

Abstract
We investigated the utility of association mapping to dissect the genetic basis of naturally occurring variation in bud phenology in European aspen (Populus tremula). With this aim, we surveyed nucleotide polymorphism in 13 fragments spanning an 80-kb region surrounding the phytochrome B2 (phyB2) locus. Although polymorphism varies substantially across the phyB2 region, we detected no signs for deviations from neutral expectations. We also identified a total of 41 single nucleotide polymorphisms (SNPs) that were subsequently scored in a mapping population consisting of 120 trees. We identified two nonsynonymous SNPs in the phytochrome B2 gene that were independently associated with variation in the timing of bud set and that explained between 1.5 and 5% of the observed phenotypic variation in bud set. Earlier studies have shown that the frequencies of both these SNPs vary clinally with latitude. Linkage disequilibrium across the region was low, suggesting that the SNPs we identified are strong candidates for being causally linked to variation in bud set in our mapping populations. One of the SNPs (T608N) is located in the “hinge region,” close to the chromophore binding site of the phyB2 protein. The other SNP (L1078P) is located in a region supposed to mediate downstream signaling from the phyB2 locus. The lack of population structure, combined with low levels of linkage disequilibrium, suggests that association mapping is a fruitful method for dissecting naturally occurring variation in Populus tremula.
A major goal of population and quantitative genetics is to identify the polymorphisms underlying phenotypic variation, particularly in traits that are important for ecological adaptations (Feder and Mitchell-Olds 2003; Stinchcombe and Hoekstra 2007). While the accumulation of functional genomics data over the last decades has provided detailed information on the genetic basis of many traits in a number of model organisms, it remains largely unknown how many of these genes that contain genetic variation segregate in natural populations. Progress in elucidating the genetic basis of ecological adaptations has been slow, partly because many of the species for which detailed information on phenotypic variation in traits of ecological relevance is available are lacking the genomic resources that allow genetic dissection of these traits. Furthermore, adaptive traits are often quantitative and are therefore unlikely to have a simple genetic basis. Nevertheless, by studying the genetic basis of ecologically important traits, hopes have been raised that it should yield insights into the number and effect sizes of genes underlying ecological adaptations and the evolutionary forces that act to maintain variation in such traits (Feder and Mitchell-Olds 2003; Stinchcombe and Hoekstra 2007).
Many organisms, such as the model species Drosophila and Arabidopsis, are suitable for “classical” genetic techniques, on the basis of the development of segregating mapping populations (Liu 1998). However, for many organisms (e.g., humans) these tools are not feasible and other methods have been developed to dissect the genetic basis of phenotypic traits. One such method is association mapping, where unrelated individuals that have undergone recombination over multiple generations are genotyped and used to connect genotype to phenotype (Risch 2000). Forest genetics is in many respects in a similar situation to human genetics, because even though segregating tree populations can in principle be constructed, the extended juvenile phases and irregular flowering of many tree species make such experiments impractical as they may take decades to complete. However, tree species have many characteristics that make them suitable for association mapping: they are predominantly outcrossing and have large, relatively unstructured populations, resulting in high levels of nucleotide diversity and low linkage disequilibrium (LD) (Neale and Savolainen 2004; González-Martínez et al. 2006). In addition, many tree species can easily be cloned, allowing for phenotyping with high precision and for replication in different environments. Incidentally, the first articles using association mapping in forest trees have recently been published (Thumma et al. 2005; González-Martínez et al. 2007).
Forest trees often have very wide geographic distributions and are ecologically dominant species in many ecosystems (Brunner et al. 2004; González-Martínez et al. 2006). Because of their perennial nature, most tree species are subjected to large seasonal variations in temperature and have as a response evolved annual growth cycles that promote long-term survival and growth (González-Martínez et al. 2006). While growth cessation and dormancy are critical to winter survival, dormancy also constrains growth by reducing the amount of time during which growth can take place (Horvath et al. 2003; González-Martínez et al. 2006). A correct tuning of phenology to changes in environmental conditions across a growing season thus represents an important ecological and evolutionary trade-off between survival and growth in most forest trees (Horvath et al. 2003; González-Martínez et al. 2006). Seasonal variation in photoperiod is an important environmental variable that many tree species use as a cue to initiate and/or terminate growth or reproduction (Howe et al. 2003; González-Martínez et al. 2006). This is clearly manifested in the adaptive response of trees to the steep latitudinal gradient in the length of the growing season that characterizes northern environments (Howe et al. 2003; González-Martínez et al. 2006). Many perennial plants also show latitudinal clines in important phenological traits, such as timing of germination, dormancy, or the onset of flowering (Howe et al. 2003; González-Martínez et al. 2006). Such clinal variation is usually interpreted as strong evidence for a balance between spatially variable selection and migration (Barton 1999; González-Martínez et al. 2006).
In many perennial plants, seasonal control of phenology is regulated by genes in the photoperiodic pathway (Wareing 1956; Mouradov et al. 2002; Horvath et al. 2003; Howe et al. 2003). Phytochromes are signal-transducing photoreceptors that are key components of the photoperiodic pathway in plants (Smith 2000) and that play an important role in light perception in plants. Known phytochrome-regulated processes include the daily entraining of the circadian clock, deetiolation, stem elongation, and seasonal developmental responses such as flowering and initiation or release of dormancy (Smith 2000). The photosensory activity of the phytochrome protein stems from its ability to convert between a biologically inactive form, Pr, absorbing light at red wavelengths and a biologically active form, Pfr, absorbing light at far-red wavelengths (Smith 2000; Quail 2002). The conversion between the Pr and Pfr forms involves conformational changes of the molecule that allows for changes in signaling activity of the phytochrome protein (Maloof et al. 2000). The ratio of red to far-red light changes over both short (a single day) and long (over a season) timescales and phytochromes can thus provide a plant with temporal signals that are used to synchronize developmental changes, such as the initiation or release of dormancy, with changing environmental conditions across a growing season (Smith 2000). Phytochromes are thus ideal candidate genes for mediating ecologically important variation in the timing of developmental processes such as dormancy or flowering (Maloof et al. 2000). Incidentally, mutations in two members of the phytochrome gene family, PHYA and PHYC have been shown to be responsible for natural variation in light sensitivity, seedling growth, and flowering time in Arabidopsis thaliana (Maloof et al. 2001; Balasubramanian et al. 2006).
Several replicated QTL mapping experiments in Populus have shown that one phytochrome gene, phytochrome B2 (phyB2), maps to a linkage group containing QTL for both bud set and bud flush. phyB2 is thus a strong candidate gene for controlling naturally occurring variation in bud phenology in Populus (Frewen et al. 2000; Chen et al. 2002). We have shown that populations of European aspen (Populus tremula) collected across a latitudinal gradient representing growing seasons ranging from 2 to 5 months show little population structure at neutral markers but nevertheless show strong adaptive population differentiation in photoperiod sensitivity (Hall et al. 2007; Luquez et al. 2007). We have also documented clinal variation with latitude at several nonsynonymous mutations at phyB2 (Ingvarsson et al. 2006). These sites are spread over ∼4 kb of the phyB2 gene (Ingvarsson et al. 2006) and are likely to represent independent clines, as linkage disequilibrium declines to negligible levels in <500 bp in P. tremula (Ingvarsson 2005). In this article we use association mapping to further study the role of phyB2 in the mediating natural variation in photoperiodic control of dormancy initiation and release in P. tremula. We show that two amino acid substitutions are associated with natural variation in bud set in P. tremula.
MATERIALS AND METHODS
Plant material and phenotypic scoring:
All data described in this article were generated using trees from the Swedish Aspen Collection (the SwAsp collection), which have been described in detail elsewhere (Ingvarsson et al. 2006; Hall et al. 2007; Luquez et al. 2007). This collection was established from 10 P. tremula clones collected from each of 12 different sites throughout Sweden in 2003. All trees were clonally replicated (four rametes per clone) and planted in 2 common garden sites in 2004 (Sävar, 63°N, and Ekebo, 56°N). A total of 480 rametes were planted at each common garden site. All data presented here are based on means from four rametes per clone at each site. During the spring, summer, and autumn of 2005 and 2006 we measured bud flush and bud set traits at the two common garden sites. Bud flush (BF) was scored every 2 days from the flush of the first tree in the spring until all trees had flushed. A tree was considered to have flushed when the first fully unfolded leaf was observed (Frewen et al. 2000). Bud set (BS) was scored twice a week starting in mid-July and was continued until trees had set terminal buds (Frewen et al. 2000). In 2006 we also scored bud flush and bud set under greenhouse conditions. At least two rametes of each clone were kept in either of two greenhouses at the Swedish University of Agricultural Sciences, Umeå, under ambient photoperiod. Bud flush and bud set were scored in the same manner as in the field planted trees. We have excluded data for bud set from Sävar in 2006 in all of our analyses, as a severe drought caused most of the trees to set buds prematurely, in the middle of the season.
SNP discovery and genotyping:
Total genomic DNA was extracted from frozen leaf tissue using the DNeasy plant mini prep kit (QIAGEN, Valencia, CA). Primers to amplify 12 fragments of ∼800 bp surrounding the Populus phyB2 gene were designed on the basis of the publicly available genome sequence of P. trichocarpa (http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html). The primer combinations were tested to ensure amplification and if the PCR reactions failed new primers were designed. An effort was made to have a roughly equal spacing between fragments, but that was sometimes not possible because of an apparent lack of conservation of intergenic regions between P. trichocarpa and P. tremula. All primer sequences used are presented in supplemental Table S1. Fragments were PCR amplified from a total of 12 individuals from the SwAsp collection. PCR products were cloned into the pCR2.1 vector using a TA-cloning kit from Invitrogen (Carlsbad, CA) and fragments were sequenced on either an ABI377 automated sequencer or a Beckman CEQ 2000 capillary sequencer at the Umeå Plant Science Centre sequencing facility. As P. tremula is highly heterozygous (Ingvarsson 2005), five or more clones from three pooled PCR reactions were sequenced in an attempt to identify the two haplotypes present within an individuals and to control for Taq polymerase errors. Sequences were verified manually and contigs were assembled using the computer program Sequencer v 4.0. Multiple sequence alignments were made using Clustal W (Thompson et al. 1994) and adjusted manually using BioEdit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). All sequences described in this article have been deposited in the GenBank database (EU478172–EU478407). SNPs were identified from the sequenced fragments and were scored by developing cleaved amplified polymorphism sequence markers (CAPS), by single base pair primer extension on a Beckman CEQ 8000 capillary sequencer, or by complete sequencing of short fragments amplified from genomic DNA (Ingvarsson et al. 2006).
Statistical analyses:
Estimates of nucleotide diversity per site from the total number of segregating sites (θ), average pairwise heterozygosity (π), and Tajima's D (Tajima 1989) were obtained from the sequence fragments using a computer program written in C++ on the basis of the publicly available C++ class library libsequence (Thornton 2003). Tests for Hardy–Weinberg equilibrium were performed for each SNP using the genetics (Warnes and Leisch 2006) package from the statistical package R (R Development Core Team 2007). This package was also used to test for pairwise linkage disequilibrium between individual SNPs using a maximum-likelihood approach since the genotype data is unphased and individual haplotypes cannot be distinguished. We also tested for clinal variation in SNP frequencies by regressing population allele frequencies on latitude of origin.
In our association analyses, we preferred single-marker tests over haplotype-based tests for several reasons. First, there are uncertainties in haplotype determination from diploid, unphased SNP data. Second, LD between markers was fairly low, yielding a large number of potential haplotypes with even a few SNPs, and haplotype-based analyses are known to rapidly loose power with increasing numbers of haplotypes. Finally, single-marker tests have equal or even higher power than haplotype tests if LD is low and/or if there is a high probability that causal SNPs have been typed (Long and Langley 1999).
Single-marker associations were tested using a model assuming additive effects of alleles within a marker locus. To control for possible spurious associations caused by genetic structuring of the sample, we used the mixed-model method of Yu et al. (2006) that allows for both population structure and more diffuse familial structure within the sample. In matrix form this model is given by
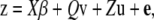 |
(1) |
where z is a vector of phenotypic observations, β is a vector of SNP effects, v is a vector of population effects, u is a vector of polygenic background effects, and e is a vector of residual effects.
Population structure and pairwise kinship coefficients were estimated using 26 putatively neutral SSR markers and 39 SNPs from four different genes (col2b, GA20ox1, hypO312, and abi1b) (Hall et al. 2007). Population structure was estimated in two different ways. We used either the program Structure (Pritchard et al. 2000) to estimate population membership for each individual for a number of different subpopulations (K = 2–5) or the principle-component (PCA) method of Price et al. (2006) to infer population structure. The effects of population structure, estimated using either Structure or PCA, were summarized by the Q matrix in Equation 1. Both methods yielded similar results, and we therefore present data only based on Structure. Similarly, a matrix of pairwise kinship coefficients, K, was calculated according to Ritland (1996), using the software package SpaGeDi (Hardy and Vekemans 2002) and was used to specify the variance of the random (clone) effects according to 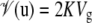 . Finally, the variance of the residuals was assumed to be
. Finally, the variance of the residuals was assumed to be 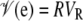 , where R is a matrix with zeros for all off-diagonal elements and the reciprocal of the number of observations along the diagonal. Vg and VR are the genetic and residual variances for the trait in question.
, where R is a matrix with zeros for all off-diagonal elements and the reciprocal of the number of observations along the diagonal. Vg and VR are the genetic and residual variances for the trait in question.
We combined data from the different sites and years by calculating best linear unbiased predictors (BLUPs) for all genotypes in the association population by fitting the model
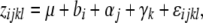 |
(2) |
where zijkl is the phenotype of the lth individual in the kth block from the jth clone from the ith population. In Equation 2 μ denotes the grand mean and ɛijkl is the residual error term. The clone (βj) and residual term (ɛijkl) were modeled as random effects, whereas site/year (bi) and block (γk) were treated as fixed effects. Equation 2 was fitted to the data using restricted maximum-likelihood techniques and BLUPs were calculated using the lmer function in R . The BLUPs were used as the dependent trait in the association analyses. We implemented the mixed-model analysis (Yu et al. 2006) using the kinship library (Atkinson and Therneau 2007) in R and fitted the models using maximum-likelihood methods. To control for multiple testing we used the method of Storey and Tibshirani (2003) to control the false discovery rate (FDR), as implemented in the qvalue package in R.
Data-perturbation simulations:
We used the data-perturbation method described in Yu et al. (2006) to create new data sets that were analyzed using the same methods as the observed phenotypic data. Briefly, a single SNP is randomly chosen and assigned a phenotypic effect which is added to the original data. By applying this to the original data, the complex correlation structure of the data is preserved. Phenotypic effects of SNPs were chosen in the range of 0.0–1.0 times the phenotypic standard deviation. The proportion of phenotypic variation explained by the causal SNP was estimated by regressing phenotype on SNP genotype (Zhao et al. 2007). We scored power across the simulated data sets for three different α-values, 0.05, 0.01, and 0.001. We also estimated the distribution of observed allelic effects conditional on the “true” allelic effect and on observing a significant effect at a SNP at a given α-value (Allison et al. 2002). For each replicate simulation where we obtained a significant result (at P < 0.001), we scored the true and estimated phenotypic effect of the SNP. This simulated distribution was then used to calculate the corrected estimates of allelic effects at the T608N and L1078P SNPs, using the method-of-moments approach outlined in Allison et al. (2002).
RESULTS
Molecular population genetics and SNP identification:
We surveyed nucleotide polymorphism in a sample of 12 short (∼800-bp) fragments spanning ∼80 kb surrounding the phyB2 gene (Figure 1 and supplemental Table S1). Nucleotide diversity (π) varies substantially across the phyB2 region; there is an almost fourfold variation in polymorphism at silent sites ranging from πS = 7.4 × 10−3 at phyLD16 to πS = 31.5 × 10−3 at phyLD18 (average πS = 18.1 × 10−3) (Figure 1). There are also large differences in the frequency spectrum of segregating mutations between the different fragments, with some fragments having an excess of mutations at low frequency (a negative Tajima's D) while others have an excess of mutations at intermediate frequencies (a positive Tajima's D, Figure 1). Nevertheless, despite the large variation seen in polymorphism and frequency spectra across the 12 fragments, there are no indications of a deviation from neutral expectations across the phyB2 region, as indicated by a nonsignificant HKA test (Hudson et al. 1987) (χ2 = 5.41, d.f. = 11, P = 0.91). This suggests that the entire 80-kb region surrounding the phyB2 locus is evolving according to neutral expectations and confirms results from a previous study (Ingvarsson et al. 2006), which showed little evidence for nonneutral evolution within the phyB2 gene itself. We identified 29 SNPs that had a minor allele frequency >0.1 in the fragments surrounding the phyB2 gene (an average of 2.4 SNPs per fragment). We scored an additional 5 SNPs from the phyB2 gene and added these to the 9 SNPs we had previously scored in the SwAsp collection (Ingvarsson et al. 2006). A total of 42 SNPs were therefore scored in all individuals from the SwAsp collection.
Figure 1.—

Polymorphism and Tajima's D across the phyB2 region. Dashed vertical lines give the 95% confidence interval for the estimates of π. The boxes at the bottom represent genes surrounding the phyB2 gene (solid box). Note that the boxes represent entire genes and that introns are not displayed.
Population structure and allele frequency clines in the phyB2 region:
Neutral markers show little evidence for population structuring in the SwAsp collection; global genetic differentiation, measured as FST from 26 SSR loci, is 0.015 (Hall et al. 2007). Although this estimate of FST is significantly greater than zero (Hall et al. 2007), such low population differentiation suggests that P. tremula is essentially panmictic across the region from which the trees were originally sampled. This is also demonstrated by our attempts to infer population structure from the SSR marker data using the program Structure (Pritchard et al. 2000). Our analyses showed patterns typical of unstructured populations, such as a roughly equal allocation of individuals to the inferred populations and with most individuals showing evidence for admixture. Also, the method outlined in Evanno et al. (2005) identified only a single population, demonstrating the lack of population structuring at neutral markers in the SwAsp collection. There was also little structuring within populations in terms of relatedness among the sampled trees, with the mean pairwise relatedness among trees being 0.01 and statistically indistinguishable from zero.
In an earlier study of polymorphisms in the phyB2 gene, we demonstrated that 4 of 9 SNPs scored showed significant clinal variation (Ingvarsson et al. 2006). That study included two populations from southern Europe and it is possible that the strong clinal variation we documented at phyB2 was partly caused by the inclusion of these outlier populations. We therefore tested for clinal variation in population frequencies at each of the 42 SNPs. We detected significant (P < 0.05) clinal variation at 8 SNPs, 3 of which remain significant after multiple-test correction. These 3 SNPs are all located within the phyB2 gene. The large number of SNPs in the phyB2 gene showing evidence for clinal variation is unlikely to be a result of chance. Clinal variation is not a common observation in P. tremula; 39 SNPs from four other candidate genes for bud phenology (GA20ox1, col2B, ABI1B, and hypO312) yielded no evidence for clinal variation (Hall et al. 2007) and the same is true for several other genes we have surveyed (our unpublished data). The pattern of clinal variation that we observe at SNPs in the phyB2 region is therefore likely to be related to adaptive differentiation to photoperiod.
Association analyses:
As suggested by the molecular population genetic analyses, LD varied across the phyB2 region, but overall the effects of LD were relatively low. A total of 110 of 861 pairwise comparisons (12.8%) between SNPs showed evidence for significant LD after multiple-test corrections, but there was no clear physical clustering of sites in LD (Figure 2). Sites in close physical proximity often showed negligible levels of LD while sites separated by several thousand kilobases sometimes showed a strong LD signal (Figure 2). However, most sites showing long-range LD are mutations occurring in relatively low frequencies (supplemental Table S2).
Figure 2.—

Linkage disequilibrium and phenotypic associations at the 42 SNPs. Strength of pairwise LD between SNPs are indicated color coding. The physical location of the SNPs in the genes surrounding the phyB2 gene are indicated with dashed lines. As in Figure 1, filled boxes represent entire genes.
We scored bud flush and bud set in two different common gardens, one in southern Sweden (Ekebo) and one in northern Sweden (Sävar) (Luquez et al. 2007), using four rametes per clone at each garden. We also scored bud flush and bud set in a greenhouse in 2006, using two rametes per clone. In addition we had phenotypic data from 2 years from Ekebo, resulting in a total of four phenotypic data sets. We combined these different data sets and calculated BLUPs for the individuals in our association mapping population for both bud set and bud flush. These BLUPs were then used as traits in our association analyses.
We did not detect any effects of population structure in our association test, as the effect of population structure, estimated using either Structure (Pritchard et al. 2000) or PCA (Price et al. 2006), was negligible. The random kinship effect was highly significant for bud set where it explained ∼7% of the observed variation, whereas for bud flush, including the kinship matrix did not explain any additional variation. We also calculated kinship using the allele-sharing method described by Zhao et al. (2007). This measure, however, explained somewhat less variation (5%) than the classical kinship estimate (Ritland 1996), so we present results based only on the latter.
After correcting for multiple testing, two SNPs showed significant associations with bud set (Table 1, supplemental Table S2). These two SNPs are nonsynonymous mutations (T608N and L1078P; Ingvarsson et al. 2006) located within the phyB2 gene itself. T608N is located in exon 1 in the vicinity of the chromophore binding site and L1078P is located in exon 4, in a region of the phyB2 protein that is hypothesized to be involved in signaling transduction (Smith 2000). To evaluate whether these SNPs represent mutations with independent effects on bud set, we fitted a model where both SNPs were included as explanatory variables. This tests for the effect of a SNP while statistically controlling for variation at the other possible causal SNP. This analysis shows that both SNPs are independently associated with bud set. This is expected, since there is no evidence for any association between T608N and L1078P, with LD being low and nonsignificant (r2 = 0.044, P > 0.5, Figure 2). There is thus strong evidence for the T608N and L1078P mutations having independent effects on bud set in P. tremula and no evidence that these associations are caused by linkage disequilibrium between the two mutations.
TABLE 1.
Associations between two nonsynonymous SNPs and bud set in P. tremula
| Factor | SNP | 2ΔL | Effect | P-value | FDR q-valuea | R2 (%) | |
|---|---|---|---|---|---|---|---|
| Bud set | Kinship (K) | 8.977 | 2.7 × 10−3 | 7.3 | |||
| Population structure (Q/P) | 0.002 | 0.977 | 0.0 | ||||
| SNP | T608N | 10.857 | 4.43 | 9.8 × 10−4 | 0.033 | 9.0 | |
| Correctedb | 1.81 | 1.4 | |||||
| SNP | L1078P | 9.977 | 5.19 | 1.6 × 10−3 | 0.033 | 8.3 | |
| Correctedb | 3.83 | 5.9 | |||||
| Bud flush | Kinship (K) | 0.000 | 0.99 | 0.0 | |||
| Population structure (Q/P) | 1.398 | 0.24 | 1.2 |
False discovery rate.
SNP effects corrected using method-of-moments approach (Allison et al. 2002).
We also tested all SNPs for associations with bud flush to determine whether the putative causal polymorphisms we identified above were specific to bud set or whether they are generally involved in regulating bud phenology. Bud flush is known to be strongly influenced by temperature in P. tremula. Temperature, on the other hand, has only a minor influence on bud set (Luquez et al. 2007), so pleiotropic effects of mutations in phyB2 are perhaps unlikely. Nevertheless, QTL for bud flush have been demonstrated to colocate with phyB2 in multiple independent mapping populations (Frewen et al. 2000; Chen et al. 2002). We did not, however, detect any SNP–phenotype associations with bud flush in any of our experiments. This is intriguing since the phyB2-associated QTL for bud flush explain ∼10% of the segregating variation in bud flush, which is ∼50% greater than the bud-set-associated QTL that were also colocated with phyB2 (Frewen et al. 2000). However, this could be explained by the fact that the mapping populations were based on interspecific crosses (Frewen et al. 2000; Chen et al. 2002) and that there is either more variation segregating in these populations than in natural populations or genetic variation segregating in the mapping populations represents variation that is normally fixed between the two species and that hence would not be found segregating within a species.
Effect sizes of SNPs associated with bud set:
The two nonsynonymous SNPs in phyB2 that we found to be associated with bud set each explain ∼8% of the phenotypic variation in bud set in our association population (Table 1, see also supplemental Table S2). The additive effects associated with the two SNPs correspond to shifts in bud set with 4.4 and 5.2 days, respectively, which correspond to 42% and 49% of the phenotypic standard deviation in bud set (Figure 3). We have, however, every reason to believe that these values are overestimating the true effects of the two mutations. Estimating the phenotypic effects of mutations from the same data that were used to establish an association leads to an ascertainment bias where the SNPs showing significant associations are also the ones that tend to be associated with the strongest phenotypic differences among genotypes [sometimes termed the “Beavis effect” (Xu 2003) or the “winner's curse” (Zöllner and Pritchard 2007)].
Figure 3.—

Genotypic effects (± SE) of phyB2 SNPs. (A) T608N and (B) L1078P on bud set. The effect is displayed as a deviation from the mean time to bud set.
In an attempt to evaluate the degree by which these estimates are upwardly biased, we performed numerical simulations to investigate the power of our association analysis. Simulations show that our power to detect associations is generally quite low, even if the causal mutation is included in the sample (supplemental Figure S1). Only when the phenotypic variation explained by a mutation is quite large (∼10%) does power reach reasonable levels. In situations where the causal mutation is not included in the sample of SNPs analyzed, power is uniformly low, consistent with the low levels of LD observed in P. tremula (Ingvarsson 2005). The degree by which the additive effect of an allele is overestimated is a function of the power of the study, such that with lower power, allelic effects will be more upwardly biased (Göring et al. 2001; Allison et al. 2002; Xu 2003; Zöllner and Pritchard 2007). This effect is apparent in our simulations, where mutations with small effects can be overestimated by more than a factor of two (supplemental Figure S2).
Allison et al. (2002) suggested an ad hoc method for correcting the ascertainment bias introduced when estimating allelic effects at loci from the same data set that were used to identify the loci. The method of Allison et al. (2002) was originally developed for QTL mapping studies, but it is easily adapted to association mapping studies. It is a method-of-moments approach that builds upon estimating the (truncated) distribution of allelic effects â, given the underlying distribution of (true) allelic effects a and a specified significance cutoff level α. Once this distribution 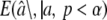 has been obtained it can be equated with the observed allelic effects aobs to get an estimate of the underlying allelic effect for a given mutation.
has been obtained it can be equated with the observed allelic effects aobs to get an estimate of the underlying allelic effect for a given mutation.
Although this distribution could be analytically derived in certain cases, Allison et al. (2002) suggested using numerical simulation to approximate the distribution. Applying this method to our association mapping data suggests that the naive estimates of the effects of the two mutations are upwardly biased by factors of 2.5 and 1.4, respectively. However, taking this reduction in effect size into account, the T608N and L1078P mutations still explain 1.4 and 5.9% of the variation in bud set.
DISCUSSION
The low LD seen in most forest trees suggests that fine-scale mapping should be possible. However, low LD currently makes full genome scans very inefficient, since several millions of markers would have to be scored to have reasonable chances to find significant associations. It has therefore been suggested that a candidate gene approach, where fine-scale mapping is applied to a selected set of candidate genes identified from, for instance, QTL mapping experiments or from functional genomic studies in model species such as Arabidopsis, could be a promising approach to dissect quantitative traits in forest trees (Neale and Savolainen 2004). In this article we have used a candidate gene approach in P. tremula to study the genetic basis of bud phenology, an ecologically important trait in long-lived tree species.
We found that two mutations (T608N and L1078P) in the photoreceptor gene phyB2 are independently associated with naturally occurring variation in bud set in P. tremula (Table 1 and supplemental Table S2). The two mutations also show parallel clines in allele frequencies (Ingvarsson et al. 2006). Despite this, these SNPs do not show stronger genetic differentiation than randomly chosen microsatellite markers or SNPs (Hall et al. 2007). This result fits theoretical expectations, which show that genetic differentiation at QTL is better approximated by genetic differentiation at neutral loci than by genetic differentiation in the quantitative traits themselves (Latta 1998; Le Corre and Kremer 2003). In the presence of high levels of gene flow, spatially variable selection generates covariances between individual quantitative trait nucleotides (QTNs) (i.e., linkage disequilibrium) that reinforce the total phenotypic effect of the QTL (Latta 1998; Le Corre and Kremer 2003). When these covariances are positive, they reinforce the total effect of the QTL and result in large population differences in the quantitative traits, despite small changes in frequencies of the underlying QTL (Latta 1998; Le Corre and Kremer 2003). These covariances reflect an among-population component of LD that develops in the face of diversifying selection and is thus distinct from the low intra-population LD that is generally observed in Populus (Ingvarsson 2005). Taken together, our results suggest that the two phyB2 mutations we observe to be associated with bud set represent independent evolutionary “solutions” to the problem of adapting bud set to seasonal differences in the length of the growing season.
Given that we found two SNPs showing significant association with bud set, a trait of critical ecological importance for perennial trees, it may seem somewhat peculiar that we did not detect any evidence of natural selection acting on phyB2. All the population surveyed must have been established within the last 10 KY, following the last glaciation, so the latitudinal cline we observe in bud set must have been established in a relatively short time. This, combined with the fact that the cline is still actively maintained today, suggests that natural selection acting on these mutations must be quite strong. However, recombination rates appear to be high in P. tremula, as there is very little LD in this species (Ingvarsson 2005). This is clearly important, since the region affected by selection is a function of both the strength of selection acting on a beneficial mutation and the recombination rate in the region surrounding the mutation (Kim and Stephan 2002). However, it is possible that these SNPs are quite old and that they have been maintained in the species over evolutionary time. If so, these mutations may have persisted at appreciable frequencies in glacial refugia populations. In this case, natural selection acting on these SNPs during postglacial colonization would leave even less of a trace in the polymorphism data in and surrounding phyB2 (Przeworski et al. 2005). This is clearly something that is worth studying further.
It is interesting to note that it likely would not be possible to identify these mutations using traditional QTL mapping, without prior knowledge of their existence. Rather, a QTL mapping experiment would confound the two mutations and estimate their combined effect. For phyB2, this would not preclude the identification of a QTL in the region, as the two mutations appear to act additively to influence bud set. However, if a QTL is composed of causal mutations having opposing effects, the probability of actually detecting the QTL in a mapping population could be drastically reduced (Kroymann and Mitchell-Olds 2005). One thing worth pointing out here is that clinal variation at a SNP does not guarantee an association with bud set, a trait that is also showing clinal variation (Ingvarsson et al. 2006; Hall et al. 2007; Luquez et al. 2007). This is evident from the fact that we did not detect significant associations with bud set for all SNPs showing clinal variation; two SNPs in phyB2 where we have shown significant clinal variation (L789M and Int3) (Ingvarsson et al. 2006) are not statistically associated with bud set.
The QTL mapping experiments, which initially demonstrated the colocation of phyB2 and a bud set QTL (Frewen et al. 2000; Chen et al. 2002), found a minimum of four QTL contributing to bud set. These mapping populations were based on interspecific crosses, however, and it is not clear whether this affects the estimation of both the number of QTL that contribute to a trait and the magnitude of the effects of these QTL. Nevertheless, the QTL that colocated with phyB2 explained 6.8% of the variation in bud set in the mapping population and none of the four QTL detected explained more than 12.2% (Frewen et al. 2000). The amount of variation explained by a QTL depends on both the quality of the phenotypic data and the size of the mapping populations used when estimating effect sizes (Xu 2003). It is therefore hard to directly compare effect sizes between experiments. The naive estimate of the phenotypic variation explained by the two mutations we identified in this study is ∼8%. Effect sizes of QTL are known, however, to be upwardly biased when they are estimated from small mapping populations, and this effect is more severe for mutations of small effect (Göring et al. 2001; Allison et al. 2002; Xu 2003). We employed an ad hoc method (Allison et al. 2002) in an effort to obtain less biased estimates of the effects of the T608N and L1078P mutations. By taking the ascertainment bias into account, the effects of the T608N and L1078P SNPs are reduced by a factor of 1.4 and 2.5, respectively. Nevertheless, even with their reduced effect sizes, the T608N and L1078P SNPs each explain a sizable fraction of the variation seen in bud set (1.4 and 5.9%). These two mutations thus appear to be important determinants of naturally occurring phenotypic variation in bud set in P. tremula. One thing worth pointing out is that the method-of-moments approach critically depends on a correct specification of the underlying genetic model (Allison et al. 2002). We have used a simple additive model in all our association analyses, because we found little evidence for dominance at any of the SNPs, and this model was also used in our data-perturbation simulations. Another drawback of the method-of-moment approach is that it only provides point estimates of the allelic effects and there is no obvious way to place confidence bounds on these estimates.
At present, we can only speculate about the possible functions of the two mutations. The phytochrome protein occurs as a homodimer in solution and is made up of two structural domains with a chromophore-bearing N-terminal half and a C-terminal half involved in determining regulatory specificity (Smith 2000). The T608N mutation is located in the hinge region, at the border between the two structural domains of the protein, close to regions that are involved in dimerization and in mediating conformational changes between the Pr and Pfr form of the phyB2 protein (Quail 2002; Chen et al. 2004). It is thus possible that mutations in this region could affect either the stability of the homodimer or the rate of conformational changes between the Pr and Pfr forms. Such changes could possibly affect phytochrome sensitivity to either red or far-red light. This is interesting, since not only day length, but also the spectral composition of light (“light quality”), varies latitudinally and studies have documented clinal responses to light quality in several tree species (Clapham et al. 1998 and references therein). The L1078P mutation, on the other hand, is located in the extreme C-terminal part of the phyB2 protein. This region of the protein is poorly characterized but has sequence similarities with prokaryotic histidine kinases. Incidentally, serine/threonine kinase activity is present in phytochromes, but it is not clear whether and how this is involved in mediating phytochrome signaling (Quail 2002; Chen et al. 2004). It is thus possible that the L1078P mutation might affect signaling to downstream components in the photoperiodic pathway. Further studies of the two phyB2 mutations are clearly needed, both to validate their association with natural variation in bud set in P. tremula and to gain further insights into their possible functional significance. The possible interplay among the three phytochromes present in Populus, and potentially other photoreceptors, allows for very intricate regulation of light-regulated development and will require both forward genetics approaches, as in this study, and reverse genetics experiments using transgenic plants to fully elucidate it. Also, although we found significant associations in a population consisting of slightly more than a hundred trees, larger mapping populations are clearly needed to get less biased estimates of the true effect sizes of the QTNs identified and to have reasonable power to identify mutations with smaller phenotypic effects. We are therefore in the process of extending our experimental population to >400 genotypes.
By identifying genes underlying ecologically important traits it should be possible to address many important questions regarding the genetic architecture of quantitative variation. How common is it that a single QTL is composed of multiple, possibly linked causal polymorphisms? Furthermore, are adaptive mutations derived from standing genetic variation or do they represent newly arisen mutations, and if so, how long have these mutations been maintained within the species? Also, are parallel adaptations in different species due to mutation in the same set of genes? Future studies on the genetic basis of ecological adaptations in Populus, and in other closely related species, should begin to shed some light on this.
Acknowledgments
This study has been funded by grants from the Swedish Research Council (VR), the Swedish Research Council for Environment, Agricultural Sciences, and Spatial Planning (Formas), Kempestiftelserna, the Swedish Foundation for Strategic Research (SSF), and the Research School in Forest Genetics and Breeding to P.K.I. or S.J.
References
- Allison, D. B., J. R. Fernadez, M. Heo, S. Zhu, C. Etzel et al., 2002. Bias in estimates of quantitative-trait-locus effect in genome scans: demonstration of the phenomenon and a method-of-moments procedure for reducing bias. Am. J. Hum. Genet. 70 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson, B., and T. Therneau, 2007. Kinship: mixed-effects Cox models, sparse matrices, and modeling data from large pedigrees. R Package, versions 1.1.0–15. http://cran.r-project.org.
- Balasubramanian, S., S. Sureshkumar, M. Agrawal, T. P. Michael, C. Wessinger et al., 2006. The PHYTOCHROME C photoreceptor gene mediates natural variation in flowering and growth response of Arabidopsis thaliana. Nat. Genet. 38 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. H., 1999. Clines in polygenic traits. Genet. Res. 74 223–236. [DOI] [PubMed] [Google Scholar]
- Brunner, A. M., V. B. Busov and S. H. Strauss, 2004. Poplar genome sequence: functional genomics in an ecologically dominant plant species. Trends Plant Sci. 9 49–56. [DOI] [PubMed] [Google Scholar]
- Chen, M., J. Chory and C. Fankhauser, 2004. Light signal transduction in higher plants. Annu. Rev. Genet. 38 87–117. [DOI] [PubMed] [Google Scholar]
- Chen, T. H. H., G. T. Howe and H. D. Bradshaw, 2002. Molecular genetic analysis of dormancy-related traits in poplars. Weed Sci. 50 232–240. [Google Scholar]
- Clapham, D. H., I. Dormling, I. Ekberg, G. Eriksson, M. Qamarudding et al., 1998. Latitudinal cline of requirement for far-red light for photoperiodic control of budset and extension of growth in Picea abies (Norway Spruce). Physiol. Plant. 102 71–78. [DOI] [PubMed] [Google Scholar]
- Evanno, G., S. Regnaut and J. Goudet, 2005. Detecting the number of clusters of individuals using the software Structure: a simulation study. Mol. Ecol. 14 2611–2620. [DOI] [PubMed] [Google Scholar]
- Feder, M. E., and T. Mitchell-Olds, 2003. Evolutionary and ecological functional genomics. Nat. Rev. Genet. 4 649–655. [DOI] [PubMed] [Google Scholar]
- Frewen, B. E., T. H. H. Chen, G. T. Howe, J. Davis, A. Rhode et al., 2000. Quantitative trait loci and candidate gene mapping of bud set and bud flush in Populus. Genetics 154 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Martínez, S. C., K. V. Krutovsky and D. B. Neale, 2006. Forest-tree population genomics and adaptive evolution. New Phytol. 170 227–238. [DOI] [PubMed] [Google Scholar]
- González-Martínez, S. C., N. C. Wheeler, E. Ersoz, C. D. Nelson and D. B. Neale, 2007. Association genetics in Pinus taeda I. wood property traits. Genetics 175 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göring, H., J. D. Terwilliger and J. Blangero, 2001. Large upward bias in estimation of locus-specific effects from genomewide scans. Am. J. Hum. Genet. 69 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, D., V. Luquez, M. V. Garcia, K. R. St. Onge, S. Jansson et al., 2007. Adaptive population differentiation in phenology across a latitudinal gradient in European aspen (Populus tremula, L.): a comparison of neutral markers, candidate genes and phenotypic traits. Evolution 61 2849–2860. [DOI] [PubMed] [Google Scholar]
- Hardy, O. J., and X. Vekemans, 2002. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2 618–620. [Google Scholar]
- Horvath, D. P., J. V. Anderson, W. S. Chao and M. E. Foley, 2003. Knowing when to grow: signals regulating bud dormancy. Trends Plant Sci. 8 534–540. [DOI] [PubMed] [Google Scholar]
- Howe, G. T., S. N. Aitken, D. B. Neale, K. D. Jermstad, N. C. Wheeler et al., 2003. From genotype to phenotype: unraveling the complexities of cold adaptation in forest trees. Can. J. Bot. 81 1247–1266. [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson, P. K., 2005. Nucleotide polymorphism and linkage disequilbrium within and among natural populations of European aspen (Populus tremula L., Salicaceae). Genetics 169 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson, P. K., M. V. Garcia, D. Hall, V. Luquez and S. Jansson, 2006. Clinal variation in phyB2, a candidate gene for day-length-induced growth cessation and bud set, across a latitudinal gradient in European aspen (Populus tremula). Genetics 172 1845–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroymann, J., and T. Mitchell-Olds, 2005. Epistasis and balanced polymorphism influencing complex trait variation. Nature 435 95–98. [DOI] [PubMed] [Google Scholar]
- Latta, R. G., 1998. Differentiation of alleleic frequencies at quantitative trait loci affecting locally adaptive traits. Am. Nat. 151 283–292. [DOI] [PubMed] [Google Scholar]
- Le Corre, V., and A. Kremer, 2003. Genetic variability at neutral markers, quantitative trait loci and trait in a subdivided population under selection. Genetics 164 1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H.-L., 1998. Statistical Genomics. Linkage, Mapping and QTL Analysis. CRC Press, Boca Raton, FL.
- Long, A. D., and C. H. Langley, 1999. The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res. 9 720–731. [PMC free article] [PubMed] [Google Scholar]
- Luquez, V., D. Hall, B. Albrectsen, J. Karlsson, P. K. Ingvarsson et al., 2007. Natural phenological variation in aspen (Populus tremula): the Swedish Aspen Collection. Tree Genet. Genomes 4 279–292. [Google Scholar]
- Maloof, J. N., J. O. Borevitz, D. Weigel and J. Chory, 2000. Natural variation in phytochrome signalling. Semin. Cell Dev. Biol. 11 523–530. [DOI] [PubMed] [Google Scholar]
- Maloof, J. N., J. O. Borevitz, T. Dabi, J. Lutes, R. B. Nehring et al., 2001. Natural variation in light sensitivity of Arabidopsis. Nat. Genet. 29 441–446. [DOI] [PubMed] [Google Scholar]
- Mouradov, A., F. Cremer and G. Coupland, 2002. Control of flowering time: interacting pathways as a basis for diversity. Plant Cell 14 S111–S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, D. B., and O. Savolainen, 2004. Association genetics of complex traits in conifers. Trends Plant Sci. 9 325–330. [DOI] [PubMed] [Google Scholar]
- Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadick et al., 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 904–909. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., G. Coop and J. D. Wall, 2005. The signature of positive selection on standing genetic variation. Evolution 59 2312–2323. [PubMed] [Google Scholar]
- Quail, P. H., 2002. Photosensory perception and signalling in plant cells: New paradigms? Curr. Opin. Cell Biol. 14 180–188. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, 2007. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna (http://www.R-project.org).
- Risch, N. J., 2000. Searching for genetic determinants in the new millennium. Nature 405 847–856. [DOI] [PubMed] [Google Scholar]
- Ritland, K., 1996. Estimators for pairwise relatedness and individual inbreeding coefficients. Genet. Res. 67 175–185. [Google Scholar]
- Smith, H., 2000. Phytochromes and light signal perception by plants - an emerging synthesis. Nature 407 585–591. [DOI] [PubMed] [Google Scholar]
- Stinchcombe, J. R., and H. E. Hoekstra, 2007. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100 158–170. [DOI] [PubMed] [Google Scholar]
- Storey, J. D., and R. Tibshirani, 2003. Statistical significance for genome-wide studies. Proc. Natl. Acad. Sci. USA 100 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignments through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K., 2003. Libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics 19 2325–2327. [DOI] [PubMed] [Google Scholar]
- Thumma, B. R., M. F. Nolan, R. Evans and G. F. Moran, 2005. Polymorphisms in Cinnamoyl CoA Reductase (CCR) are associated with variation in microfibril angle in Eucalyptys spp. Genetics 171 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareing, P. F., 1956. Photoperiodism in woody plants. Ann. Rev. Plant Physiol. 7 191–214. [Google Scholar]
- Warnes, G., and F. Leisch, 2006. Genetics: population genetics. R Package, version 1.2.1. http://cran.r-project.org.
- Xu, S., 2003. The theoretical basis of the Beavis effect. Genetics 165 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J., G. Pressoir, W. H. Briggs, I. Vroh Bi, M. Yamasaki et al., 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38 203–208. [DOI] [PubMed] [Google Scholar]
- Zhao, K., M. J. Aranzana, S. Kim, C. Lister, C. Shindo et al., 2007. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 3 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöllner, S., and J. K. Pritchard, 2007. Overcoming the winner's curse: estimating penetrance parameters from case-control data. Am. J. Hum. Genet. 80 605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]