restricted to the oxidation of NAD in support of GDH degradation of glutamate to OG. For the GGMC and the GGCC, the oxidative catabolism of neurotransmitter glutamate also includes TCA cycle enzyme activity in astrocytes.
For all of the proposed cycles for functional glutamate trafficking considered in the present analysis, two specific transport processes directly associated with intercellular trafficking of neurotransmitter glutamate, proton transport associated with acidification of, and glutamate packaging into, vesicles, and electrogenic sodium-potassium ionic exchanges (driven by Na-K ATPases) in support of sodium-coupled glutamate transport into astrocytes via GLAST, consume ATP. Each cycle requires the same total amount of ATP to accomplish these ATP-driven transport processes per mole of neurotransmitter glutamate trafficked. Furthermore, for all of the proposed cycles considered, GS consumes ATP in astrocytes. However, the amount of ATP consumed in GS and in support of sodium-coupled glutamine transport into presynaptic neurons, per mole of neurotransmitter glutamate trafficked, differs between variants of the glutamateglutamine cycle and newly proposed cycles based on transport of TCA cycle intermediates. For each variant of the glutamate-glutamine cycle, astrocytes produce and transport 1 mole of glutamine per mole of neurotransmitter glutamate trafficked. However, for each of the newly proposed cycles based on the transport of a TCA cycle intermediate, astrocytes produce and transport only 1/2 moles of glutamine per mole of neurotransmitter glutamate trafficked. Therefore, cycles based on transport of a TCA cycle intermediate are energetically more efficient than each of the variants of the glutamate-glutamine cycle, consuming 2/3 mole less ATP (1/2 mole less ATP in GS in astrocytes and 1/6 mole less ATP in support of glutamine transport to presynaptic neurons) per mole of neurotransmitter glutamate trafficked.
Carboxylation of pyruvate
Neurons and astrocytes differ with respect to the expression and function of enzymes capable of catalyzing the carboxylation of pyruvate, an essential process in the de novo synthesis of TCA cycle metabolites and glutamate from glucose or lactate. Astrocytes express mitochondrial pyruvate carboxylase (mPC) (Yu et al., 1983; Shank et al., 1985) and cytosolic malic enzyme (cME) (Kurz et al., 1993). Because mPC is irreversible, its activity is committed exclusively to the de novo synthesis of TCA cycle metabolites and their derivatives. However, cME has the potential to catalyze not only the carboxylation of pyruvate to form malate, but also the decarboxylation of malate to form pyruvate (Bukato et al., 1995a). Neurons express neither mPC nor cME (Yu et al., 1983; Shank et al., 1985; Kurz et al., 1993). Although neurons express mitochondrial malic enzyme (mME) (Vogel et al., 1998), mME activity heavily favors the decarboxylation of malate to form pyruvate and likely serves a cataplerotic, as opposed to an analperotic, function (Bukato et al., 1995a). Consequently, the capacity for carboxylation of pyruvate, an essential step in the process of de novo synthesis of glutamate from glucose or lactate, is most likely functionally present in astocytes and absent in neurons (Yu et al., 1983; Shank et al., 1985; Kurz et al., 1993; Bukato et al., 1995a).
In the proposed cycles for functional glutamate trafficking based on the transport of TCA cycle intermediates from astrocytes to presynaptic neurons, astrocytes perform an anaplerotic function for presynaptic neurons, providing material for the de novo synthesis of glutamate, without a net anaplerotic reaction. In these cycles, astrocytes convert glutamate to OG via GDH, and OG to another TCA intermediate via TCA cycle reactions. The net production of TCA intermediates by astrocytes is supported by the net consumption of glutamate and requires no net anaplerotic reaction. However, the fact that these proposed cycles require no net anaplerosis does not preclude the possibility that they may be mediated by an anaplerotic process, provided that this process is effectively reversed during the execution of the complete cycle.
Among the proposed cycles for functional glutamate trafficking elaborated in the current analyses, the GGMC may be unique in that it is potentially mediated by an anaplerotic process. The hypothesized phases of the proposed GGMC represent an elaboration of this possibility: An anaplerotic process executed during the glutamate production phase of the cycle, the production of malate in the carboxylation of pyruvate by means of cME in astrocytes, is reversed in the glutamate consumption phase of the cycle. In the glutamate production phase of the GGMC, de novo synthesis of glutamate from lactate and glucose is a composite process that begins with the production of malate from lactate via LDH and cME in astrocytes, proceeds with the production of OG from glucose and malate via glycolysis, mPDH, mMDH, mCS, mAH, and mIDH in presynaptic neurons, and results in the production of neurotransmitter glutamate from OG via GDH in presynaptic neurons. In the glutamate consumption phase of the GGMC, oxidative degradation of neurotransmitter glutamate is a composite process in astrocytes that begins with the production of OG from glutamate via GDH, includes TCA cycle activity to convert OG to malate, and results in the production of lactate from malate via cME and LDH.
Within the context of the biphasic representation of the proposed GGMC, cME in astrocytes may serve at least two functions. First, bi-directional activity of cME in astrocytes may allow lactate to serve as a reservoir for malate derived from the partial oxidation of glutamate, and thereby minimize disruption in the homeostasis of TCA cycle intermediates and glutamate during transient changes in rates of functional glutamate trafficking. According to this hypothesis, during transient increases in rates of functional glutamate trafficking, cME activity favors the production of malate (and the depletion of lactate) in support of increasing rates of glutamate production in presynaptic neurons relative to rates of glutamate consumption in astrocytes. Accordingly, during transient decreases in rates of functional glutamate trafficking, cME favors the consumption of malate (and the accumulation of lactate) in support of decreasing rates of glutamate production in presynaptic neurons relative to rates of glutamate consumption in astrocytes. This interpretation of the biphasic representation of the proposed GGMC integrates basic elements of Hassel and Sonnewald’s (1995) hypothesized glutamate-lactate cycle with basic elements of the glutamate-glutamine cycle. Transient imbalances in the relative rates of glutamate production in presynaptic neurons and glutamate consumption in astrocytes may be reflected in transient changes in concentrations of lactate and glutamine as opposed to transient changes in concentrations of TCA cycle intermediates and glutamate.
Second, bi-directional activity of cME in astrocytes may modulate the availability of malate for glutamate production in presynaptic neurons, and thereby modulate the function of mitochondria in presynaptic neurons. Neurotransmitter trafficking, one component of a larger process of synaptic neurotransmission, must be coordinated with other component processes, including those involved in the modulation of ionic potentials across plasma membranes of presynaptic nerve endings. In the biphasic representation of the proposed GGMC, glutamate production in presynaptic neurons is dependent on material inputs provided by astrocytes. During the hypothesized glutamate production phase of the cycle, astrocytes transport malate (as well as glutamine) to presynaptic neurons in support of glutamate production and packaging into vesicles. During this phase of the cycle, mitochondria in presynaptic neurons produce OG from glucose and malate. Depolarization of presynaptic nerve endings precipitates the release of glutamate into synaptic clefts and initiates the glutamate consumption phase of the GGMC cycle. During this phase, mitochondria in presynaptic nerve terminals are unable to produce glutamate (in the absence of a source of malate) and therefore completely oxidize glucose or lactate in support of ATP production, which drives the restoration of ionic membrane potentials of presynaptic nerve terminals. That is, according to this hypothesis, mitochondria in presynaptic neurons shift between two essential functions, OG production in support of glutamate synthesis during the glutamate production phase, and complete substrate (glucose or lactate) oxidation in support of the restoration of ionic membrane potentials during the glutamate consumption phase, of the proposed GGMC, in coordination with the availability and transport of malate from astrocytes to presynaptic neurons. This interpretation of the biphasic representation of the GGMC is compatible with Kaufman and Driscoll’s (1993) observation that astrocytes increase release of organic acid products of CO2 fixation (presumably including malate) in response to increases in extracellular potassium (presumably a consequence of neuronal depolarization). It is compatible with Waagepetersen et al.’s (2000b) observation that depolarization increases oxidative metabolism in neurons, and hypothesis that neurons possess distinct, heterogeneous mitochondria (Sonnewald et al., 1998), specialized for neurotransmitter synthesis and energy metabolism, that are influenced by depolarization. It is also compatible with Bak et al.’s (2006) observations that depolarization increases neuronal glucose metabolism and that glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons.
In the overall GGMC, there is no net anaplerosis, so that carboxylation of pyruvate in the production of a TCA cycle intermediate metabolite during one phase of the GGMC must be balanced by the decarboxylation of a TCA cycle intermediate in the production of pyruvate during a second phase of the GGMC. In the current analyses, both of these transformations are proposed to be accomplished in astrocytes by a single enzyme, cME, whose bi-directional kinetic properties have been established in molecular studies (Frenkel, 1972; Simpson and Estabrook, 1969; Bukato et al. 1995a). There is considerable evidence that carboxylation of pyruvate is active (Hassel and Brathe, 2000; Lieth et al., 2001; Gonzalez et al., 2005) and activity-dependent (Oz et al., 2004) in vivo. Although carboxylation of pyruvate in astrocytes is commonly attributed to mPC, it may be due to cME. Consistent with Bukato’s (1995a) suggestion that cME is reversible under in vivo conditions, Gonzales et al. (2005) report that exogenous pyruvate undergoes reversible carboxylation in brain and indicate that glial cME cannot be excluded as a explanation for this phenomenon. In principle, carboxylation of pyruvate in astrocytes during the glutamate production phase of the GGMC could be accomplished by mPC rather than cME However, because mPC is an irreversible, mitochondrial enzyme that consumes ATP, carboxylation of pyruvate in the production of a TCA cycle intermediate in astrocytes for export to presynaptic neurons is less direct and less efficient energetically by the mPC pathway as compared to the cME pathway. Studies of astrocytes in cell culture demonstrate cME activity in the direction of the decarboxylation of malate based on experimental conditions that favor the production of pyruvate from malate (McKenna et al., 1995; Alves et al., 2000).
Integration of insights derived from cell culture studies
Studies in cell culture provide insight into the functional potentialities of different cell types, possibilities which may be operative in vivo predominantly, only under certain circumstances, or perhaps not at all (Hertz et al., 1998). The present study provides a framework within which apparently disparate functional potentialities of neurons and astrocytes, established in cell culture, can be seen as essential elements of potential cycles for functional glutamate trafficking in synaptic neurotransmission in vivo. For example, within the biphasic representation of the proposed GGMC, astrocytes convert exogenous glutamate to glutamine (Waniewski and Martin, 1986), metabolize exogenous glutamate through TCA cycle reactions to generate lactate from malate (Sonnewald et al., 1993; Schousboe et al., 1993; Westergaard et al., 1995), export glutamine and malate (Waniewski and Martin, 1986; McKenna et al., 1996a; McKenna et al., 1989; Sonnewald et al., 1993; Westergaard et al., 1994a), and utilize ammonium derived from GDH to support glutamine synthesis (Sonnewald et al., 1997). In this way, these proposed cycles for functional glutamate trafficking integrate specific functional potentialities of neurons and astrocytes into a larger functional whole. Consequently, it may be possible, some day, to demonstrate that specific sets of functional potentialities of neurons and astrocytes, established in cell culture, are operative in vivo, based on a demonstration that particular cycles for functional glutamate trafficking that are operative in vivo.
Possibility of other cycles for functional glutamate trafficking
The present study has focused on previously and newly proposed cycles for functional glutamate trafficking that incorporate the coordinated, compartmentalized activities of PAG in presynaptic neurons and GS in astrocytes. Studies in cell culture clearly demonstrate that neurons have the capacity to produce glutamate from combinations of TCA cycle intermediates and amino acids other than glutamine, e.g., from OG and alanine (Peng et al., 1991; Peng et al., 1993; Peng et al., 1994). Therefore, some cycles for functional glutamate trafficking that depend on neither PAG in presynaptic neurons nor GS in astrocytes may exist. For example, OG and alanine, produced from neurotransmitter glutamate and pyruvate via ALAT in astrocytes, might be transported from astrocytes to presynaptic neurons and converted back to pyruvate and neurotransmitter glutamate via ALAT in presynaptic neurons. In this case, glycolysis might produce pyruvate consumed by ALAT in astrocytes, and TCA cycle processes might consume pyruvate produced by ALAT in presynaptinc neurons.
Conclusion
Each of the hypothetical, proposed cycles for functional glutamate trafficking elaborated in the current study identifies a set of molecular mechanisms by which PAG and GS activities may be coordinated in synaptic function in vivo. These proposed cycles, which represent only part of the metabolism and should not be interpreted as models for total cell or brain metabolism, are neither mutually exclusive nor exhaustive. The extent to which any variant of the glutamateglutamine cycle, any of the hypothetical, newly proposed alternative cycles, or some other cycle is operative in synaptic function in vivo is at present an open question. Presumably, for a given physiological state, spatially and temporally localized chemical environments within various intracellular and intercellular compartments favor chemical reactions and transport processes associated with one cycle for functional glutamate trafficking as opposed to another. It may be that different cycles play more prominent roles in different physiological states. For example, the ammonia diffusion variant of the glutamate-glutamine cycle, which apparently does not require oxygen to function, may play a greater role in hypoxic states than do cycles, such as the newly proposed cycles based on the transport of TCA cycle intermediate metabolites from astrocytes to presynaptic neurons, that do require oxidative processes to function. Alternatively, during intense synaptic activity, the intrinsic properties of these other, newly proposed cycles may provide significant functional advantages.
The current study serves as a guide to future research in functional glutamate trafficking in vivo. To date, data from magnetic resonance spectroscopy isotopic label transfer studies have been used primarily to estimate in vivo rates of metabolism and neurotransmitter trafficking within the context of a specific cycle for functional glutamate trafficking, i.e., the glutamateglutamine cycle (e.g., Gruetter et al., 1998; Shen et al., 1999). However, the question of which cycle for functional glutamate trafficking predominates in vivo for a given physiological state must be answered before one can obtain any meaningful estimates of cycle parameters, which have meaning only within the context of a particular model. At a minimum, the current investigation suggests that it may be premature to presume that in vivo functional glutamate trafficking proceeds via some variant of the glutamate-glutamine cycle alone. The immediate challenge will be to design in vivo experiments to discriminate between variants of the glutamate-glutamine cycle, between the glutamate-glutamine cycle and cycles based on the transport of TCA cycle intermediates from astrocytes to presynaptic neurons, and between cycles based on the transport of one TCA intermediate as opposed to another. The quantitative stoichiometry derived for each cycle presented here will provide a guide for future experimental determinations of the degree to which each of these proposed cycles contributes to functional glutamate trafficking in vivo.
Acknowledgements
This work was supported by NIH NS-044316 (PKM) and NS-035527 (DLR). The authors also acknowledge helpful discussions with Kevin L. Behar on metabolic compartmentalization.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alves PM, Nunes R, Zhang C, Maycock CD, Sonnewald U, Carrondo MJ, Santos H. Metabolism of 3-(13)C-malate in primary cultures of mouse astrocytes. Dev Neurosci. 2000;22:456–462. doi: 10.1159/000017475. [DOI] [PubMed] [Google Scholar]
- Aoki C, Kaneko T, Starr A, Pickel VM. Identification of mitochondrial and non-mitochondrial glutaminase within select neurons and glia of rat forebrain by electron microscopic immunocytochemistry. J Neurosci Res. 1991;28:531–548. doi: 10.1002/jnr.490280410. [DOI] [PubMed] [Google Scholar]
- Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS. Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J Cereb Blood Flow Metab. 2006 doi: 10.1038/sj.jcbfm.9600281. [DOI] [PubMed] [Google Scholar]
- Bak LK, Sickmann HM, Schousboe A, Waagepetersen HS. Activity of the lactate-alanine shuttle is independent of glutamate-glutamine cycle activity in cerebellar neuronal-astrocytic cultures. J Neurosci Res. 2005;79:88–96. doi: 10.1002/jnr.20319. [DOI] [PubMed] [Google Scholar]
- Bakken IJ, White LR, Aasly J, Unsgard G, Sonnewald U. Lactate formation from [U-13C]aspartate in cultured astrocytes: compartmentation of pyruvate metabolism. Neurosci Lett. 1997;237:117–120. doi: 10.1016/s0304-3940(97)00834-3. [DOI] [PubMed] [Google Scholar]
- Benjamin AM, Quastel JH. Metabolism of amino acids and ammonia in rat brain cortex slices in vitro: a possible role of ammonia in brain function. J Neurochem. 1975;25:197–206. doi: 10.1111/j.1471-4159.1975.tb06953.x. [DOI] [PubMed] [Google Scholar]
- Broer S, Brookes N. Transfer of glutamine between astrocytes and neurons. J Neurochem. 2001;77:705–719. doi: 10.1046/j.1471-4159.2001.00322.x. [DOI] [PubMed] [Google Scholar]
- Bukato G, Kochan Z, Swierczynski J. Different regulatory properties of the cytosolic and mitochondrial forms of malic enzyme isolated from human brain. Int J Biochem Cell Biol. 1995a;27:1003–1008. doi: 10.1016/1357-2725(95)00080-9. [DOI] [PubMed] [Google Scholar]
- Bukato G, Kochan Z, Swierczynski J. Purification and properties of cytosolic and mitochondrial malic enzyme isolated from human brain. Int J Biochem Cell Biol. 1995b;27:47–54. doi: 10.1016/1357-2725(94)00057-3. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kraig RP. Intracellular pH of astrocytes increases rapidly with cortical stimulation. Am J Physiol. 1987;253:R666–R670. doi: 10.1152/ajpregu.1987.253.4.R666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, Kraig RP. Intracellular pH transients of mammalian astrocytes. J Neurosci. 1989;9:2011–2019. doi: 10.1523/JNEUROSCI.09-06-02011.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF. Neighborly interactions of metabolically-activated astrocytes in vivo. Neurochem Int. 2003;43:339–354. doi: 10.1016/s0197-0186(03)00021-4. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Hertz L. Glucose and lactate metabolism during brain activation. J Neurosci Res. 2001;66:824–838. doi: 10.1002/jnr.10079. [DOI] [PubMed] [Google Scholar]
- Faff-Michalak L, Albrecht J. Hyperammonemia and hepatic encephalopathy stimulate rat cerebral synaptic mitochondrial glutamate dehydrogenase activity specifically in the direction of glutamate oxidation. Brain Res. 1993;618:299–302. doi: 10.1016/0006-8993(93)91279-2. [DOI] [PubMed] [Google Scholar]
- Frenkel R. Isolation and some properties of a cytosol and a mitochondrial malic enzyme from bovine brain. Arch Biochem Biophys. 1972;152:136–143. doi: 10.1016/0003-9861(72)90201-9. [DOI] [PubMed] [Google Scholar]
- Gonzalez SV, Nguyen NH, Rise F, Hassel B. Brain metabolism of exogenous pyruvate. J Neurochem. 2005 doi: 10.1111/j.1471-4159.2005.03365.x. [DOI] [PubMed] [Google Scholar]
- Gruetter R, Seaquist ER, Kim S, Ugurbil K. Localized in vivo 13C-NMR of glutamate metabolism in the human brain: initial results at 4 tesla. Dev Neurosci. 1998;20:380–388. doi: 10.1159/000017334. [DOI] [PubMed] [Google Scholar]
- Gruetter R, Seaquist ER, Ugurbil K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab. 2001;281:E100–E112. doi: 10.1152/ajpendo.2001.281.1.E100. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J. 1999;343(Pt 2):281–299. [PMC free article] [PubMed] [Google Scholar]
- Hamberger AC, Chiang GH, Nylen ES, Scheff SW, Cotman CW. Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res. 1979;168:513–530. doi: 10.1016/0006-8993(79)90306-8. [DOI] [PubMed] [Google Scholar]
- Hassel B, Brathe A. Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J Neurosci. 2000;20:1342–1347. doi: 10.1523/JNEUROSCI.20-04-01342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Sonnewald U. Glial formation of pyruvate and lactate from TCA cycle intermediates: implications for the inactivation of transmitter amino acids? J Neurochem. 1995;65:2227–2234. doi: 10.1046/j.1471-4159.1995.65052227.x. [DOI] [PubMed] [Google Scholar]
- Hertz L. Intercellular metabolic compartmentation in the brain: past, present and future. Neurochem Int. 2004;45:285–296. doi: 10.1016/j.neuint.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res. 1999;57:417–428. [PubMed] [Google Scholar]
- Hertz L, Hertz E. Cataplerotic TCA cycle flux determined as glutamate-sustained oxygen consumption in primary cultures of astrocytes. Neurochem Int. 2003;43:355–361. doi: 10.1016/s0197-0186(03)00022-6. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Lai JC. Functional studies in cultured astrocytes. Methods. 1998;16:293–310. doi: 10.1006/meth.1998.0686. [DOI] [PubMed] [Google Scholar]
- Hogstad S, Svenneby G, Torgner IA, Kvamme E, Hertz L, Schousboe A. Glutaminase in neurons and astrocytes cultured from mouse brain: kinetic properties and effects of phosphate, glutamate, and ammonia. Neurochem Res. 1988;13:383–388. doi: 10.1007/BF00972489. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zielke CL, Tildon JT, Zielke HR. Monitoring in vivo oxidation of 14C-labelled substrates to 14CO2 by brain microdialysis. Dev Neurosci. 1993;15:233–239. doi: 10.1159/000111339. [DOI] [PubMed] [Google Scholar]
- Hutson SM, Berkich D, Drown P, Xu B, Aschner M, LaNoue KF. Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J Neurochem. 1998;71:863–874. doi: 10.1046/j.1471-4159.1998.71020863.x. [DOI] [PubMed] [Google Scholar]
- Inoue K, Fei YJ, Zhuang L, Gopal E, Miyauchi S, Ganapathy V. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem J. 2004;378:949–957. doi: 10.1042/BJ20031261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam PL, Lin CC, Li JC, Meng CL, Chang GG. High malic enzyme activity in tumor cells and its cross-reaction with anti-pigeon liver malic enzyme serum. Mol Cell Biochem. 1988;79:171–179. doi: 10.1007/BF02424560. [DOI] [PubMed] [Google Scholar]
- Kaufman EE, Driscoll BF. Evidence for cooperativity between neurons and astroglia in the regulation of CO2 fixation in vitro. Dev Neurosci. 1993;15:299–305. doi: 10.1159/000111348. [DOI] [PubMed] [Google Scholar]
- Kurz GM, Wiesinger H, Hamprecht B. Purification of cytosolic malic enzyme from bovine brain, generation of monoclonal antibodies, and immunocytochemical localization of the enzyme in glial cells of neural primary cultures. J Neurochem. 1993;60:1467–1474. doi: 10.1111/j.1471-4159.1993.tb03309.x. [DOI] [PubMed] [Google Scholar]
- Kvamme E, Roberg B, Torgner IA. Phosphate-activated glutaminase and mitochondrial glutamine transport in the brain. Neurochem Res. 2000;25:1407–1419. doi: 10.1023/a:1007668801570. [DOI] [PubMed] [Google Scholar]
- Kvamme E, Svenneby G, Hertz L, Schousboe A. Properties of phosphate activated glutaminase in astrocytes cultured from mouse brain. Neurochem Res. 1982;7:761–770. doi: 10.1007/BF00965528. [DOI] [PubMed] [Google Scholar]
- Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL. Astroglial contribution to brain energy metabolism in humansrevealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieth E, LaNoue KF, Berkich DA, Xu B, Ratz M, Taylor C, Hutson SM. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J Neurochem. 2001;76:1712–1723. doi: 10.1046/j.1471-4159.2001.00156.x. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004;447:784–795. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- Marcaggi P, Coles JA. Ammonium in nervous tissue: transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog Neurobiol. 2001;64:157–183. doi: 10.1016/s0301-0082(00)00043-5. [DOI] [PubMed] [Google Scholar]
- Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. 1977;195:1356–1358. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR. Exogenous glutamateconcentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem. 1996a;66:386–393. doi: 10.1046/j.1471-4159.1996.66010386.x. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Stevenson JH, Huang X, Hopkins IB. Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem Int. 2000;37:229–241. doi: 10.1016/s0197-0186(00)00042-5. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Tildon JT, Stevenson J, Couto R, Caprio FJ. Stimulation of malate release from astrocytes by glutamate. Transact. Am. Soc. Neurochem. 1989;20:A191. [Google Scholar]
- McKenna MC, Tildon JT, Stevenson JH, Huang X. New insights into the compartmentation of glutamate and glutamine in cultured rat brain astrocytes. Dev Neurosci. 1996b;18:380–390. doi: 10.1159/000111431. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Tildon JT, Stevenson JH, Huang X, Kingwell KG. Regulation of mitochondrial and cytosolic malic enzymes from cultured rat brain astrocytes. Neurochem Res. 1995;20:1491–1501. doi: 10.1007/BF00970599. [DOI] [PubMed] [Google Scholar]
- Mellergard P, Ou-Yang Y, Siesjo BK. Relationship between intra- and extracellular pH in primary cultures of rat astrocytes. Am J Physiol. 1994;267:C581–C589. doi: 10.1152/ajpcell.1994.267.2.C581. [DOI] [PubMed] [Google Scholar]
- Nagaraja TN, Brooks N. Intracellular acidification induced by passive and active transport of ammonium ions in astrocytes. Am J Physiol. 1998;274:C883–C891. doi: 10.1152/ajpcell.1998.274.4.C883. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. The glutamatergic nerve terminal. Eur J Biochem. 1993;212:613–631. doi: 10.1111/j.1432-1033.1993.tb17700.x. [DOI] [PubMed] [Google Scholar]
- Orlowski J, Grinstein S. Na+/H+ exchangers of mammalian cells. J Biol Chem. 1997;272:22373–22376. doi: 10.1074/jbc.272.36.22373. [DOI] [PubMed] [Google Scholar]
- Oz G, Berkich DA, Henry PG, Xu Y, LaNoue K, Hutson SM, Gruetter R. Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J Neurosci. 2004;24:11273–11279. doi: 10.1523/JNEUROSCI.3564-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Hull C, Vigh J, von Gersdorff H. Synaptic cleft acidification and modulation of short-term depression by exocytosed protons in retinal bipolar cells. J Neurosci. 2003;23:11332–11341. doi: 10.1523/JNEUROSCI.23-36-11332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Mason GF, Kanamatsu T, Rothman DL, Shulman RG, Behar KL. Glutamatergic neurotransmission and neuronal glucose oxidation are coupled during intense neuronal activation. J Cereb Blood Flow Metab. 2004;24:972–985. doi: 10.1097/01.WCB.0000126234.16188.71. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Hunt A, Gordon RD, Balazs R. The activities in different neural cell types of certain enzymes associated with the metabolic compartmentation glutamate. Brain Res. 1982;256:3–11. doi: 10.1016/0165-3806(82)90091-8. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bergersen LH, Halestrap AP, Pierre K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res. 2005;80:739. doi: 10.1002/jnr.20307. [DOI] [PubMed] [Google Scholar]
- Peng L, Hertz L, Huang R, Sonnewald U, Petersen SB, Westergaard N, Larsson O, Schousboe A. Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev Neurosci. 1993;15:367–377. doi: 10.1159/000111357. [DOI] [PubMed] [Google Scholar]
- Peng L, Swanson RA, Hertz L. Effects of L-glutamate, D-aspartate, and monensin on glycolytic and oxidative glucose metabolism in mouse astrocyte cultures: further evidence that glutamate uptake is metabolically driven by oxidative metabolism. Neurochem Int. 2001;38:437–443. doi: 10.1016/s0197-0186(00)00104-2. [DOI] [PubMed] [Google Scholar]
- Peng L, Zhang X, Hertz L. Alteration in oxidative metabolism of alanine in cerebellar granule cell cultures as a consequence of the development of the ability to utilize alanine as an amino group donor for synthesis of transmitter glutamate. Brain Res Dev Brain Res. 1994;79:128–131. doi: 10.1016/0165-3806(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Peng LA, Schousboe A, Hertz L. Utilization of alpha-ketoglutarate as a precursor for transmitter glutamate in cultured cerebellar granule cells. Neurochem Res. 1991;16:29–34. doi: 10.1007/BF00965824. [DOI] [PubMed] [Google Scholar]
- Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94:1–14. doi: 10.1111/j.1471-4159.2005.03168.x. [DOI] [PubMed] [Google Scholar]
- Rothman DL, Behar KL, Hyder F, Shulman RG. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu Rev Physiol. 2003;65:401–427. doi: 10.1146/annurev.physiol.65.092101.142131. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Drejer J, Hertz L. Uptake and release of glutamate and glutamine in neurons and astrocytes in primary cultures. In: Kvamme E, editor. Glutamate and Glutamine in Mammals. Boca Raton, FL: CRC Press; 1988. pp. 21–39. [Google Scholar]
- Schousboe A, Westergaard N, Sonnewald U, Petersen SB, Huang R, Peng L, Hertz L. Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev Neurosci. 1993;15:359–366. doi: 10.1159/000111356. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Westergaard N, Waagepetersen HS, Larsson OM, Bakken IJ, Sonnewald U. Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia. 1997;21:99–105. [PubMed] [Google Scholar]
- Shank RP, Bennett GS, Freytag SO, Campbell GL. Pyruvate carboxylase: an astrocytespecific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985;329:364–367. doi: 10.1016/0006-8993(85)90552-9. [DOI] [PubMed] [Google Scholar]
- Shank RP, Campbell GL. Glutamine and alpha-ketoglutarate uptake and metabolism by nerve terminal enriched material from mouse cerebellum. Neurochem Res. 1982;7:601–616. doi: 10.1007/BF00965126. [DOI] [PubMed] [Google Scholar]
- Shank RP, Campbell GL. Alpha-ketoglutarate and malate uptake and metabolism by synaptosomes: further evidence for an astrocyte-to-neuron metabolic shuttle. J Neurochem. 1984a;42:1153–1161. doi: 10.1111/j.1471-4159.1984.tb12724.x. [DOI] [PubMed] [Google Scholar]
- Shank RP, Campbell GL. Glutamine, glutamate, and other possible regulators of alpha-ketoglutarate and malate uptake by synaptic terminals. J Neurochem. 1984b;42:1162–1169. doi: 10.1111/j.1471-4159.1984.tb12725.x. [DOI] [PubMed] [Google Scholar]
- Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, Petroff OA, Shulman GI, Shulman RG, Rothman DL. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Natl Acad Sci U S A. 1999;96:8235–8240. doi: 10.1073/pnas.96.14.8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci U S A. 1998;95:316–321. doi: 10.1073/pnas.95.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo B. Brain energy metabolism. New York: Wiley & Sons; 1978. [Google Scholar]
- Simpson ER, Estabrook RW. Mitochondrial malic enzyme: the source of reduced nicotinamide adenine dinucleotide phosphate for steroid hydroxylation in bovine adrenal cortex mitochondria. Arch Biochem Biophys. 1969;129:384–395. doi: 10.1016/0003-9861(69)90190-8. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Hertz L, Schousboe A. Mitochondrial heterogeneity in the brain at the cellular level. J Cereb Blood Flow Metab. 1998;18:231–237. doi: 10.1097/00004647-199803000-00001. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Schousboe A, Qu H, Waagepetersen HS. Intracellular metabolic compartmentation assessed by 13C magnetic resonance spectroscopy. Neurochem Int. 2004;45:305–310. doi: 10.1016/j.neuint.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Jones P, Taylor A, Bachelard HS, Schousboe A. Metabolism of [U-13C5] glutamine in cultured astrocytes studied by NMR spectroscopy: first evidence of astrocytic pyruvate recycling. J Neurochem. 1996;67:2566–2572. doi: 10.1046/j.1471-4159.1996.67062566.x. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Petersen SB, Unsgard G, Schousboe A. Metabolism of [U-13C]glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J Neurochem. 1993;61:1179–1182. doi: 10.1111/j.1471-4159.1993.tb03641.x. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Schousboe A. Glutamate transport and metabolism in astrocytes. Glia. 1997;21:56–63. doi: 10.1002/(sici)1098-1136(199709)21:1<56::aid-glia6>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Takanaga H, Mackenzie B, Peng JB, Hediger MA. Characterization of a branched-chain amino-acid transporter SBAT1 (SLC6A15) that is expressed in human brain. Biochem Biophys Res Commun. 2005;337:892–900. doi: 10.1016/j.bbrc.2005.09.128. [DOI] [PubMed] [Google Scholar]
- Talley EM, Sirois JE, Lei Q, Bayliss DA. Two-pore-Domain (KCNK) potassium channels: dynamic roles in neuronal function. Neuroscientist. 2003;9:46–56. doi: 10.1177/1073858402239590. [DOI] [PubMed] [Google Scholar]
- van den Berg CJ, Garfinkel D. A stimulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem J. 1971;123:211–218. doi: 10.1042/bj1230211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R, Jennemann G, Seitz J, Wiesinger H, Hamprecht B. Mitochondrial malic enzyme: purification from bovine brain, generation of an antiserum, and immunocytochemical localization in neurons of rat brain. J Neurochem. 1998;71:844–852. doi: 10.1046/j.1471-4159.1998.71020844.x. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Hansen GH, Fenger K, Lindsay JG, Gibson G, Schousboe A. Cellular mitochondrial heterogeneity in cultured astrocytes as demonstrated by immunogold labeling of alpha-ketoglutarate dehydrogenase. Glia. 2006;53:225–231. doi: 10.1002/glia.20276. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Qu H, Hertz L, Sonnewald U, Schousboe A. Demonstration of pyruvate recycling in primary cultures of neocortical astrocytes but not in neurons. Neurochem Res. 2002;27:1431–1437. doi: 10.1023/a:1021636102735. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Larsson OM, Schousboe A. A possible role of alanine for ammonia transfer between astrocytes and glutamatergic neurons. J Neurochem. 2000a;75:471–479. doi: 10.1046/j.1471-4159.2000.0750471.x. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Larsson OM, Schousboe A. Compartmentation of TCA cycle metabolism in cultured neocortical neurons revealed by 13C MR spectroscopy. Neurochem Int. 2000b;36:349–358. doi: 10.1016/s0197-0186(99)00143-6. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Larsson OM, Schousboe A. Multiple compartments with different metabolic characteristics are involved in biosynthesis of intracellular and released glutamine and citrate in astrocytes. Glia. 2001;35:246–252. doi: 10.1002/glia.1089. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Exogenous glutamate is metabolized to glutamine and exported by rat primary astrocyte cultures. J Neurochem. 1986;47:304–313. doi: 10.1111/j.1471-4159.1986.tb02863.x. [DOI] [PubMed] [Google Scholar]
- Westergaard N, Drejer J, Schousboe A, Sonnewald U. Evaluation of the importance of transamination versus deamination in astrocytic metabolism of [U-13C]glutamate. Glia. 1996;17:160–168. doi: 10.1002/(SICI)1098-1136(199606)17:2<160::AID-GLIA7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Westergaard N, Sonnewald U, Schousboe A. Release of alpha-ketoglutarate, malate and succinate from cultured astrocytes: possible role in amino acid neurotransmitter homeostasis. Neurosci Lett. 1994a;176:105–109. doi: 10.1016/0304-3940(94)90882-6. [DOI] [PubMed] [Google Scholar]
- Westergaard N, Sonnewald U, Schousboe A. Metabolic trafficking between neurons and astrocytes: the glutamate/glutamine cycle revisited. Dev Neurosci. 1995;17:203–211. doi: 10.1159/000111288. [DOI] [PubMed] [Google Scholar]
- Westergaard N, Sonnewald U, Unsgard G, Peng L, Hertz L, Schousboe A. Uptake, release, and metabolism of citrate in neurons and astrocytes in primary cultures. J Neurochem. 1994b;62:1727–1733. doi: 10.1046/j.1471-4159.1994.62051727.x. [DOI] [PubMed] [Google Scholar]
- Wurdig S, Kugler P. Histochemistry of glutamate metabolizing enzymes in the rat cerebellar cortex. Neurosci Lett. 1991;130:165–168. doi: 10.1016/0304-3940(91)90388-a. [DOI] [PubMed] [Google Scholar]
- Yu AC, Drejer J, Hertz L, Schousboe A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem. 1983;41:1484–1487. doi: 10.1111/j.1471-4159.1983.tb00849.x. [DOI] [PubMed] [Google Scholar]
- Yu AC, Schousboe A, Hertz L. Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J Neurochem. 1982;39:954–960. doi: 10.1111/j.1471-4159.1982.tb11482.x. [DOI] [PubMed] [Google Scholar]
- Yudkoff M. Brain metabolism of branched-chain amino acids. Glia. 1997;21:92–98. doi: 10.1002/(sici)1098-1136(199709)21:1<92::aid-glia10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Grunstein L, Nissim I, Stern J, Pleasure D. Astrocyte leucine metabolism: significance of branched-chain amino acid transamination. J Neurochem. 1996;66:378–385. doi: 10.1046/j.1471-4159.1996.66010378.x. [DOI] [PubMed] [Google Scholar]
- Zaganas I, Waagepetersen HS, Georgopoulos P, Sonnewald U, Plaitakis A, Schousboe A. Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J Neurosci Res. 2001;66:909–913. doi: 10.1002/jnr.10058. [DOI] [PubMed] [Google Scholar]
- Zwingmann C, Leibfritz D. Regulation of glial metabolism studied by 13C-NMR. NMR Biomed. 2003;16:370–399. doi: 10.1002/nbm.850. [DOI] [PubMed] [Google Scholar]