

Abstract
The c-Myc transcription factor is a key regulator of cell proliferation and cell fate decisions. c-Myc overexpression is observed in a variety of human tumors, revealing the importance of maintaining normal levels of c-Myc protein. c-Myc protein stability in mammalian cells is controlled by interdependent and sequential phosphorylation and dephosphorylation events on two highly conserved residues, serine 62 and threonine 58. Here we show that these sequential phosphorylation and dephosphorylation events and their effect on c-Myc stability also occurs in the model system Saccharomyces cerevisiae. These results suggest the presence of a conserved pathway in yeast that controls protein turnover in response to a specific phospho-degron sequence. These findings have implications regarding conserved pathways for regulated protein degradation, and they validate the use of genetically tractable yeast for the study of the turnover of proteins such as c-Myc that contain this motif.
The c-myc proto-oncogene encodes a helix-loop helix transcription factor that is involved in a number of crucial cellular processes, including cell proliferation, cell growth, differentiation, and apoptosis. c-Myc heterodimerizes with its partner protein, Max, and together they regulate transcription at E box sequences (CAC(A/G)TG) of a variety of important genes. Recently, it has been reported that c-Myc also regulates transcription of polymerase I- and III-dependent genes (1, 2). Given that c-Myc is involved in many vital cellular activities, it is not surprising that it is highly regulated at several levels, including transcription, mRNA stability, translation, and protein stability (3-6). A number of animal models have shown that misregulation of c-Myc can result in tumorigenesis (7). Indeed, overexpression of c-Myc is observed in more than 70% of human cancers. This can involve amplification or translocation of the c-myc gene (8). However, these genetic changes are observed only in a minority of the cases, suggesting that other mechanisms, such as a change in c-Myc protein stability, may play a role in tumorigenesis.
We and others have previously reported that the stability of c-Myc protein in mammalian cells is controlled by sequential phosphorylation and dephosphorylation events on two highly conserved residues, threonine 58 and serine 62 (9-11). Phosphorylation at these sites has opposing effects on c-Myc protein stability. An initial event, phosphorylation at residue serine 62 by Ras-activated ERKs,2 stabilizes c-Myc, whereas a subsequent phosphorylation at residue threonine 58 by the glycogen synthase kinase 3β (GSK3β), destabilizes c-Myc. Before degradation, a cis to trans isomerization at the bond proceeding serine 62 is catalyzed by the peptidylprolyl isomerase, Pin1, allowing the stabilizing serine 62 phosphate to be removed by the trans-specific phosphatase, protein phosphatase 2A (PP2A). Singly, threonine 58-phosphorylated c-Myc can then be targeted for multiubiquitination by the E3 ligase SCFFbw7 and degraded by the 26 S proteasome (12, 13).
Saccharomyces cerevisiae is frequently used as a model system to study mammalian proteins because of the ease of genetic manipulation, rapid doubling time, and the presence of conserved orthologs between a number of yeast and mammalian proteins, including a number of proteins required for proteasome-mediated degradation. For these reasons it is an excellent model system to study interactions, function, and turnover of mammalian proteins. Indeed, a number of groups have used the budding yeast, S. cerevisiae, to study the ubiquitination and subsequent destruction of important mammalian cell cycle proteins (14-17). Likewise, c-Myc half-life has been studied in yeast cells and found to be very short (14, 18). Additionally, it has been reported that c-Myc protein can be stabilized in this system by mutating core components of the yeast SCF E3 ubiquitin ligase complex. Specifically, c-Myc half-life has been shown to be increased in yeast strains containing mutations in Cdc53, Skp1, or the F-box protein, Grr1 (18). Interestingly, many of the mammalian proteins that regulate c-Myc phosphorylation and protein stability are conserved in yeast.
In this study we show that yeast orthologs of key proteins in the signaling pathways that control c-Myc phosphorylation are functionally conserved. These proteins are involved in the same sequential and interdependent phosphorylation and dephosphorylation events at serine 62 and threonine 58 in c-Myc that occur in mammalian cells. Additionally, these phosphorylation events have the same consequence on c-Myc protein stability. This is an important finding because it supports discoveries made in mammalian cells, it emphasizes the importance of this signaling pathway in regulating protein turnover through a conserved phospho-degron motif, it validates the use of the yeast S. cerevisiae as a model system to study c-Myc turnover, and it implicates this pathway in the degradation of yeast proteins and other mammalian proteins.
MATERIALS AND METHODS
Yeast Media and Reagents
All strains used were haploid and isogenic with BY4741 (Mata his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) or W303 (Mata ade2-1 his3-1,15 ura3-1 leu2-3,112 trp1-1 can1-100). Cells were grown in selective media or 1% yeast extract, 2% peptone, adenine, 2% glucose at 23, 30, or 37 °C as indicated. Yeast strains were transformed with c-Myc expression constructs by lithium acetate-mediated transformation (19). To arrest cells in G1 or M phases, cells were treated with α factor (2 μg/ml) or nocodazole (15 μg/ml), respectively.
Strains and Plasmids
Strains used in this study were BY4741, Δrim11, Δpph21, Δrts1, TAP-Rim11, TAP-Kss1 (Open Biosystems), W303, cdc4-1, Δgrr1 (generous gift of S. Lanker) ess1H164R, and ess1A144T (generous gifts of S. Hanes). The 2μ plasmids pYESDEST52-MycWT, pYESDEST52-MycT58A, and pYESDEST52-MycS62A containing V5-His6 epitope-tagged c-Myc or c-Myc mutants under the control of the GAL1 promoter were generated using TOPO cloning (Invitrogen).
Determination of Protein Stability
Cells were grown in a 2% raffinose synthetic complete medium overnight. Cells were then diluted to an optical density at 600 nm (A600) of 0.3 and grown for an additional 2–4 h at the temperature indicated. A sample was removed as a negative control before the addition of galactose. Galactose was added to the media to a final concentration of 2% to induce expression of the c-myc gene from the GAL1 promoter for 1–3 h at the indicated temperatures. Glucose was added to a final concentration of 5% to stop gene expression, and samples were taken at the indicated time points. Protein extraction and Western blotting were performed as described below. The optical density at 600 nm (A600) of all samples was measured, and the volume of each sample was adjusted to ensure that equal cell numbers were used.
Antibodies
The monoclonal V5 antibody used to detect total c-Myc protein was from Invitrogen. The c-Myc serine 62 phospho-specific antisera were raised against the chemically synthesized phosphopeptides CKFELLP(A/T)PPLpSPSRRSG (pS is phosphoserine) in rabbits. The antisera were purified against this phosphopeptide (T only) conjugated to Sulfolink coupling gel (Pierce) as described (20). To deplete antibodies that recognized unphosphorylated c-Myc, the affinity-purified antibodies were re-purified by passing through Sulfolink coupling gel conjugated with the corresponding unphosphorylated peptide. The c-Myc threonine 58 phospho-specific antibody was purchased from Cell Signaling Technology (Beverly, MA). The polyclonal Cdc28 antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Western Blotting and Quantitation
Yeast extracts were prepared by using the rapid protein extraction procedure (21). Protein from equal yeast cell numbers were separated by SDS-PAGE gel and transferred to Immobilon-FL membrane (Millipore, Billerica, MA). Membranes were blocked with Odyssey Blocking buffer (LI-COR Biosciences, Lincoln, NE) or 5% nonfat milk in PBS (P-Thr-58 antibody). Primary antibodies were diluted in 1:1 Odyssey Blocking buffer:PBS with 0.05% Tween 20 or in 2.5% nonfat milk PBS, 0.05% Tween (P-Thr-58 antibody). Primary antibodies were detected with secondary antibodies labeled with the near-infrared fluorescent dyes IRDye800 (Rockland, Philadelphia, PA) and Alexa Fluor 680 (Molecular Probes, Eugene, OR) to allow two-color imaging and band overlay. Secondary antibodies were diluted 1:10,000 in 1:1 Odyssey Blocking buffer:PBS, 0.05% Tween or in 1.25% nonfat milk PBS, 0.05% Tween (P-Thr-58 antibody). Blots were scanned with a LI-COR Odyssey Infrared Imager (Lincoln, NE) to visualize proteins. c-Myc and Cdc28 protein levels were quantitated using LI-COR Odyssey Infrared Imager software Version 1.2. For degradation assays c-Myc total protein levels were normalized to total protein as measured by Cdc28 protein levels. Normalized c-Myc protein levels at each time point were calculated as a percentage of the first time point and graphed on a semi-log graph. Half-lives were calculated based on best-fit lines drawn using Microsoft Excel. Mean half-lives ± S.D. were calculated based on three or more independent experiments.
In Vitro Kinase Assay
c-Myc expression was induced in the Δkss1 or the Δrim11 strains as described above. Cells were lysed in Ni-NTA lysis buffer (5 mm imidazole, 5 mm βME and 0.5% Nonidet P-40), and c-Myc was extracted using Ni-NTA agarose (Qiagen, Valencia, CA). c-Myc was eluted from agarose in Ni-NTA elution buffer (1:1 Ni-NTA lysis buffer:1 m imidazole). The TAP-Kss1 and TAP-Rim11 strains were grown overnight in 1% yeast extract, 2% peptone, adenine, 2% glucose to an optical density at 600 nm of 0.5. TAP-tagged proteins were extracted using calmodulin affinity resin (Stratagene, La Jolla, CA.) in IPP150 calmodulin binding buffer (22). Beads were washed 3 times in calmodulin binding buffer and one time in kinase reaction buffer (50 mm Tris-HCL, pH 7.5, 0.1 mm EDTA, 15 mm dithiothreitol). The immobilized kinases were incubated with eluted c-Myc, kinase buffer, and 12 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) in a final volume of 60 μl. Reactions were incubated at 30 °C for 30 min. Unbound proteins were mixed with an equal volume of 2× SDS-PAGE sample buffer. Proteins were analyzed by SDS-PAGE and autoradiography.
RESULTS
Interdependent Phosphorylation of c-Myc at Threonine 58 and Serine 62 Occurs in Yeast
We and others have previously used phospho-peptide mapping and phospho-specific antibodies to demonstrate an interrelationship between phosphorylation at serine 62 and threonine 58 (9, 10). Specifically, Ser-62 phosphorylation is required before Thr-58 phosphorylation, and Thr-58 phosphorylation facilitates subsequent Ser-62 dephosphorylation (9, 11). To perform a yeast two-hybrid assay to identify new proteins that interact with phospho-Thr-58 and phospho-Ser-62, we first investigated whether mammalian c-Myc is phosphorylated at either of these sites in yeast. We expressed c-MycWT or one of two c-Myc phosphorylation mutants, c-MycT58A or c-MycS62A, in a wild type yeast strain using the GAL1 promoter. This promoter is induced by the addition of galactose to the yeast media. After a 1-h induction of c-MycWT or the c-Myc mutants from the GAL1 promoter, total cell lysates from yeast expressing c-MycWT, c-MycT58A, or c-MycS62A were visualized by Western blot using antibodies specific for phosphorylated threonine 58 or phosphorylated serine 62. Specificity of the Thr-58 phospho-specific antibody has been previously documented (9). We recently generated new Ser-62 phospho-specific antibodies (see “Materials and Methods”). Specificity of these antibodies were demonstrated by enzyme-linked immunosorbent assay (data not shown) and Western blotting with wild type and mutant c-Myc expressed in mammalian cells (23) and yeast (here). Using LI-COR technology we can double label for total c-Myc and phospho-c-Myc. This technology allows us to accurately quantitate the ratio of phospho-Ser-62 or phospho-Thr-58 c-Myc to total c-Myc (see “Materials and Methods”). As shown in Fig. 1A, c-MycWT is both Thr-58 and Ser-62 phosphorylated, with a weaker signal detected by the anti-phosopho-Ser-62 antibody (lane 1). The relative difference in Thr-58 and Ser-62 phosphorylation detected with the phospho-specific antibodies is consistent with phosphopeptide mapping results showing a reduced amount of Ser-62 phosphorylation and increased Thr-58 phosphorylation under conditions where c-Myc is unstable (9). In contrast, c-MycT58A, which lacks phosphorylation at Thr-58, showed a large increase, >10-fold, in Ser-62 phosphorylation levels compared with c-MycWT (Fig. 1A, lane 2). This increase in Ser-62 phosphorylation with the c-MycT58A mutant is also observed in mammalian cells both by phospho-peptide mapping and phospho-specific antibodies and is due to Thr-58 phosphorylation-dependent Ser-62 dephosphorylation (9-11). Last, c-MycS62A does not appear to be phosphorylated on either site in yeast cells (Fig. 1A, lane 3). This is again consistent with observations in mammalian cells in which c-MycS62A lacks phosphorylation at Thr-58, forming the basis for the reported dependent relationship between Ser-62 and Thr-58 (9, 10, 24). These results are striking because they demonstrate the conservation of a pathway controlling two interdependent relationships between the Ser-62 and Thr-58 phosphorylation sites; 1) the hierarchical phosphorylation of Ser-62 followed by Thr-58 and 2) the role of Thr-58 phosphorylation in promoting Ser-62 dephosphorylation.
FIGURE 1. Interdependent phosphorylation of c-Myc at Thr-58 and Ser-62 control stability in yeast.
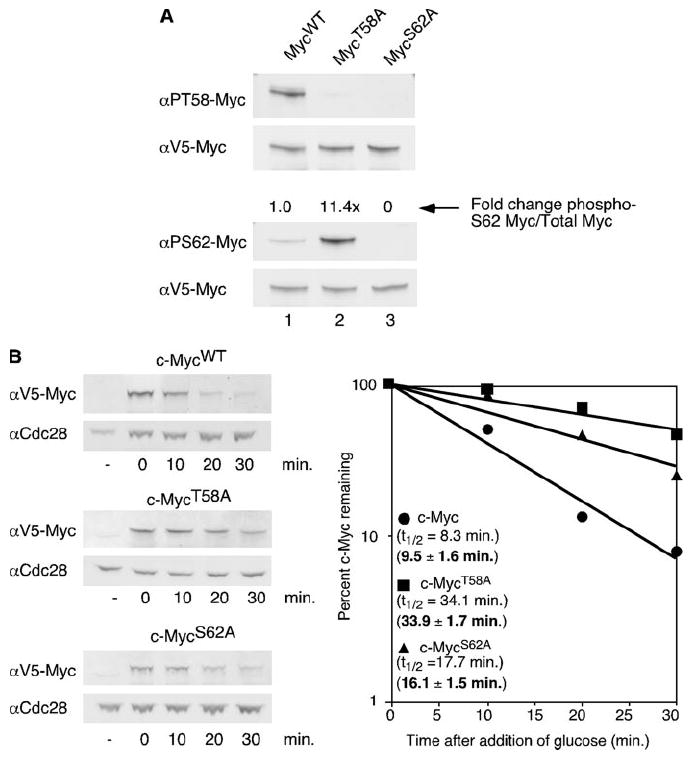
A, V5-tagged MycWT, MycT58A, or MycS62A expression was induced from a GAL1 promoter in the BY4741 yeast strain by the addition of galactose for 1 h at 30 °C. Equal cell numbers were visualized by Western blot analysis with αV5, α-Ser(P)-62 (αPS62), or α-Thr(P)-58 (αPT58) using dual probing and overlay with the Odyssey Imaging System. Protein levels were quantitated, and ratios of phosphorylated c-Myc to total c-Myc were calculated as described under “Materials and Methods”. -Fold change compared with c-MycWT is shown. B, V5-tagged MycWT, MycT58A, or MycS62A expression was induced for 1 h in the BY4741 yeast strain at 30 °C by the addition of galactose. Glucose was added to inhibit expression of c-Myc. Cells were harvested at the time points indicated after glucose addition and lysed in SDS sample buffer. Equal cell numbers were visualized by Western blot analysis with αV5 and αCdc28. c-Myc protein levels and Cdc28p levels were quantitated by the Odyssey Imaging System, and c-Myc levels were normalized to total protein as determined by the amount of Cdc28 protein. c-Myc levels at each time point are shown as a percent of the first time point and are plotted on a semi-log graph. Best-fit lines were calculated using Microsoft Excel. Experiments were repeated three or more times, and representative data are shown. Mean half-lives ± S.D. are indicated in bold.
Given that the interdependent phosphorylation of c-Myc at Ser-62 and Thr-58 occurs in yeast, we next asked if phosphorylation at these sites also controls c-Myc protein stability in yeast as it does in mammalian cells. To study the stability of c-Myc protein in yeast, we used a galactose induction/glucose shut off system. Specifically, the addition of galactose to yeast cells activates expression of genes under control of the GAL1 promoter, whereas the addition of glucose rapidly shuts down the promoter. Galactose was added to yeast cells carrying a plasmid with either c-mycWT or the phosphorylation mutants under the control of this promoter to induce expression. After 1 h, expression was terminated by the addition of glucose, and samples were taken at various time points for Western blot analysis. c-Myc protein levels were quantified and normalized to the control Cdc28 protein, and c-Myc half-life was calculated. As shown in Fig. 1B, c-MycWT exhibited a short half-life in a wild type yeast strain of 8.3 min, with a mean half-life of 9.5 ± 1.6 min based on multiple independent experiments. This short half-life has previously been reported for c-Myc in both yeast cells and mammalian cells (14, 15, 18, 25), and it is consistent with the levels of detected Thr-58 and Ser-62 phosphorylation seen in Fig. 1A. In contrast, c-MycT58A showed a marked increase in half-life to 34.1 min, with a mean half-life of 33.9 ± 1.7 min. This approximate 4-fold increase over wild type c-Myc is consistent with results in mammalian cells where c-MycT58A is 4–6 times more stable compared with c-MycWT (9, 14). The c-MycS62A mutant demonstrated an intermediate mean half-life of 16.1 ± 1.5 min. The half-life of c-MycS62A in mammalian cells is either reported to be short like c-MycWT, or somewhat longer, but less than c-MycT58A, consistent with our observations in yeast (9, 13). Taken together our results not only show that the interdependent phosphorylation of Thr-58 and Ser-62 occurs in yeast but also that phosphorylation of c-Myc at these sites appears to control c-Myc protein stability in yeast cells. That there are no known functional yeast homologues to mammalian c-Myc suggests the presence of a conserved pathway that controls protein degradation through a conserved phospho-degron.
Proteins Controlling c-Myc Ser-62 and Thr-58 Phosphorylation and c-Myc Stability in Mammalian Cells Are Conserved in Yeast
The previous results suggest that functional homologues to mammalian proteins that target c-Myc for degradation are conserved in yeast. Indeed, many of the key proteins that have been implicated in controlling c-Myc Thr-58 and Ser-62 phosphorylation and regulating c-Myc protein stability in mammalian cells have S. cerevisiae orthologs (Table 1). One of the advantages to working in yeast is its easy genetic manipulation. Thus, strains that are mutant in each of these proteins are available. We made use of these strains to ask whether deletion or mutation of yeast orthologs of proteins which destabilize c-Myc in mammalian cells affect c-Myc phosphorylation in yeast and whether this correlates with changes in protein stability.
TABLE 1.
Mammalian proteins known to regulate c-Myc protein stability and their S. cerevisiae orthologs
| Mammalian protein | Effect on c-Myc protein stability | S. cerevisiae orthologs |
|---|---|---|
| ERK kinase | Stabilizing (6) | Kss1 |
| GSK3β kinase | Destabilizing (9) | Rim11 |
| Pin1 isomerase | Destabilizing (11) | Ess1 |
| PP2A C subunit | Destabilizing (11, 23) | Ppph21 |
| PP2A B subunit (B′ type) | Destabilizing (23) | Rts1 |
| F-box hCdc4 (Fbw7) | Destabilizing (12, 13) | Cdc4 |
| F-box Skp2 | Destabilizing (18, 38) | Grr1 |
A Role for Yeast Kinases Kss1p and Rim11p in Controlling c-Myc Phosphorylation and c-Myc Stability
Kss1p is a yeast mitogen-activated protein kinase that has high sequence homology to the mammalian ERK kinase, and Rim11p is the yeast serine/threonine kinase that shares a conserved function to the GSK3β kinase (26, 27). In mammalian cells, ERK can phosphorylate c-Myc on Ser-62, stabilizing the protein, whereas GSK3β is responsible for phosphorylating c-Myc on Thr-58, thereby destabilizing c-Myc protein. Both KSS1 and RIM11 are nonessential yeast genes, presumably due the presence of additional mitogen-activated protein kinase-like and GSK3-like kinases (28-31).
We asked whether the yeast kinase, Kss1p, affects the phosphorylation of c-Myc at Ser-62 in yeast. To examine this, we expressed V5-tagged c-Myc in a wild type strain and a strain lacking the Kss1p gene (Δkss1) from the GAL1 promoter for 1 h. Cells were lysed in SDS sample buffer, and whole cell lysates were run on an SDS-PAGE gel. c-Myc protein was visualized by double labeling with one of the c-Myc phospho-antibodies as well as an antibody to the V5 tag to measure total c-Myc. The intensity of each band was quantified, and ratios of phosphorylated c-Myc to total c-Myc were calculated relative to c-Myc phosphorylation in the wild type strain. As shown in Fig. 2A, c-Myc expressed in the Δkss1 strain is decreased for phosphorylation at Ser-62 compared with that of c-Myc expressed in a wild type strain. This decrease in Ser-62 phosphorylation is presumably due to the loss of Kss1p. Residual phosphorylation of Ser-62 is likely due to other mitogen-activated protein-like kinases is the cell. This result suggests that Kss1p plays a role in phosphorylating c-Myc at Ser-62 in yeast cells, as ERK does in mammalian cells.
FIGURE 2. The yeast kinases Kss1 and Rim11 phosphorylate c-Myc and thereby affect c-Myc protein stability.
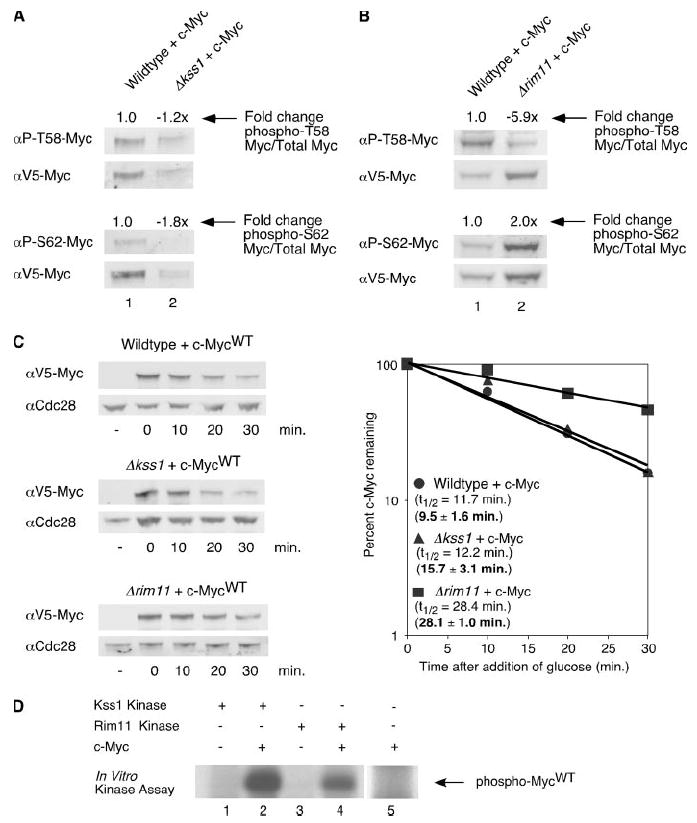
A, V5-tagged c-MycWT was expressed in the BY4741 and Δkss1 yeast strains for 1 h at 30 °C. Cells were lysed in SDS sample buffer. Equal cell numbers were analyzed by Western blotting with the indicated antibodies as described in Fig. 1A. Ratios of phosphorylated c-MycWT to total c-MycWT were calculated. -Fold change of ratios in the Δkss1 strain compared with the BY4741 strain are shown. B, V5-tagged c-MycWT was expressed in the BY4741 and Δrim11 yeast strains for 1 h at 30 °C. Cells were lysed in SDS sample buffer. Equal cell numbers were analyzed by Western blotting as described in Fig. 1A. Ratios of phosphorylated c-MycWT to total c-MycWT were calculated. -Fold change of ratios in the Δrim11 strain compared with the BY4741 strain are shown. C, V5-tagged c-MycWT expression was induced in the BY4741, Δkss1, or Δrim11 strain for 1 h at 30 °C. After the addition of glucose, cells were harvested at the time points indicated, and cells were lysed in SDS sample buffer. Western blotting and quantitation were performed as described in Fig. 1B. Experiments were repeated three or more times, and representative data are shown. Mean half-lives ± S.D. are indicated in bold. D, lysates from Δkss1 or Δrim11 strains expressing or not expressing V5-His6-tagged c-Myc protein were incubated with nickel-agarose to purify c-Myc. Kss1 and Rim11 kinases were purified from yeast strains with knocked-in TAP tags using calmodulin purification as described under “Materials and Methods”. c-Myc or lysates not expressing c-Myc were eluted from the nickel-agarose and then incubated with either the immobilized TAP-Kss1 or TAP-Rim11 kinases in the presence of 32P-labeled ATP (lanes 1– 4). As an additional control, c-Myc protein was incubated with 32P-labeled ATP in the absence of either TAP-tagged kinase (lane 5). Representative results are shown. αP-S62, α-Ser(P)-62; αP-T58, α-Thr(P)-58.
We next examined whether the presence of Rim11p affects phosphorylation of Thr-58 in yeast. To test this, V5-tagged c-MycWT was again expressed from the GAL1 promoter for 1 h in both a yeast strain lacking RIM11 (Δrim11) and an isogenic wild type control strain. Whole cell lysates were prepared in SDS sample buffer and run on an SDS-PAGE gel. c-Myc protein was visualized by double labeling with one of the c-Myc phospho-specific antibodies and anti-V5 (Fig. 2B). We observed a substantial decrease in Thr-58 phosphorylation and an increase in Ser-62 phosphorylation in the Δrim11 strain compared with the wild type control. This decrease in Thr-58 phosphorylation is consistent with observations in mammalian cells where inhibition of GSK3β results in a decrease in phospho-Thr-58 c-Myc (9, 32). Likewise, the increase in Ser-62 phosphorylation is consistent with the role of Thr-58 phosphorylation in facilitating PP2A-mediated dephosphorylation of Ser-62 seen in mammalian cells (11). Thus the inter-relationship between the two phosphorylation sites was again observed. There was some residual phosphorylation of Thr-58 in the Δrim11 yeast strain. This is presumably due to the presence of three other GSK3-like kinases in the yeast (28-30). This result suggests that Rim11p plays a significant role in phosphorylating c-Myc at residue 58 in yeast just as GSK3β does in mammalian cells.
We next asked whether loss of Kss1p and/or Rim11p affects the half-life of c-Myc in yeast. As shown in Fig. 1, mutation of the Ser-62 residue of c-Myc results in a small increase in half-life compared with c-MycWT. Therefore, if the Kss1 kinase phosphorylates c-Myc at Ser-62 we would expect a similar result from loss of Kss1p. To test this, c-Myc expression was induced in the Δkss1 strain for 1 h, and after termination of induction, cells were collected at the indicated time points. As shown in Fig. 2C, the half-life of c-Myc in a wild type strain was 11.7 min, whereas the half-life in the Δkss1 strain was 12.2 min. Based on multiple experiments, the mean half-life of c-Myc in the Δkss1 yeast strain was 15.7 ± 3.1 min. This is similar to the c-MycS62A mutant mean half-life, which was measured to be 16.1 ± 1.5 min (see Fig. 1B). The small increase in stability with loss of phosphorylation at Ser-62 is also consistent with previously published results in mammalian cells and suggests that without phosphorylation at Ser-62 or at Thr-58 c-Myc is degraded by an alternate E3 ligase (13).
On the other hand, mutation of Thr-58 to a non-phosphorylatable residue results in enhanced Ser-62 phosphorylation and substantial stabilization of c-Myc (Fig. 1, A and B). Thus, loss of Rim11p may result in similar stabilization of c-Myc. To test this, c-Myc was expressed from a GAL1 promoter for 1 h in the Δrim11 strain. After the addition of glucose to stop induction of the promoter, samples were taken at various time points. c-Myc protein levels and half-lives are shown in Fig. 2C. The half-life of c-Myc expressed in the Δrim11 background was 28.4 min. Based on multiple independent experiments, the mean half-life of c-Myc in the Δrim11 strain is 28.1 ± 1.0 min, a 3-fold increase when compared with mean c-Myc half-life in a wild type strain. These results demonstrate that in S. cerevisiae, Rim11p participates in the phosphorylation of c-Myc at Thr-58 and that this phosphorylation leads to c-Myc destabilization, similar to the activity of GSK3β on c-Myc in mammalian cells.
Because c-Myc is not a yeast protein, it is important to ask whether these yeast kinases are able to directly phosphorylate mammalian c-Myc protein. To answer this question we preformed an in vitro kinase assay. Briefly, V5-His6-c-Myc expression was induced in either the Δkss1 strain or the Δrim11 strain, and c-Myc protein was extracted using nickel agarose. We used c-Myc protein from these strains because 1) we have already shown them to have low Ser(P)-62 or low Thr(P)-58, respectively (see Figs. 2, A and B), and 2) in the case of Rim11-mediated phosphorylation, to ensure that c-Myc was properly primed with a phosphate at Ser-62, since GSK3β kinases are reported to be processive kinases. c-Myc protein was eluted from the nickel agarose and incubated with either the Kss1 or Rim11 kinase in the presence of 32P-labeled ATP. As a negative control nickel agarose was incubated with non-induced yeast lysates, and those elutions were also incubated with either the Kss1 or Rim11 protein (Fig. 2D, lanes 1 and 3). The samples were analyzed by SDS-PAGE and autoradiography. As shown in Fig. 2D, c-Myc extracted from the Δkss1 strain was phosphorylated in the presence of purified Kss1 (lane 2), whereas c-Myc extracted from the Δrim11 strain was phosphorylated in the presence of purified Rim11 (lane 4). As an additional control, c-Myc alone was incubated with 32P-labeled ATP to ensure that additional kinases were not being co-purified with Myc (lane 5). These data demonstrate that the Kss1 and Rim11 yeast kinases can directly phosphorylate mammalian c-Myc protein.
c-Myc Isomerization by Ess1 Is Not a Significant Limiting Step in c-Myc Degradation in Yeast
Ess1p is the yeast homologue of the peptidylprolyl isomerase, Pin1. In mammalian cells Pin1 recognizes c-Myc phosphorylated at Thr-58 and catalyzes a cis to trans isomerization of the bond preceding proline 63 in c-Myc. This isomerization is thought to underlie the role of Thr-58 phosphorylation in facilitating the dephosphorylation of Ser-62 by the PP2A phosphatase, which dephosphorylates residues when the proceeding proline is in trans. In Pin1-null mouse embryo fibroblasts c-Myc Ser-62 phosphorylation is increased, and degradation is inhibited (11). ESS1 is an essential yeast gene, and cells mutated for this gene arrest in M phase of the cell cycle (33). It is also a highly conserved gene, and mammalian Pin1 can substitute for Ess1 protein function in yeast (33).
We asked if the yeast peptidylprolyl isomerase, Ess1p, affects phosphorylation and, consequently, protein stability of c-Myc in yeast. Because ESS1 is an essential yeast gene, a temperature-sensitive yeast mutant strain with an H164R mutation in the active site of the enzyme was used (34). This strain arrests in M phase; however, with slow kinetics. c-Myc expression was induced in the ess1H164R strain at the restrictive temperature of 37 °C for 3 h. Approximately 85% of the cells showed mutant phenotype microscopically (data not shown). Whole cell lysates were run on a gel, and the Western blot was duel-labeled with antibodies specific for phospho-Ser-62 or phospho-Thr-58 and anti-V5 (Fig. 3A). Interestingly, c-Myc expressed in the ess1H164R strain showed only a small increase in Thr-58 or Ser-62 phosphorylation compared with the similarly treated wild type strain. We also examined c-Myc half-life in the ess1H164R at the restrictive temperature, 37 °C, and compared this to an isogenic wild type control strain under the same temperature but with the addition of the microtubule inhibitor, nocodazole, to control for M phase arrest in the ess1H164R strain. c-Myc half-life overall did not appear to be significantly longer in the ess1H164R strain (13.8 min) compared with the wild type strain arrested with nocodazole (12.5 min, Fig. 3B). Based on multiple experiments, the mean half-life of c-Myc in a nocodazole-arrested wild type strain was 13.0 ± 2.0 min, whereas the mean overall half-life of c-Myc in the ess1H164R strain was 13.8 ± 1.3 min. Although the overall half-life for c-Myc did not appear to be significantly affected by loss of Ess1p, it is interesting to note that we consistently observed a biphasic decay where there is an initial decrease in c-Myc levels, which then levels off between 10 and 20 min in the ess1H164R strain (Fig. 3B, dashed lines). This suggests that loss of Ess1p does affect a subset of c-Myc protein, which could explain why we only saw a small increase in phosphorylation in c-Myc in the ess1H164R strain. It may be that a significant portion of c-Myc already exists in the trans conformation in the yeast cells and, therefore, does not require Ess1p activity for dephosphorylation or degradation. However, after this portion was degraded, a remaining subset of c-Myc protein existed in the cis conformation, and it is this population that continues to be stable out to at least 60 min in the ess1H164R strain (data not shown).
FIGURE 3. Mutation of the yeast peptidylprolyl isomerase, Ess1, does not significantly affect c-Myc phosphorylation or overall stability.
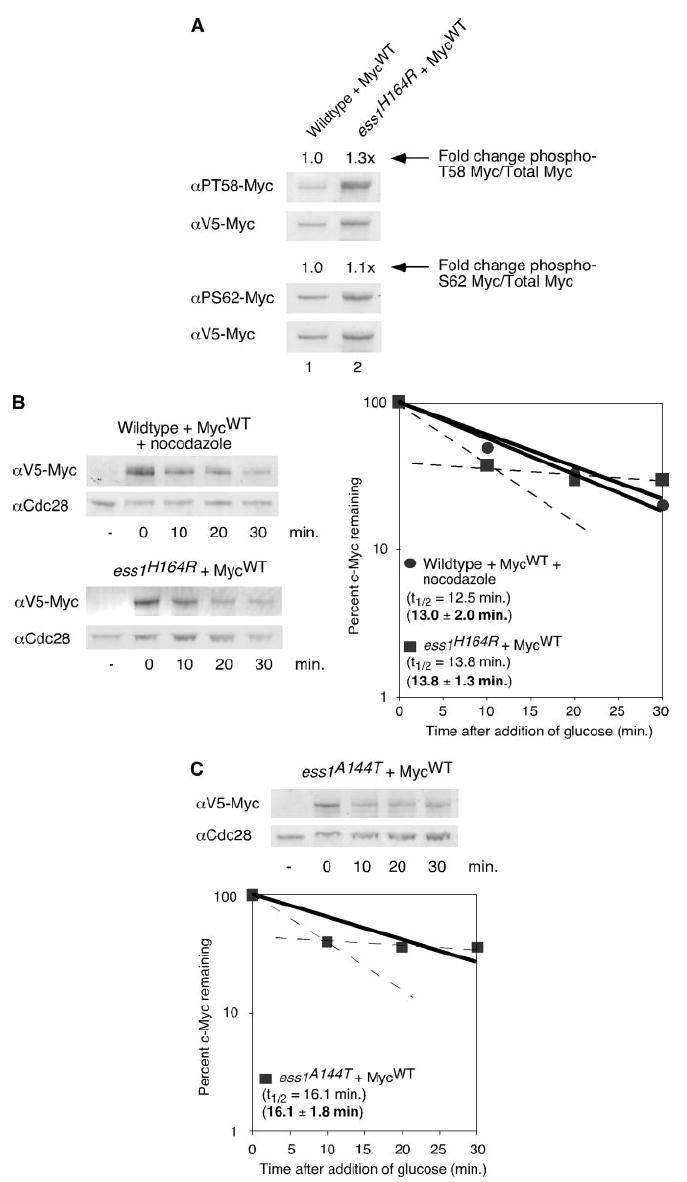
A, W303 and ess1H164R cells were grown in 2% raffinose medium at 30 °C overnight. Cells were diluted to A600 = 0.3, nocodazole was added to the W303 strain, and cells were grown at 37 °C for 4 h. Galactose was added, and c-MycWT expression was induced for 3 h at 37 °C. After the addition of glucose, cells were harvested and lysed in SDS sample buffer. Equal cell numbers were visualized by Western blot analysis with the indicated antibodies and quantitated as described in Fig. 1A. Ratios of phosphorylated c-MycWT to total c-MycWT were calculated. -Fold change of ratios in the ess1H164R strain compared with the W303 strain are shown. B, c-MycWT expression in the W303 or ess1H164R yeast strain was induced as described above. After the addition of glucose, cells were harvested at the indicated time points and lysed in SDS sample buffer. Western blot analysis and quantitation were performed as described in Fig. 1B. Experiments were repeated three or more times, and representative data are shown. Mean half-lives ± S.D. are indicated in bold. Dashed lines indicate the biphasic degradation of c-Myc in the ess1H164R strain. C, c-Myc expression was induced in the ess1A144T yeast strain as described in A, after the addition of glucose, cells were harvested at the indicated time points and lysed in SDS sample buffer. Western blot analysis and quantitation were performed as described in Fig. 1B. Experiments were repeated three or more times, and representative data are shown. Mean half-life ± S.D. is indicated in bold. Dashed lines indicate the biphasic degradation of c-Myc in the ess1A144T strain. αPS62, α-Ser(P)-62; αPT58, α-Thr(P)-58.
To confirm the previous results and ensure that the biphasic nature of c-Myc decay was not specific to the ess1H164R yeast strain, we also tested c-Myc half-life in another ESS1 temperature-sensitive strain with a A144T mutation in the substrate binding pocket (34). As shown in Fig. 3C, c-Myc expressed in the ess1A144T stain has a half-life of 16.1 min with an overall mean half-life of 16.6 ± 1.8 min. Consistent with our observations in the ess1H164R strain, this is not a significant increase compared with c-Myc half-life in a wild type strain under the same conditions (13.0 ± 2.0 min). However, again we observed a biphasic decay of c-Myc in this yeast strain (Fig. 3C, dashed lines), suggesting that this phenotype may be common to all ESS1 mutants and reflects a requirement for ESS1 to degrade a subset of c-Myc.
Yeast PP2A Activity Facilitates c-Myc Ser-62 Dephosphorylation and Degradation
PP2A is a heterotrimeric enzyme that dephosphorylates serine 62 of c-Myc, thereby destabilizing the protein in mammalian cells (11, 23). PP2A is a trans-specific phosphatase, which explains the role of prior isomerization by Pin1 (35). The PP2A holoenzyme is composed of a catalytic (C) subunit that is encoded by two genes in S. cerevisiae, PPH21 and PPH22, a scaffolding (A) subunit, encoded by the TPD3 gene in S. cerevisiae, and a substrate-recognizing regulatory (B) subunit, of which there are two in yeast. RTS1 encodes the B′ family ortholog, whereas CDC55 encodes the B family ortholog (36, 37).
In mammalian cells Ser-62 phosphorylation is associated with c-Myc stabilization, and removal of the Ser-62 phosphate by PP2A occurs before polyubiquitination and degradation. Previous studies in mammalian cells have shown that inhibiting PP2A activity by the addition of okadaic acid or SV40 small T antigen results in increased c-Myc half-life (11). To determine whether yeast PP2A activity affects the phosphorylation of serine 62, we used a mutant strain lacking one of the two main PP2A catalytic subunits in S. cerevisiae, PPH21. We asked whether loss of Pph21p (Δpph21) changes the amount of c-Myc phosphorylation at serine 62 compared with that of c-Myc in a wild type yeast strain. As shown in Fig. 4A, c-Myc in the mutant strain has increased serine 62 phosphorylation compared with c-Myc expressed in the wild type strain. This is presumably due to a reduction in the ability of the cell to dephosphorylate serine 62. The increase in Ser-62 phosphorylation in the Δpph21 strain is modest, likely due to redundancy by the other PP2A C subunit.
FIGURE 4. Loss of yeast PP2A activity increases c-Myc Ser-62 phosphorylation and c-Myc half-life.
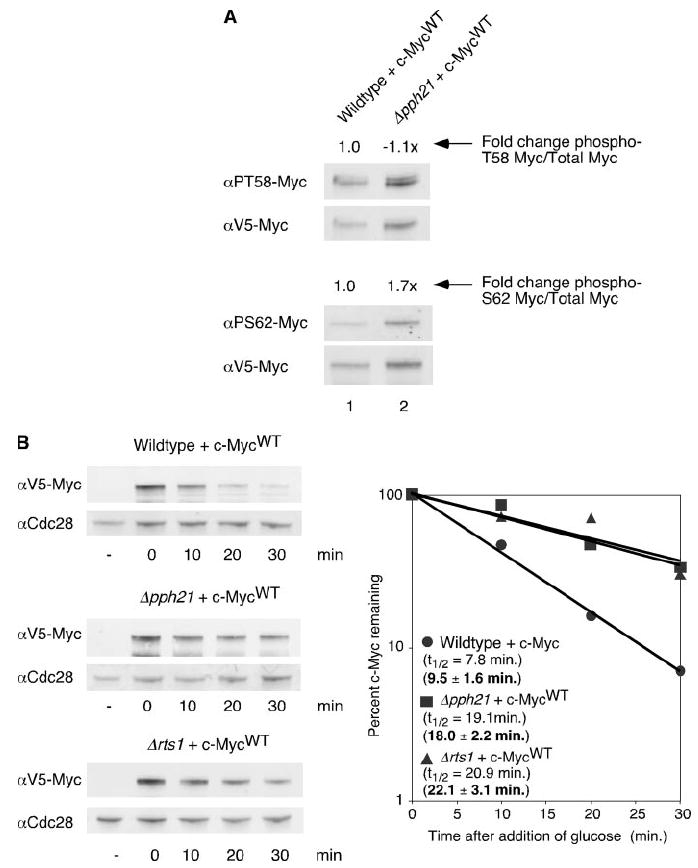
A, c-MycWT expression was induced at 30 °C for 1 h in the BY4741 or Δpph21 yeast strains. After the addition of glucose, cells were harvested and lysed in SDS sample buffer. Equal cell numbers were visualized by Western blot analysis with the indicated antibodies and quantitated as described in Fig. 1A. Ratios of phosphorylated c-MycWT to total c-MycWT were calculated. -Fold change of ratios in the Δpph21 strain compared with the BY4741 strain are shown. B, c-MycWT expression was induced for 1 h at 30 °C in the BY4741, Δpph21, or Δrts1 yeast strain. After the addition of glucose, cells were harvested at the indicated time points and lysed in SDS sample buffer. Western blot analysis and quantitation were performed as described in Fig. 1B. Experiments were repeated three or more times, and representative data are shown. Mean half-lives ± S.D. are indicated in bold. αPS62, α-Ser(P)-62; αPT58, α-Thr(P)-58.
We next examined c-Myc half-life in the Δpph21 strain. As shown in Fig. 4B, c-Myc half-life in this mutant strain was increased ~2-fold (19.1 min) compared with c-Myc half-life in a wild type control strain (7.8 min). Based on multiple independent experiments, the mean half-life of c-Myc in the Δpph21 strain was 18.0 ± 2.2 min compared with a mean half-life of 9.5 ± 1.6 min in the isogenic control. We also examined c-Myc protein stability in a strain lacking the yeast ortholog to the PP2A substrate-recognizing subunit from the B′ family, RTS1 (Δrts1). We have recently reported that the B′ family subunit, B56α, is responsible for targeting PP2A to c-Myc for dephosphorylation in mammalian cells (23). As shown in Fig. 4B, c-Myc expressed in the Δrts1 strain had a little more than a 2-fold increase in half-life compared with c-Myc expressed in the wild type control, with a mean half-life of 22.1 ± 3.1 min. The modest effect in these PP2A mutant yeast strains is likely due to multiple PP2A C and B subunits. Taken together, these data suggest that PP2A in yeast does dephosphorylate c-Myc at Ser-62, and this leads to destabilization of c-Myc protein. We also tested c-Myc half-life in a strain deleted for the PP2A A subunit. However, we obtained variable results from quite stable (34.3 min) to unstable (data not shown). This is likely due to spurious activity by the C subunit in the absence of the structural A subunit as previously reported (23).
The Yeast F-box Protein, Cdc4p, Does Not Appear to Be Involved in Controlling c-Myc Degradation
The ubiquitination machinery is highly conserved from yeast to mammals. The ubiquitin ligase complex, SCF, which targets phosphoproteins for ubiquitination, has been shown in yeast to target c-Myc and other cell cycle proteins for ubiquitination and subsequent degradation (17, 18). The SCF complex is composed of four subunits, Skp1, Cdc53, Roc1, and a variable F-box protein, which determines substrate specificity. In mammalian cells, the F-box protein, Skp2, is reported to target c-Myc through MBII and the C-terminal domains in a phosphorylation-independent manner (18, 38). In contrast, the F-box protein, Fbw7, has been shown to target c-Myc that is phosphorylated at Thr-58, suggesting that it is the E3 ligase which degrades c-Myc in response to regulated phosphorylation of Ser-62 and Thr-58 (12, 13).
Fbw7 is also referred to as hCdc4 since it is the structural homologue of the S. cerevisiae F-box protein, Cdc4 (17). Therefore, to examine c-Myc half-life in the absence of functional Cdc4, we expressed c-Myc in the temperature-sensitive cdc4-1 strain from the GAL1 promoter for 2 h at the restrictive temperature. It has been reported that cdc4-1 cells arrest at the G1/S transition; therefore, c-Myc protein stability was also measured in an isogenic wild type control strain at the restrictive temperature with the addition of α -factor to arrest the cells at the G1 phase of the cell cycle (39). Mutant phenotype at the restrictive temperature was verified microscopically (data not shown). As shown in Fig. 5A, c-Myc is equally unstable in both the cdc4-1 strain and the wild type control strain with the half-lives of 11.0 and 12.6 min, respectively. This result is consistent with previous reports showing that c-Myc is not stabilized in a yeast strain mutant for Cdc4p; however, this is a surprising result given the structural homology with Fbw7/hCdc4 (18).
FIGURE 5. Mutation of the Fbw7 structural homolog, Cdc4, does not result in c-Myc stabilization.
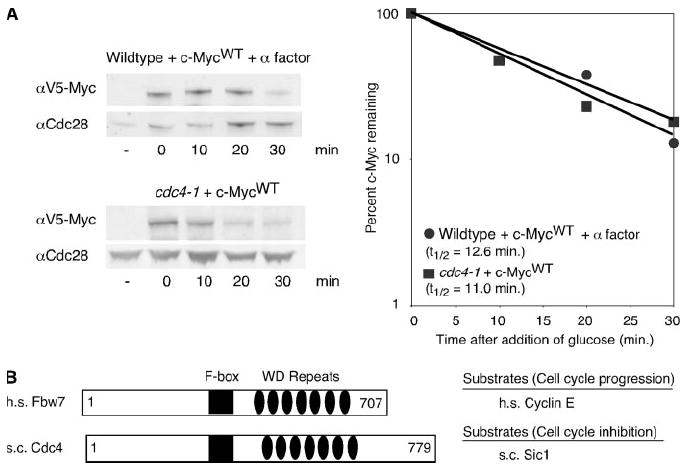
A, W303 cells, in the presence of α factor, and cdc4-1 cells were grown in 2% raffinose medium at 37 °C for 2 h. c-Myc expression was induced for 1 h at 37 °C by the addition of galactose. After termination of expression by the addition of glucose, cells were harvested at the indicated time points and lysed in SDS sample buffer. Western blotting and quantitation were performed as described in Fig. 1B. B, schematics of the Homo sapiens (h.s.) F-box protein, Fbw7, and the S. cerevisiae (s.c.) F-box protein, Cdc4. Key structural domains are indicated as well as target proteins of the F-boxes that are important in regulating the cell cycle (see “Results”).
The structural homology between Fbw7 and yeast Cdc4 is based on similar WD40 repeats (Fig. 5B) (17). However, despite the fact that these proteins are structurally related, their substrates do not have similar cell cycle roles. In mammalian cells, Fbw7 targets cyclin E and c-Jun in addition to c-Myc, all of which are proteins known to drive cell cycle progression (17, 40). In contrast, yeast Cdc4p targets the yeast cell cycle inhibitor, Sic1p, for ubiquitin-mediated degradation (41, 42) (Fig.5B). This suggests that although these proteins are structurally similar, they may not be functional homologues, and it also argues for the presence of another yeast F-box protein capable of targeting c-Myc.
DISCUSSION
Previously, we and others have shown that phosphorylation of c-Myc at serine 62 and threonine 58 is sequential and interdependent and that these phosphorylation events have opposing effects on c-Myc protein stability, presumably by influencing the ability of c-Myc to bind its E3 ligase (9, 10, 12, 13). Many investigators have used the genetically tractable budding yeast S. cerevisiae as a model system to study the ubiquitin-mediated degradation of mammalian proteins. This is because the ubiquitin-proteasome pathway is highly conserved from mammals to yeast. Several groups have examined the half-life of c-Myc in the S. cerevisiae and found it to be very short. However, it has never been determined whether c-Myc could be phosphorylated at either Ser-62 or Thr-58 in yeast. Additionally, whereas there are yeast orthologs to the mammalian proteins that phosphorylate c-Myc, it was unknown whether these proteins could phosphorylate mammalian c-Myc in the complex manner observed in mammalian cells. In this report we have shown that c-Myc is phosphorylated at Thr-58 and Ser-62 in yeast, and these phosphorylation events are interrelated like they are in mammalian cells. Specifically, phosphorylation of Thr-58 required prior phosphorylation of Ser-62, and Ser-62 dephosphorylation was facilitated by Thr-58 phosphorylation, just as in mammalian cells. This is a striking observation and points to the importance and conserved nature of the signaling pathway that can control phosphorylation of the Thr-58–Ser-62 phospho-domain found in mammalian c-Myc.
Phosphorylation by Conserved Proteins Controls c-Myc Stability in Yeast
We have shown that phosphorylation of c-Myc at Ser-62 and Thr-58 occurs in yeast through a conserved signaling pathway that is likely to target a specific phospho-degron motif similar to that found in c-Myc (Fig. 6A). As in mammalian cells, phosphorylation at Ser-62 stabilizes c-Myc, and phosphorylation at Thr-58 destabilizes c-Myc. This suggests that the mode of recognition of c-Myc by an E3 ubiquitin ligase and its subsequent proteasomal degradation is conserved in yeast. The Thr-58 and Ser-62 residues are located in a highly conserved region of c-Myc designated Myc Box I (MBI). Consistent with our findings, it has previously been reported that deletion of MBI or a region encompassing MBI results in increased c-Myc protein stability in yeast (14, 15, 43).
FIGURE 6. A conserved pathway to control protein degradation.
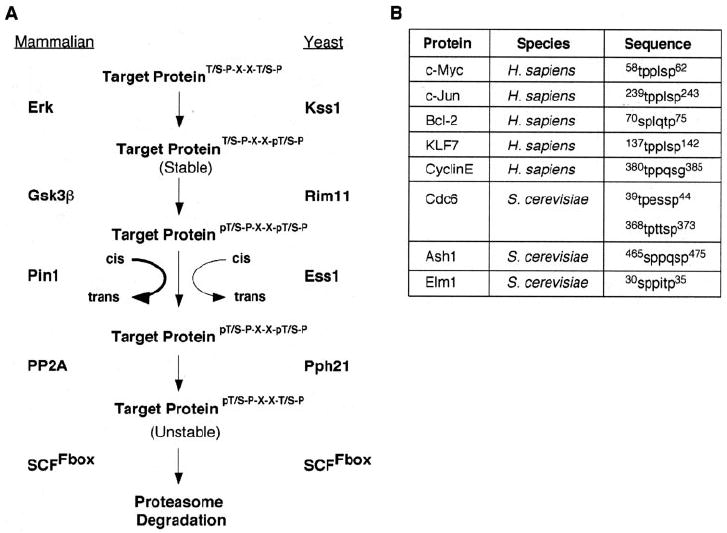
A, summary of the conserved pathway that controls protein stability through a specific phospho-degron present in c-Myc, mammalian cells, and yeast cells. B, other mammalian and yeast proteins with predicted phospho-degron sequence that would be affected by this pathway (see “Discussion” for details).
Serine 62 phosphorylation by ERK stabilizes c-Myc in mammalian cells. However, without Ser-62 phosphorylation Thr-58 cannot be phosphorylated, and c-Myc degradation is likely mediated by an alternate E3 ligase not involving these phosphorylation sites; thus, a somewhat longer half-life is observed in mammalian cells with the S62A mutant as well as in Δkss1 yeast with wild-type c-Myc. Our data support a role for yeast Rim11p in mediating Thr-58 phosphorylation similar to GSK3β, leading to c-Myc destabilization. Likewise, Pph21p/Rts1p appears to dephosphorylate Ser-62 similar to PP2A-B56α and destabilizes c-Myc. Our data, however, did not support a strong role for Ess1p in regulating c-Myc turnover in yeast nor do we observe a role for the Fbw7 structural homologue, Cdc4p, in regulating c-Myc ubiquitin-mediated degradation. The F-box protein, Grr1p, is, however, reported to play a role in c-Myc degradation in yeast, as described below.
The Requirement for c-Myc Isomerization Is Not Conserved in Yeast
Although the phosphorylation and dephosphorylation events that control c-Myc protein stability appear to be conserved from yeast to mammals, isomerization by a peptidylprolyl isomerase does not appear to be a limiting step in c-Myc degradation in yeast cells, as it is in mammalian cells. Because the dephosphorylation step leading toward c-Myc degradation is conserved in yeast, there are two possibilities that would explain why Ess1p does not appear to be required for c-Myc destruction; 1) yeast PP2A does not require c-Myc to be in the trans conformation before dephosphorylation, or 2) c-Myc is already in the trans conformation, allowing for dephosphorylation of Ser-62 by PP2A without Ess1p activity. The former is a less likely explanation since it has been previously shown that Pin1 and PP2A have a reciprocal genetic interaction in yeast in that Pin1 has been able to partially rescue a yeast temperature-sensitive mutant that lacks PP2A activity, and overexpression of one of the yeast PP2A catalytic subunits almost completely rescues an Ess1 temperature-sensitive mutation (35). Thus, although there is accumulation of the cis conformation of c-Myc in mammalian cells before its degradation, in yeast cells the majority of c-Myc may remain in the trans conformation. Given that in mammalian cells mitogen-activated protein kinases phosphorylate Ser/Thr-Pro motifs in the trans conformation and PP2A requires Pin1 to efficiently dephosphorylate Ser-62, it is likely that an intermediary trans to cis isomerization step exists in mammalian cells but not in yeast (35, 44). At this time it is unknown which enzyme catalyzes this initial conversion, although Pin1 is a likely candidate; however, it appears that this step may not occur in yeast, perhaps due to the fact that c-Myc is not functioning as a transcription factor in yeast.
Structurally Related E3 Ligase F-box Proteins in Mammals, Fbw7, and Yeast, Cdc4, Are Not Functionally Conserved
Another interesting aspect of this work is the lack of stabilization of c-Myc in a yeast strain mutated for the F-box Cdc4, the reported yeast homologue to mammalian Fbw7 (17). This observation has been previously reported; Kim et al. (18) has shown that mutation of the F-box protein Grr1, but not Cdc4, results in the stabilization of c-Myc in yeast. This led to the finding that the mammalian F-box, Skp2, which has a similar structure to Grr1, can target c-Myc for ubiquitination in mammalian cells. At the same time, Skp2 can also enhance c-Myc transcriptional activity (18, 38). However, it was also shown that the Skp2-c-Myc interaction was via MBII, a second highly conserved region in c-Myc, and through the c-Myc C-terminal domain, and recognition was not phosphorylation-dependent (18, 38). Shortly after this, Fbw7 was identified as a second mammalian F-box that targets c-Myc for ubiquitination, and binding of Fbw7 to c-Myc was dependent on the phosphorylation of Thr-58 in MBI (12, 13). Because we now have shown that these phosphorylation events are conserved, and they are important for controlling c-Myc stability in yeast, the question arises, Why does mutation of the Fbw7 homologue, Cdc4, not result in c-Myc stability in yeast? Although c-Myc contains a reported optimal Cdc4 phospho-degron) (45), some of Cdc4p substrates in yeast, like Sic1p, require multiple phosphorylation events for recognition, and perhaps a singly Thr-58 phosphorylated c-Myc is not well recognized (46, 47).
In yeast cells Grr1p targets the G1 cyclin, Cln2p, for degradation (48). Because mammalian Fbw7 has similar substrates, such as cyclin E, it is possible that Grr1p, but not Cdc4p, may be more functionally homologous to Fbw7. At this time it is unknown whether recognition of c-Myc for ubiquitination by Grr1p occurs through phosphorylation at Thr-58 in yeast cells, but our data strongly suggest that either Grr1p or another yet unknown F-box targets c-Myc for ubiquitination through this phospho residue in yeast cells.
A Conserved Pathway to Control Protein Degradation
In this work we have shown a number of similarities between the complex control of c-Myc protein stability in mammalian cells and in the model system S. cerevisiae. This conservation supports the importance of this pathway that targets c-Myc for degradation, and it validates the use of S. cerevisiae as a model system to study c-Myc. Additionally, this work suggests that this pathway may act on other target proteins with a similar phosphorylation consensus sequence of (T/S)PXX(T/S)P in both mammalian and yeast cells, as illustrated in Fig. 6A.
Fig. 6B shows a selected list of mammalian and yeast proteins with this potential phospho-degron sequence. As we and others have shown in mammalian cells and now in yeast cells, the sequence TPPLSP of c-Myc controls c-Myc stability presumably by changing its affinity for its E3 ligase (11-13). In yeast cells the transcription factor Ash1, the kinase Elm1, and the pre-replication complex protein, Cdc6, all contain a similar phospho-degron sequence. Although little work has been done elucidating the control of stability of Elm1 and Ash1, there has been a great deal of work dedicated to describing the mechanism of Cdc6 degradation. It has been shown that there are three modes of degradation for Cdc6 (49). Two of the modes, which occur in either S Phase or M phase, appear to be controlled by the two sequences indicated in Fig. 6B. For example, Perkins et al. (50) demonstrated that mutation of Thr-368 of Cdc6 completely stabilizes the Cdc6 protein, whereas mutation of Ser-372 of Cdc6 resulted in a partially stable protein (50). These results are very similar to our observations of mutations of the corresponding residues of c-Myc (see Fig. 1B), suggesting that this pathway in yeast may normally target Cdc6 for degradation.
There are also other proteins that may be targeted by this degradation pathway in mammalian cells; these include Kruppel-like factor KLF7, the anti-apoptotic protein Bcl-2, the cell cycle protein cyclin E, and the proto-oncogene c-Jun. Although we were unable to find any data concerning regulation of KLF7 stability, it has been shown that ERK-dependent phosphorylation of Thr-74 of Bcl-2 has a stabilizing effect on the protein (51). This is similar to the effect of the ERK-dependent phosphorylation of Ser-62, the corresponding residue of c-Myc in the phospho-degron sequence. Cyclin E does not have a perfect consensus sequence; however, its degradation is controlled by many of the same players as c-Myc, including GSK3β and FBW7 (17, 52). Additionally, it has recently been shown that PIN1 may be important in regulating cyclin E levels, although at this time it appears to require phosphorylated Ser-284, the residue that corresponds to c-Myc Ser-62, for binding to PIN1 (53). This is in contrast to c-Myc, which requires a phosphorylation at Thr-58 for PIN1 binding (11). Like c-Myc, c-Jun contains the sequence TPPLSP. Recently it was shown that Thr-239 of c-Jun is phosphorylated by GSK3β, and mutation of this site results in a stabilized protein, just as when the corresponding Thr-58 is mutated in c-Myc (35). Both proteins have also been reported to be substrates of the F-box SCFFbw7 (12, 13, 54). Although testing all of these proteins is not within the scope of this paper, currently studies are under way to determine whether this degradation pathway controls the stability of other mammalian and yeast proteins.
Footnotes
The abbreviations used are: ERK, extracellular signal-regulated kinase; GST, glutathione S -transferase; Ni-NTA, nickel-nitrilotriacetic acid; PP2A, protein phosphatase 2A; PBS, phosphate-buffered saline; WT, wild type; MBI, Myc Box I; GSK, glycogen synthase kinase; S.D., standard deviation.
References
- 1.Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 2.Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, Larsson LG, Wright AP. Nat Cell Biol. 2005;7:303–310. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 3.Kelly K, Cochran BH, Stiles CD, Leder P. Cell. 1983;35:603–610. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- 4.Jones TR, Cole MD. Mol Cell Biol. 1987;7:4513–4521. doi: 10.1128/mcb.7.12.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luscher B, Eisenman RN. Genes Dev. 1990;4:2025–2035. doi: 10.1101/gad.4.12a.2025. [DOI] [PubMed] [Google Scholar]
- 6.Sears R, Leone G, DeGregori J, Nevins JR. Mol Cell. 1999;3:169–179. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- 7.Arvanitis C, Felsher DW. Cancer Lett. 2005;226:95–99. doi: 10.1016/j.canlet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 8.Popescu NC, Zimonjic DB. J Cell Mol Med. 2002;6:151–159. doi: 10.1111/j.1582-4934.2002.tb00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lutterbach B, Hann SR. Mol Cell Biol. 1994;14:5510–5522. doi: 10.1128/mcb.14.8.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 12.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. Proc Natl Acad Sci U S A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salghetti SE, Kim SY, Tansey WP. EMBO J. 1999;18:717–726. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flinn EM, Busch CM, Wright AP. Mol Cell Biol. 1998;18:5961–5969. doi: 10.1128/mcb.18.10.5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herbst A, Salghetti SE, Kim SY, Tansey WP. Oncogene. 2004;23:3863–3871. doi: 10.1038/sj.onc.1207492. [DOI] [PubMed] [Google Scholar]
- 17.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Nature. 2001;413:316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 18.Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Mol Cell. 2003;11:1177–1188. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- 19.Sherman F, Fink GR, Hicks JB Cold Spring Harbor Laboratory. Laboratory Course Manual for Methods in Yeast Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1986. [Google Scholar]
- 20.Shieh SY, Ikeda M, Taya Y, Prives C. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 21.Horvath A, Riezman H. Yeast. 1994;10:1305–1310. doi: 10.1002/yea.320101007. [DOI] [PubMed] [Google Scholar]
- 22.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 23.Arnold HK, Sears RC. Mol Cell Biol. 2006;26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang DW, Claassen GF, Hann SR, Cole MD. Mol Cell Biol. 2000;20:4309–4319. doi: 10.1128/mcb.20.12.4309-4319.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hann SR, Eisenman RN. Mol Cell Biol. 1984;4:2486–2497. doi: 10.1128/mcb.4.11.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puziss JW, Hardy TA, Johnson RB, Roach PJ, Hieter P. Mol Cell Biol. 1994;14:831–839. doi: 10.1128/mcb.14.1.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boulton TG, Yancopoulos GD, Gregory JS, Slaughter C, Moomaw C, Hsu J, Cobb MH. Science. 1990;249:64–67. doi: 10.1126/science.2164259. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi MW, Plyte SE, Kreis M, Woodgett JR. Gene (Amst) 1993;134:51–56. doi: 10.1016/0378-1119(93)90173-z. [DOI] [PubMed] [Google Scholar]
- 29.Casamayor A, Khalid H, Balcells L, Aldea M, Casas C, Herrero E, Arino J. Yeast. 1996;12:1013–1020. doi: 10.1002/(SICI)1097-0061(199609)12:10B%3C1013::AID-YEA980%3E3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 30.Hardy TA, Wu D, Roach PJ. Biochem Biophys Res Commun. 1995;208:728–734. doi: 10.1006/bbrc.1995.1398. [DOI] [PubMed] [Google Scholar]
- 31.Elion EA, Brill JA, Fink GR. Cold Spring Harbor Symp Quant Biol. 1991;56:41–49. doi: 10.1101/sqb.1991.056.01.007. [DOI] [PubMed] [Google Scholar]
- 32.Gregory MA, Qi Y, Hann SR. J Biol Chem. 2003;278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 33.Lu KP, Hanes SD, Hunter T. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 34.Wu X, Wilcox CB, Devasahayam G, Hackett RL, Arevalo-Rodriguez M, Cardenas ME, Heitman J, Hanes SD. EMBO J. 2000;19:3727–3738. doi: 10.1093/emboj/19.14.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 36.Csortos C, Zolnierowicz S, Bako E, Durbin SD, DePaoli-Roach AA. J Biol Chem. 1996;271:2578–2588. doi: 10.1074/jbc.271.5.2578. [DOI] [PubMed] [Google Scholar]
- 37.Healy AM, Zolnierowicz S, Stapleton AE, Goebl M, DePaoli-Roach AA, Pringle JR. Mol Cell Biol. 1991;11:5767–5780. doi: 10.1128/mcb.11.11.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG. Mol Cell. 2003;11:1189–1200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]
- 39.Goh PY, Surana U. Mol Cell Biol. 1999;19:5512–5522. doi: 10.1128/mcb.19.8.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 42.Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 43.Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP. EMBO Rep. 2005;6:177–183. doi: 10.1038/sj.embor.7400333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weiwad M, Kullertz G, Schutkowski M, Fischer G. FEBS Let. 2000;478:39–42. doi: 10.1016/s0014-5793(00)01794-4. [DOI] [PubMed] [Google Scholar]
- 45.Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 46.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 47.Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- 48.Barral Y, Jentsch S, Mann C. Genes Dev. 1995;9:399–409. doi: 10.1101/gad.9.4.399. [DOI] [PubMed] [Google Scholar]
- 49.Drury LS, Perkins G, Diffley JF. Curr Biol. 2000;10:231–240. doi: 10.1016/s0960-9822(00)00355-9. [DOI] [PubMed] [Google Scholar]
- 50.Perkins G, Drury LS, Diffley JF. EMBO J. 2001;20:4836–4845. doi: 10.1093/emboj/20.17.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Breitschopf K, Haendeler J, Malchow P, Zeiher AM, Dimmeler S. Mol Cell Biol. 2000;20:1886–1896. doi: 10.1128/mcb.20.5.1886-1896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM. Mol Cell. 2003;12:381–392. doi: 10.1016/s1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 53.Yeh ES, Lew BO, Means AR. J Biol Chem. 2006;281:241–251. doi: 10.1074/jbc.M505770200. [DOI] [PubMed] [Google Scholar]
- 54.Nateri AS, Riera-Sans L, Da Costa C, Behrens A. Science. 2004;303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]