

Abstract
Glial modulation of synaptic transmission and neuronal excitability in the mammalian retina is mediated by several mechanisms. Stimulation of glial cells evokes Ca2+ waves, which propagate through the network of retinal astrocytes and Müller cells and result in the modulation of the activity of neighboring ganglion cells. Light-evoked spiking is enhanced in some ganglion cells and depressed in others. A facilitation or depression of light-evoked excitatory postsynaptic currents is also seen in ganglion cells following glial stimulation. In addition, stimulation of glial cells evokes a sustained hyperpolarizing current in ganglion cells which is mediated by ATP release from Müller cells and activation of neuronal A1 adenosine receptors. Recent studies reveal that light-evoked activity in retinal neurons results in an increase in the frequency of Ca2+ transients in Müller cells. Thus, there is two-way communication between neurons and glial cells, suggesting that glia contribute to information processing in the retina.
Keywords: Müller cell, astrocyte, neuron, synapse, enhancement, depression, ATP, glutamate, calcium wave
INTRODUCTION
Glial modulation of synaptic function was initially described in cocultures of neurons and astrocytes. These studies demonstrated that neurotransmitters can evoke Ca2+ increases in astrocytes and that transmitters released from astrocytes can modulate synaptic transmission between neurons (see Araque and Perea in this issue). More recently, experiments have been extended from culture to in situ systems, where neuronglia interactions have been characterized in greater detail (see Evanko et al., Oliet et al., and Colomar and Robitaille in this issue).
The mammalian retina has proved to be a valuable preparation for examining neuronglia interactions in intact CNS tissue. The isolated retina preparation offers several advantages. The retina can be removed from the eye and maintained in a chamber in an intact state. It can be activated by its natural stimulus, light, as well as by electrical stimulation. The electrical activity of ganglion cells, the output neurons of the retina, can be monitored with both extracellular and intracellular recordings. Notably, retinal glial cells form a two-dimensional network at the retinal surface, where their Ca2+ activity as well as their electrical responses are easily monitored.
There are two types of macroglial cells in the mammalian retina, astrocytes and Müller cells (Fig. 1) (Newman, 2001a; Sarthy and Ripps, 2001). Astrocytes are restricted to the vitreal surface of the retina, where they are located largely within the nerve fiber layer. Müller cells, specialized radial glial cells, extend deep into the retina. Their endfeet form the inner (vitreal) border of the retina while their apical processes extend into the photoreceptor layer. Retinal glial cells are chemically and electrically coupled together by gap junctions (Zahs and Newman, 1997; Ceelen et al., 2001).
Fig. 1.

Astrocytes and Müller cells, the two macroglial cells of the retina, are shown in this schematic of the mammalian retina. Astrocytes are confined largely to the nerve fiber layer at the vitreal (inner) border of the retina. Müller cells, specialized radial glial cells found only in the retina, span much of the retinal depth, from the vitreal border to the photoreceptor layer. The principal neurons of the retina are illustrated, as are the recording and stimulating electrodes used in experiments to monitor glial cell modulation of neuronal spiking.
GLIA-GLIA COMMUNICATION
Studies of neuronglia interactions in the retina have, to date, focused primarily on changes in neuronal excitability elicited by activation of glial cells. Before discussing these experiments, it will be useful to describe the experimental techniques used to activate retinal glia. The mechanisms by which glial cells communicate with each other will also be discussed in this section.
Glial Ca2+ Increases
Application of many neurotransmitters results in a rise in intracellular Ca2+ in both astrocytes and Müller cells at the surface of the retina. ATP and the ATP analog ATPγS are most effective in eliciting Ca2+ increases, typically a sustained Ca2+ rise in astrocytes and transient Ca2+ increases and oscillations in Müller cells (Newman and Zahs, 1997; Newman, 2003). Carbachol, phenylephrine, dopamine, thrombin, and lysophosphatidic acid also elicit Ca2+ increases, although these Ca2+ responses are normally smaller than those evoked by ATP (Newman, 2003). Calcium increases in retinal glial cells are generated by the release of Ca2+ from thapsigargin-sensitive internal stores (Newman and Zahs, 1997).
Glutamate, even at high concentration (1 mM), is ineffective in producing Ca2+ increases in retinal astrocytes and Müller cells (Newman and Zahs, 1997), even though it evokes large Ca2+ increases in brain astrocytes. It is not clear what accounts for the insensitivity of retinal glial cells to glutamate, although functionally this insensitivity seems reasonable as extracellular glutamate levels are normally high (~10 μM) at the retinal surface in vivo (Dreyer et al., 1996). Increases in glial Ca2+ are also evoked by mechanical and electrical stimuli applied to single astrocyte somata (Newman and Zahs, 1997).
Glial Ca2+ Waves
Activation of retinal glial cells with chemical, mechanical, or electrical stimuli often initiate propagated Ca2+ waves (Newman and Zahs, 1997; Newman, 2001b). These Ca2+ waves travel through both astrocytes and Müller cells, even when the wave is initiated by stimulating a single astrocyte soma. The Ca2+ waves travel at a velocity of ~23 μm/s and up to 180 μm from the site of initiation.
Calcium waves propagate between glial cells in the retina by two mechanisms, via diffusion of an intracellular messenger through gap junctions and by release of an extracellular messenger (Newman, 2001b). Several lines of evidence confirm the existence of these two mechanisms. When an astrocyte soma is stimulated mechanically, the resulting Ca2+ wave propagates continuously through the processes of the astrocyte and into adjacent astrocytes. After a delay of ~ 1 s, the wave also propagates into neighboring Müller cells (Fig. 2A). When glial purinergic receptors are blocked with suramin or PPADS, or when apyrase is added to the bath, propagation from astrocytes to Müller cells is either delayed or blocked entirely, while propagation between astrocytes is largely unaffected (Fig. 2B). The purinergic antagonists also block Ca2+ wave propagation from Müller cell to Müller cell when a wave is initiated by stimulating Müller cells.
Fig. 2.

Propagation of Ca2+ waves through astrocytes and Müller cells in the rat retina. A: A Ca2+ wave propagates through both astrocytes and Müller cells following mechanical stimulation of an astrocyte soma (asterisk). A as well as B, D, and E are pseudocolor Ca2+ ratio images that show Ca2+ increases within glial cells. B: In the presence of 100 μM suramin, a purinergic receptor antagonist, stimulation of an astrocyte (asterisk) evokes a Ca2+ wave that propagates through several astrocytes but fails to propagate into neighboring Müller cells. C: Stimulation of glial cells on the retinal surface evokes ATP release, detected by the luciferin-luciferase chemiluminescence assay. Five line scans show the spatial pattern of ATP chemiluminescence at the indicated times after stimulation. D: Superfusate flow across the retinal surface (from left to right) causes asymmetric propagation of a Ca2+ wave. E: Addition of suramin eliminates the asymmetry despite continued superfusate flow. A, B, D, and E reproduced from Newman (2001b); C from Newman (2003).
These experiments indicate that Ca2+ waves are propagated in part by the release of ATP from glial cells. This mechanism has been confirmed by imaging ATP release using the luciferin-luciferase chemiluminescence assay (Newman, 2001b). When a glial Ca2+ wave propagates across the retinal surface, a wave of ATP release precedes the Ca2+ wave by approximately 1 s (Fig. 2C). It is likely that ATP release occurs from the glial cells as the ATP chemiluminescence signal persists in the presence of Cd2+ (Newman, 2003), which blocks Ca2+-dependent ATP release from neurons.
When superfusate flows across the retinal surface, Ca2+ wave propagation is asymmetric, as released ATP is carried by the bath (Fig. 2D). If a purinergic antagonist is added to the flowing superfusate, this asymmetric propagation is eliminated (Fig. 2E), demonstrating that only ATP (or a related purinergic agonist) contributes to wave propagation as an extracellular messenger (Newman, 2001b).
Taken together, these experiments demonstrate that Ca2+ waves are propagated through glial cells in the retina by two mechanisms. Propagation between astrocytes is mediated largely by diffusion of an intracellular messenger, most likely IP3 (Newman and Zahs, 1997), through gap junctions. Propagation from astrocytes to Müller cells, and from one Müller cell to others, is mediated by the release of ATP. ATP release also contributes to wave propagation between astrocytes.
NEURON-GLIA COMMUNICATION
The experiments described above demonstrate that mechanical stimulation or experimental application of neurotransmitters can activate retinal glial cells. An essential but unanswered question is whether retinal glial cells are also activated by intrinsic neuronal activity, as are glial cells in brain slice preparations (see Schipke and Kettenmann in this issue). Unpublished observations by this author indicate that retinal glial cells are indeed activated by neurons when the retina is stimulated by light.
In freshly isolated retinas or in the presence of low levels of ATP (0.5–2 μM), Müller cells display transient Ca2+ increases that occur at a low frequency (approximately 0.3–0.6 transients/s/100 cells). Stimulation of the retina with repetitive light flashes significantly increases the frequency of these Ca2+ transients. These evoked Ca2+ transients are most prominent in Müller cell endfeet at the retinal surface, but are also observed in Müller cell processes in the inner plexiform layer. The light-evoked increase in Müller cell Ca2+ transients is blocked by tetrodotoxin (TTX), suggesting that the increases are generated by the spiking of retinal ganglion cells. Ganglion cells are the only retinal neurons that consistently generate action potentials, although some amacrine cells also spike (Bloomfield, 1996).
The importance of ganglion cells in mediating the light-evoked activation of Müller cells is supported by experiments in which ganglion cell axons are stimulated electrically. Antidromic activation of ganglion cells results in an increase in the frequency of Ca2+ transients in Müller cells that is even larger than the increase seen during light stimulation. The increase in glial Ca2+ transients following ganglion cell stimulation is blocked by TTX. The mechanism by which neurons evoke glial Ca2+ increases in the retina remains to be elucidated. Nevertheless, the demonstration of neuron-to-glia communication suggests that glial Ca2+ transients are physiological responses that occur in vivo.
GLIA-NEURON COMMUNICATION
Direct Modulation: Glial Release of Transmitters
Modulation of neuron spiking
Stimulated glial cells can modulate the electrical activity of retinal neurons by several mechanisms, leading either to an enhancement or to a depression of neuronal spiking. In one study of glia-neuron interactions, the light-evoked spike activity of ganglion cells was monitored with extracellular recordings as neighboring glial cells were activated by mechanical stimuli (Newman and Zahs, 1998). When a glial Ca2+ wave was initiated and propagated past a ganglion cell, action potential generation in that neuron was altered; 57% of the neurons showed a significant change in spike activity (Fig. 3). Of these, 17% of the neurons showed an increase in spiking while 83% showed a decrease. Glial Ca2+ waves failed to modulate neuronal activity if they died out before reaching the neuron.
Fig. 3.
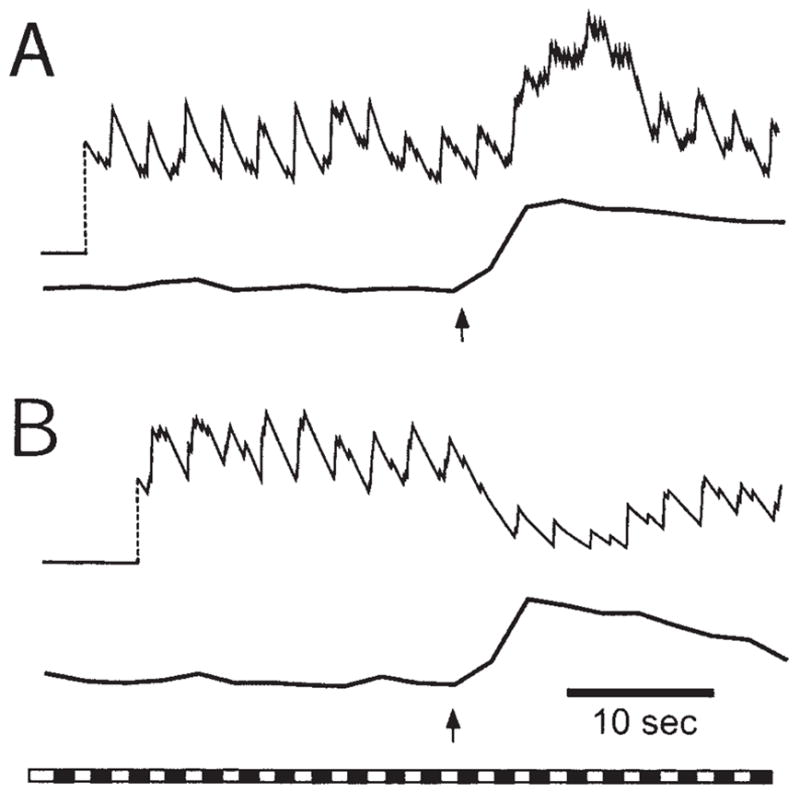
Glial modulation of light-evoked spiking in rat ganglion cells. Enhancement of neuron spiking (A) and depression of spiking (B) are illustrated in recordings from two different neurons. A frequency plot of spike activity (top trace) and Ca2+ levels within glial cells adjacent to the neuron (bottom trace) are shown for each trial. Arrows indicate initiation of the glial Ca2+ wave. The bar at the bottom shows the repetitive light stimulus that evoked neuronal spiking. Reproduced with modification from Newman and Zahs (1998).
Inhibitory glial modulation of neuronal spiking may be Ca2+ -dependent as the magnitude of neuronal modulation was found to be proportional to the amplitude of the Ca2+ increase in neighboring glial cells. In addition, thapsigargin substantially reduced the magnitude of neuronal modulation as well as reducing Ca2+ increases in glial cells.
The results of pharmacological experiments (Newman and Zahs, 1998) indicate that several neurotransmitters may be involved in glial modulation of neuronal spiking. Depression of neuronal spiking by glia was reduced by the AMPA and NMDA glutamate antagonists NBQX and D-AP7 as well as by the GABA and glycine antagonists bicuculline and strychnine.
Modulation of synaptic transmission
Unpublished observations by this author demonstrate that light-evoked excitatory posysnaptic currents (EPSCs) recorded from ganglion cells in whole-cell voltage-clamp experiments are also modulated by glial cells. In some ganglion cells, EPSCs are facilitated during a brief period beginning ~ 2 s following glial cell stimulation and lasting for 10–25 s. In other ganglion cells, EPSCs are depressed following glial cell activation. The mechanisms by which glial cells modulate synaptic transmission in the retina remain to be elucidated.
Inhibition of ganglion cells
In addition to facilitation and depression of synaptic transmission, glial cells can modulate neuronal activity in the retina by a third mechanism (Newman, 2003). When glial cells are activated by agonist application (ATP, ATPγS, dopamine, thrombin, lysophosphatidic acid) or by mechanical stimulation, an outward hyper-polarizing current is evoked in many ganglion cells (Fig. 4A). This glial-evoked hyperpolarization reduces the spiking of those neurons with spontaneous activity. Selective stimulation of either astrocytes or Müller cells demonstrates that neuronal inhibition is mediated by Müller cells; Müller cell activation is both necessary and sufficient to evoke inhibition. The inhibition is not reduced when synaptic transmission is blocked with Cd2+, demonstrating that glial inhibition is not mediated by glial activation of inhibitory interneurons.
Fig. 4.
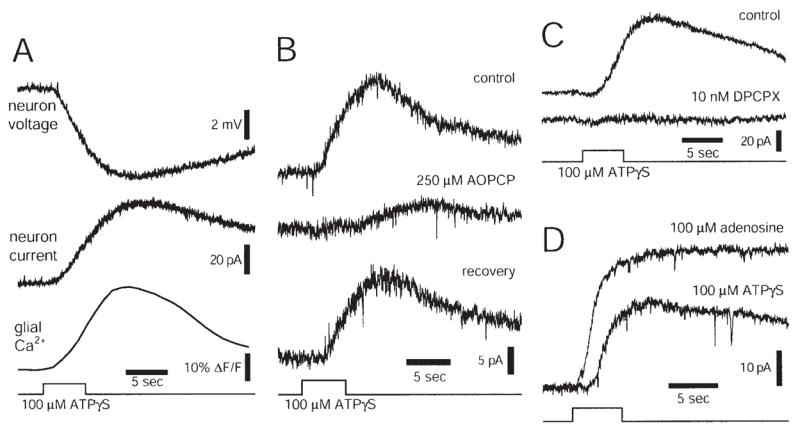
Glial release of ATP inhibits rat ganglion cells. A: Ejection of ATPγS onto the retinal surface evokes a Ca2+ increase in glial cells and a hyperpolarization (current-clamp recording) and an outward current (voltage-clamp recording) in a neighboring ganglion cell. B: Stimulation of glial cells with ATPγS evokes an inhibitory outward current in a ganglion cell. Addition of AOPCP, an ectonucleotidase inhibitor that blocks conversion of AMP to adenosine, reduces and slows the time course of the current. The effect is largely reversible. C: Addition of DPCPX, an A1 adenosine receptor antagonist, abolishes the outward neuronal current evoked by ATPγS stimulation of glial cells. D: Adenosine ejection evokes a larger, shorter latency current in a neuron than does ATPγS ejection at the same retinal location. Reproduced from Newman (2003).
Müller cell inhibition of ganglion cells is mediated by glial release of ATP, which is rapidly converted by ectoenzymes to adenosine. Adenosine in turn activates ganglion cell A1 receptors, resulting in an increase in neuronal K+ conductance. This mechanism of glial inhibition of neurons is supported by a number of findings (Newman, 2003). Müller cell release of ATP into the inner plexiform layer has been directly imaged using the luciferin-luciferase chemiluminescence assay. In addition, ganglion cell inhibition is reduced by blockers of the ectoenzymes which convert ATP to adenosine (Fig. 4B) and inhibition is blocked completely by the A1 receptor antagonist DPCPX (Fig. 4C). Direct ejection of adenosine onto ganglion cells produces neuronal inhibition similar to that observed following glial cell stimulation (Fig. 4D).
In summary, glial cells can modulate neuronal activity in the retina by at least three mechanisms. In some ganglion cells, glial cell activation results in a facilitation of synaptic transmission and to an enhancement of light-evoked spiking. In other ganglion cells, a depression of synaptic transmission and a decrease in spiking is observed. Glial cell activation can also result in the hyperpolarization of ganglion cells mediated by activation of A1 receptors and the opening of neuronal K+channels.
Indirect Modulation
Glutamate transport
The types of glial modulation of neurons described above are mediated by the release of transmitters from glial cells and the subsequent activation of neuronal receptors. These mechanisms are forms of direct modulation of neurons by glia. In addition, glial cells can modulate neurons in the retina by indirect mechanisms.
The best characterized form of indirect modulation is mediated by the uptake of glutamate by Müller cells at synapses (see Marcaggi and Attwell in this issue). Glutamate transport into glia is well characterized and is mediated by GLAST (EAAT1) and GLT-1 (EAAT2) transporters in Müller cells (Lehre et al., 1997; Eliasof et al., 1998).
When glutamate transport in the retina is blocked with the competitive inhibitor L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC), both the amplitude and the duration of ganglion cell EPSCs are increased dramatically (Fig. 5A) (Higgs and Lukasiewicz, 2002). Both AMPA and NMDA components of the current are enhanced. This increase in synaptic transmission is substantially larger than the enhancement seen when glutamate transport is blocked in other CNS regions (see Marcaggi and Attwell in this issue). The dominant role of glutamate transport in shaping synaptic currents in the retina arises because EPSCs in retinal ganglion cells normally decay slowly due to sustained release of glutamate from presynaptic terminals. Diffusion away from the synaptic cleft is not sufficient to clear released glutamate, which must be removed by transporters.
Fig. 5.

Glial modulation of synaptic transmission by glutamate transport and release of D-serine. A: Inhibition of the glial glutamate transporter by PDC greatly increases the amplitude and duration of a light-evoked EPSC recorded from a ganglion cell in the salamander retina. Both the ON and the OFF components of the light response are potentiated. B: Addition of D-serine, an NMDA receptor coagonist, potentiates the inward current recorded from a rat ganglion cell in response to NMDA ejection. C: Addition of D-amino acid-oxidase, which degrades D-serine, reduces the inward current evoked by NMDA ejection. A reproduced from Higgs and Lukasiewicz (2002); B and C from Stevens et al. (2003).
It is likely that Müller cell glutamate transporters, rather than neuronal transporters, shape synaptic responses in the retina. Glutamate uptake occurs primarily in glial cells rather than neurons in the CNS (Rothstein et al., 1996; Bergles and Jahr, 1998). In addition, when neuronal transport in the retina is blocked with dihydrokainate (DHK), little change in synaptic current is observed (Higgs and Lukasiewicz, 2002).
D-serine release
Synaptic currents in the retina may also be modulated by the release of D-serine from glial cells (see Miller in this issue). Activation of glutamatergic NMDA receptors requires, in addition to glutamate, the presence of a coagonist that binds to the glycine binding site of the receptor. D-serine, rather than glycine, may be the endogenous ligand that binds to this site (Wolosker et al., 1999; Mothet et al., 2000). In the retina, glial cells appear to be the primary source of D-serine (Stevens et al., 2003). D-serine and serine racemase, the synthesizing enzyme for the amino acid, are localized to Müller cells and astrocytes in the retina. In culture, D-serine is released from astrocytes when they are stimulated with glutamate (Schell et al., 1995).
Physiological experiments demonstrate the importance of D-serine for NMDA transmission in the retina (Stevens et al., 2003). Addition of D-serine potentiates light-evoked EPSCs mediated by NMDA receptors and heightens ganglion cell responses to NMDA ejections (Fig. 5B), demonstrating that the glycine binding site of the receptor is not normally saturated. Addition of D-amino acid oxidase, which degrades D-serine, decreases light-evoked EPSCs and reduces responses to NMDA ejections (Fig. 5C), demonstrating that D-serine is present in the retina and normally potentiates NMDA transmission.
Regulation of extracellular ion levels
Glia may also modulate synaptic transmission in the retina through the regulation of extracellular K+ and H+ levels. Neuronal activity leads to substantial variations in the concentration of K+ and H+ in the extracellular space (Kelly and Van Essen, 1974; Karwoski et al., 1985; Chesler and Kaila, 1992; Newman, 1995). These variations can alter synaptic transmission; K+ increases depolarize synaptic terminals (Raushe et al., 1990) while H+ block presynaptic Ca2+ channels (Prod’hom et al., 1989; Barnes and Bui, 1991) and NMDA receptors (Traynelis and Cull-Candy, 1990). Müller cells play an essential role in regulating extracellular K+ and H+ concentrations (Newman, 1995, 1996), thus influencing the effect of these ions on synaptic transmission.
FUTURE DIRECTIONS AND IMPLICATIONS
As summarized in this review, retinal glial cells and neurons communicate and mutually influence each other. Neurons signal glial cells, evoking Ca2+ increases in Müller cells when the retina is stimulated with light. Glial cells in turn signal neurons by several mechanisms. Müller cells release ATP into the inner plexiform layer, activating ganglion cell adenosine receptors and eliciting neuronal hyperpolarization. Glial cells also facilitate and depress synaptic transmission in the retina. Synaptic modulation could be mediated by the release of ATP from Müller cells and the activation of neuronal purine receptors (Cunha, 2001; Zhang et al., 2003). Alternately, synaptic modulation could be mediated by the release of glutamate and the activation of neuronal ionotropic or metabotropic glutamate receptors, as it is in other CNS systems (Araque et al., 1998; Kang et al., 1998).
Many important questions concerning glia-neuron communication in the retina remain to be answered. How does neuronal activity evoke Ca2+ increases in retinal glial cells? Are light-evoked Ca2+ increases large enough to elicit the release of transmitters from glia or to trigger propagated intercellular glial Ca2+ waves? Which transmitters released from glial cells are responsible for modulating synaptic transmission in the retina? Do these gliotransmitters act pre- or postsynaptically? Most importantly, does glial modulation of neuronal activity occur in vivo? Only after these questions are answered will we be able to assess the importance of glial cells to information processing in the retina.
Acknowledgments
The author thanks Janice Gepner, Kathleen R. Zahs, and Monica R. Metea for comments on the manuscript.
Grant sponsor: the National Institutes of Health; Grant number: EY04077.
References
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Barnes S, Bui Q. Modulation of calcium-activated chloride current via pH-induced changes of calcium channel properties in cone photoreceptors. J Neurosci. 1991;11:4015–4023. doi: 10.1523/JNEUROSCI.11-12-04015.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield SA. Effect of spike blockade on the receptive-field size of amacrine and ganglion cells in the rabbit retina. J Neurophys. 1996;75:1878–1893. doi: 10.1152/jn.1996.75.5.1878. [DOI] [PubMed] [Google Scholar]
- Ceelen PW, Lockridge A, Newman EA. Electrical coupling between glial cells in the rat retina. Glia. 2001;35:1–13. doi: 10.1002/glia.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol. 1996;114:299–305. doi: 10.1001/archopht.1996.01100130295012. [DOI] [PubMed] [Google Scholar]
- Eliasof S, Arriza JL, Leighton BH, Kavanaugh MP, Amara SG. Excitatory amino acid transports of the salamander retina: identification, localization, and function. J Neurosci. 1998;18:698–712. doi: 10.1523/JNEUROSCI.18-02-00698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs MH, Lukasiewicz PD. Glutamate uptake limits synaptic excitation of retinal ganglion cells. J Neurosci. 2002;19:3691–3700. doi: 10.1523/JNEUROSCI.19-10-03691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Newman EA, Shimazaki H, Proenza LM. Light-evoked increases in extracellular K+ in the plexiform layers of amphibian retinas. J Gen Physiol. 1985;86:189–213. doi: 10.1085/jgp.86.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JP, Van Essen DC. Cell structure and function in the visual cortex of the cat. J Physiol. 1974;238:515–547. doi: 10.1113/jphysiol.1974.sp010541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Davanger S, Danbolt NC. Localization of the glutamate transporter protein GLAST in rat retina. Brain Res. 1997;744:129–137. doi: 10.1016/s0006-8993(96)01022-0. [DOI] [PubMed] [Google Scholar]
- Mothet J-P, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial cell regulation of extracellular potassium. In: Kettenmann H, Ransom BR, editors. Neuroglia. New York: Oxford University Press; 1995. pp. 717–731. [Google Scholar]
- Newman EA. Acid efflux from retinal glial cells generated by sodium-bicarbonate cotransport. J Neurosci. 1996;16:159–168. doi: 10.1523/JNEUROSCI.16-01-00159.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847. doi: 10.1126/science.275.5301.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. In: Glia of the retina. Ryan SJ, editor. Retina. St. Louis: Mosby; 2001a. pp. 89–103. [Google Scholar]
- Newman EA. Propagation of intercellular calcium waves in retinal astrocytes and Müller cells. J Neurosci. 2001b;21:2215–2223. doi: 10.1523/JNEUROSCI.21-07-02215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. 2003;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prod’hom B, Pietrobon D, Hess P. Interactions of protons with single open L-type calcium channels: location of protonation site and dependence of proton-induced current fluctuations on concentration and species of permeant ion. J Gen Physiol. 1989;94:23–42. doi: 10.1085/jgp.94.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raushe G, Igelmund P, Heinemann U. Effects of changes in extracellular potassium, magnesium and calcium concentration on synaptic transmission in area CA1 and the dentate gyrus of rat hippocampal slices. Pflugers Arch. 1990;415:588–593. doi: 10.1007/BF02583510. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kunci RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sarthy V, Ripps H. The retinal Müller cell. New York: Kluwer Academic/Plenum Publishers; 2001. [Google Scholar]
- Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci USA. 1995;92:3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens ER, Esquerra M, Kim P, Newman EA, Snyder SH, Zahs KR, Miller RF. D-serine and serine racemase are present in the vertebrate retina and contribute to the functional expression of NMDA receptors. Proc Natl Acad Sci USA. 2003;100:6789–6794. doi: 10.1073/pnas.1237052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature. 1990;345:347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc Natl Acad Sci USA. 1999;96:13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahs KR, Newman EA. Asymmetric gap junctional coupling between glial cells in the rat retina. Glia. 1997;20:10–22. [PubMed] [Google Scholar]
- Zhang J, Wang H, Ye C, Jiang Z, Wu C, Poo M, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]