

Abstract
We have determined by 15N, 1H, and 13C NMR, the chemical behavior of the six histidines in subtilisin BPN′ and their PMSF and peptide boronic acid complexes in aqueous solution as a function of pH in the range of from 5 to 11, and have assigned every 15N, 1H, Cɛ1, and Cδ2 resonance of all His side chains in resting enzyme. Four of the six histidine residues (17, 39, 67, and 226) are neutrally charged and do not titrate. One histidine (238), located on the protein surface, titrates with pKa = 7.30 ± 0.03 at 25°C, having rapid proton exchange, but restricted mobility. The active site histidine (64) in mutant N155A titrates with a pKa value of 7.9 ± 0.3 and sluggish proton exchange behavior, as shown by two-site exchange computer lineshape simulation. His 64 in resting enzyme contains an extremely high Cɛ1-H proton chemical shift of 9.30 parts per million (ppm) owing to a conserved Cɛ1-H. . .O=C H-bond from the active site imidazole to a backbone carbonyl group, which is found in all known serine proteases representing all four superfamilies. Only His 226, and His 64 at high pH, exist as the rare Nδ1-H tautomer, exhibiting 13Cδ1 chemical shifts ~9 ppm higher than those for Nɛ2-H tautomers. His 64 in the PMSF complex, unlike that in the resting enzyme, is highly mobile in its low pH form, as shown by 15N-1H NOE effects, and titrates with rapid proton exchange kinetics linked to a pKa value of 7.47 ± 0.02.
Keywords: Histidine, tautomerism, serine protease, hydrogen bond, NMR spectroscopy, peptide boronic acid inhibitor, 15N NMR
Subtilisin BPN′ (EC 3.4.21.14, also known as Novo, or nagarse) from Bacillus amyloliquefaciens consists of a single chain of 275 amino acids (MW 27.5 kD) with no disulfide linkages, but having two tight, EF hand calcium-binding sites (Kretsinger 1976a,b). Subtilisin, used worldwide by many tons annually in commercial detergents, is a member of one of four superfamilies of independently evolved serine proteases (trypsin, Clp protease, and wheat-germ carboxypeptidase representing the other three families). All serine protease superfamilies share the three-dimensional arrangement of the Asp–His–Ser catalytic triad and at least two H-bond donating groups in the oxyanion hole at their active sites, but no other structural similarities or sequence homologies are conserved by members of other superfamilies (Rawlings and Barrett 1994; Krem et al. 2000). The catalytic mechanism of serine proteases after more than 30 yr of active scrutiny is still very much in dispute (Bachovchin 2001). Central to the function of the catalytic triad are issues of active site histidine tautomerism, H-bonding, microscopic acid-base equilibria, and proton exchange kinetics.
The protonated or low pH form of the imidazole side chain 1 (see below) exists in acid-base equilibrium described by constant Ka with two unprotonated species. These unprotonated or high pH forms of imidazole, the more common Nɛ2-H tautomer 2 and the rare Nδ1-H tautomer 3 exist in equilibrium described by constant KT.
In 1969, the first X-ray crystal structure of subtilisin BPN′ at 2.5 Å, 1SBT.pdb; (Alden et al. 1971) was published, demonstrating the existence of the catalytic triad in this serine protease family (Wright et al. 1969). Three years later, using classical potentiometry and electrophoretic studies of the enzyme and its PMSF complex, Ottesen and Ralston concluded that the enzyme probably has four of its six histidines buried, that is, shielded from solvent, and the other two histidines titrating with pKa values of ~7.5 (Ottesen and Ralston 1972). In 1974, Robillard and Shulman (1974a) reported the broad, low-field [~17.8 parts per million (ppm)] Nδ1-H 1H NMR resonance observed at low temperature (3°C) and pH 6.0 for subtilisin BPN′, which they attributed to the proton involved in the Asp 32–His 64 H-bond, thus providing evidence of the small Asp–His distance between these side chains in solution and further implicating them in the proteolytic process. These authors also reported low-field 1H resonances at ~17.2 ppm for subtilisin BPN′ complexes with phenyl boronates (Robillard and Shulman 1974b). NMR studies of subtilisin BPN′ since then have shed little light on the nature of its histidine groups.
In 1979, 1H NMR titrations by Omar and colleagues of the PMSF complex of subtilisin BPN′ from Novo Industries revealed four titratable His groups having pKa values of 5.4, 5.7, 6.0, and 6.4 (Omar et al. 1979). Repeated cycles of sample pre-exchange with D2O helped reduce the amide -NH background, but it must be noted that no Ca2+ was supplied to aid in stabilizing the structure. In 1981, Jordan and Polgar (1981) failed to observe the low-field Nδ1-H 1H resonance of subtilisin BPN′ purchased from the Sigma Chemical Co., contrary to the observations of Robillard and Shulman (1974a), whose enzyme had been obtained from the Enzyme Development Corp. The difference was attributed by Jordan and Polgar (1981), although not confirmed experimentally, to an alleged inhibitory effect of the 0.1 M N-acetyl-L-tryptophan present in Robillard and Shulman’s sample (Robillard and Shulman 1974a). Recently, Stratton et al. (2001) confirmed the presence at ~17.8 ppm of the low-field, low pH active site Nδ1-H proton in wild-type subtilisin BPN′ in the absence of N-acetyl-L-tryptophan or any other inhibitors at 11°C, a result that we have also duplicated at 5°C. In 1985, Jordan et al. performed 1H and 31P NMR studies similar to that of Omar et al. (1979) on both subtilisin BPN and subtilisin Carlsberg (both from Sigma) and their inactivated DIFP complexes (Jordan et al. 1985). Pre-exchange of N-D for the amide N-H resonances was achieved by treatment at pH 9 for 16 h, with Ca2+ deliberately removed to speed up the proton exchange rate. Jordan et al. (1985) found that five of the six histidines in BPN and four of the five histidines in Carlsberg titrated, all with pKa values between 6.3 and 7.2. The active site His was found to have a pKa value of 7.40 for both types of subtilisin.
In 1986, Russell and Fersht reported in a letter to Nature (see also Russell and Fersht 1987) that the subtilisin BPN supplied by Sigma and possibly other vendors for the previous decade had been mislabeled, and was actually subtilisin Carlsberg. In 1988, Bycroft and Fersht attempted to improve on previous 1H NMR titrations methods for subtilisin BPN by using a spin-echo technique, in effect suppressing all broad resonances, which they assumed to include most N-H resonances and to exclude the desired Cɛ1-H histidine resonances. These authors also dissolved their lyophilized samples in D2O containing EDTA, thus removing Ca2+, but they omitted the more drastic pre-exchange procedure of Jordan et al. (1985). Thus, Bycroft and Fersht (1988) obtained six titratable histidines having pKa values in the range of from 6.72 to 7.45. Site-directed mutagenesis together with comparisons from Jordan’s previous Carlsberg study were used to deduce the assignments of the six titrating histidines.
However, as pointed out in 1992 by Consonni et al., all 1H NMR titrations of resting subtilisin BPN′ and Carlsberg published as of that time were due to the sharp resonances from degradation products of these enzymes, whose autolysis half life is measured in hours. In the intervening years, most NMR studies of subtilisin have focused on inhibited subtilisin complexes, or low-activity, chemically modified sulfur- or selenium-containing derivatives and their perturbations upon pH/activity profiles (House et al. 1993; Stratton et al. 2001; Kahyaoglu and Jordan 2002). There has been no previous direct measure of the microscopic pKa value of the active site His 64. Instead, what has become accepted dogma is the notoriously unreliable (Knowles 1976) substitution of pH/activity profiles, with a pKa value of 7.17 ± 0.02 evidently being the accepted value (Thomas et al. 1985; Stratton et al. 2001).
In an earlier report, we introduced the oxyanion hole mutation N155A of subtilisin BPN′97 (O’Connell et al. 1997), which leads to a 300-fold decrease in catalytic efficiency. This method of increasing the stability of subtilisin BPN′ has the advantage that the mutation alters no electrostatic charge, and the closest distance of approach between His 64 and Asn 155 (or Ala 155) is a sizeable 5.5 Å. No chemical shift difference compared with wild-type subtilisin BPN′ has yet been observed on any histidine resonance owing to the stabilizing N155A or BPN′97 mutations. Nor have the BPN′97 surface mutations exhibited any noticeable effect upon enzyme activity.
NMR, particularly 15N NMR, has made important contributions to the elucidation of the chemical behavior of histidine in α-lytic protease, a member of the trypsin superfamily (Bachovchin 2001). The present study was undertaken in order to extend this knowledge of the chemical behavior of histidine, particularly the catalytic group, to the subtilisin family. 15N NMR is exquisitely sensitive to the local magnetic environment, and thus capable of revealing the presence of H-bonding on both sides of the imidazole ring as well as pKa values and tautomeric states. 1H NMR, when correctly applied, can also reveal the presence of histidine Cɛ1-H-donated H-bonds in serine protease active sites, as we first presented in 1996 (E.L. Ash, M.P. Vincent, J.L. Sudmeier, and W.W. Bachovchin, unpubl.; Ash et al. 2000), in addition to the often reported determination of histidine pKa values in proteins by 1H NMR. In a 1994 survey of serine hydrolase X-ray crystal structures, Derewenda et al. 1994 postulated the presence of an additional conserved Cɛ1-H. . .O=C H-bond from the active site His to a backbone carbonyl group on the basis of favorable distances and geometry. We confirmed the presence of this H-bond by discovering and interpreting the anomolously large His Cɛ1-H 1H chemical shifts present in α-lytic protease, subtilisin, and their boronic acid-inhibited complexes. Similar reports for other serine proteases, including a boronic acid complex of subtilisin E soon followed (Bao et al. 1998, 1999; Lin et al. 1998). The additional H-bond suggested to us a novel reaction-driven imidazole ring flip mechanism that could solve a long-standing dilemma regarding how such enzymes catalyze both the formation and productive breakdown of the tetrahedral intermediates (Ash et al. 2000). Acting as a mechanical device, the enzyme is thus able to present multiple templates for stabilizing multiple intermediates, thus ratcheting the catalyzed reaction forward. This theory of a dynamic active site imidazole, which is under active investigation in our laboratory, is in stark contrast to competing theories, which envision a histidine completely immobilized by an ultra-strong Asp–His H-bond (Gerlt and Gassman 1993; Frey et al. 1994; Cassidy et al. 2000; Neidhart et al. 2001).
Here, we present in detail the 1H, 15N, and 13C NMR assignments of the histidines that gave rise to Otteson and Ralston’s fundamentally correct presumption of two titrating and four nontitrating histidines (Ottesen and Ralston 1972), and provide the groundwork for continuing NMR investigations.
Results and discussion
One-dimensional 15N NMR spectra
Figure 1 ▶ shows one-dimensional 15N NMR spectra of wild-type subtilisin BPN′ prepared from singly (Nδ1, Fig. 1B,F ▶) and doubly (Nδ1 and Nɛ2, Fig. 1A,C–E ▶) 15N-labeled histidine. There are 12 distinct resonances in Figure 1A ▶, as would be expected from six histidine residues. Histidine model studies showed that the chemical shift of the 15N signal is sensitive to its protonation state, its participation in hydrogen bonds, and the hydrophobicity of its environment (Witanowski et al. 1972; Blomberg et al. 1977; Bachovchin 1986). The nitrogen atoms within the tautomeric structures of the histidine imidazole ring can assume three possible bonding states. Two of these occur in the neutral imidazole ring. These are the protonated or pyrolle-like (>N-H) and unprotonated or pyridine-like (>N:) nitrogen types. A third nitrogen type occurs in the positively charged imidazolium ring – the pyrolle-like (+>N-H) protonated nitrogen. Typical chemical shift values for these nitrogen types, referred to as α, β, and α+, have been determined in aqueous solutions:
Figure 1.

One-dimensional 15N NMR spectra (40 MHz) with 1H Waltz16 decoupling of singly [Nδ1] and doubly [Nδ1,Nɛ2] 15N histidine-labeled wild-type subtilisin BPN′ and inhibited complexes at various pH values and 25°C.
Table t.
| Nitrogen Type | 15N Chemical Shift (ppm) | H-bonded (ppm) |
| >N-H (type α) | 168 (−210) | 178 (−200) |
| >N: (type β) | 249 (−128) | 239 (−138) |
| +>N-H (type α+) | 176 (−202) | 186 (−192) |
(The negative 15N chemical shifts in parentheses throughout this manuscript, referenced to 1M HNO3, are supplied for the convenience of readers who are more comfortable with the large body of seminal 15N NMR work published using this older standard, whose values are obtained by adding −377.5 ppm to the modern IUPAC values versus liq. NH3).
The resonance of an >N-H or +>N-H nitrogen has been observed previously to increase as much as 10 ppm when donating an H-bond to a carboxylate group (Bachovchin and Roberts 1978). The resonance of a >N: nitrogen can decrease as much as 10 ppm as the acceptor of an H-bond from a hydroxyl group. This 15N spectrum (Fig. 1A ▶) indicates by the cluster of four low-field, unprotonated (type β) resonances that at low pH (5.6), four of the histidines are neutral, whereas two histidines must be protonated. The identities of Nδ1 and Nɛ2 resonances are revealed by the 15N spectra of single Nδ1-labeled enzyme, such as those shown in Figure 1, B and F ▶.
Complex formation; assignment of His 64 active site 15N resonances
The active site residue His 64 is revealed by changes in the 15N chemical shifts upon complex formation with phenylmethanesulfonyl fluoride (PMSF) (Fig. 1D ▶) and the peptide boronic acid inhibitor methoxysuccinyl-Ala-Ala-ProboroPhe [AAPbF, in which the carboxy-terminal carboxylate group of F is replaced by the -B(OH)2 group] (Fig. 1E,F ▶) (chemical shifts shown in Table 1). PMSF inhibition of serine proteases results in specific covalent modification of the active site serine, resulting in disruption of His 64 side chain H-bonds at low pH and expulsion from its resting position, which alters the Nδ1 chemical shift and allows its assignment. Comparison of resting and PMSF-bound BPN′ spectra shows that the 15N chemical shift of one nitrogen decreases from 191.9 (−185.6) ppm (a shift which corresponds to an H-bonded α+ nitrogen, Fig. 1A ▶) to 178.1 (−199.4) ppm (a shift which corresponds to an α+ nitrogen without an H-bond, Fig. 1D ▶). Experiments with singly labeled enzyme revealed this to be an Nδ1 resonance (Fig. 1B ▶). On the basis of this result and those of the peptide boronic acid complex shifts next described, we have assigned the resonances indicated by the labels and brackets in Figure 1 ▶ to the active site His 64 of subtilisin BPN′.
Table 1.
Chemical shift assignments, spin-coupling constants, and p Kavalues of histidines affected in inhibited complexes of subtilisin BPN′ and subtilisin N155A at 25°C
| Histidine | pH | Cɛ1H (ppm) | Cδ2H (ppm) | Nδ1 (ppm) | Nɛ2 (ppm) | Cɛ1 (ppm) | Cδ2 (ppm) | Nδ1H (ppm) | Nɛ2H (ppm) | 1JCɛ1-H (Hz) |
| PMSF | ||||||||||
| 64 (active site) | 5.5 | 8.13 | — | 176.5 (−201.0)a | 173.2 (−204.3)a | 133.4 | — | exch | exch | 222 |
| pKa = 7.47 ± 0.02 | 10.5 | 7.31 | — | 180b (−198)a,b | 249b (−129)a,b | 136.0 | — | exch | — | 207 |
| AAbF | ||||||||||
| 67, pKa < 3 | 5.5 | 7.50 | 6.13 | 259.5 (−118.0)a | 166.7 (−210.8)a | 136.5 | 114.9 | — | 11.00 | — |
| 64, pKa > 10.5 | 5.5 | 9.40 | 7.30 | 188.8 (−188.7)a | 178.8 (−198.7)a | 134.9 | 118.2 | 17.21c | 15.34c | — |
| AAPbF | ||||||||||
| 67, pKa < 3 | 5.5 | 7.50 | 6.13 | 259.9 (−117.6)a | 165.6 (−211.9)a | — | — | — | 11.00 | |
| 64, pKa ~ 11 | 5.5 | 9.50 | 7.36 | 188.3 (−189.2)a | 179.5 (−198.0)a | 135.1 | — | 16.90c | 15.30c | — |
a Referenced to 1.0 M HNO3 for historical comparison, values obtained by adding −377.5 ppm.
b Tentative, from 15N CP-MAs solid-state 40 MHz NMR at 200°K (frozen solution; Fig. 3A ▶).
c Obtained at 5°C.
We have shown previously that peptide boronic acid inhibitors form covalent transition state-like tetrahedral adducts in the enzyme active site when the amino acid sequence of the inhibitor is analogous to a good protease substrate (Bachovchin et al. 1988). NMR experiments with these complexes have shown that the boronic inhibitor forms a covalent adduct with the active site serine and results in strong H-bonds to both nitrogens of the active site histidine in a pH-independent manner (Bachovchin et al. 1988). Work by others identified succinyl-Ala-Ala-Pro-Phe-paranitroanilide as a well-catalyzed substrate for subtilisin BPN′ (Wells et al. 1987a,b), and here we use the corresponding boronic acid inhibitor to help identify the Nɛ2 of His 64. The boronic inhibitor AAPbF induces an increased chemical shift of one Nɛ2 from 175.3 (−202.2) ppm to 182.2 (−195.3) ppm, indicating the presence of a strong H-bond induced by the boronic acid inhibitor. The 15N chemical shifts of His 64 bound to AAPbF were quite constant with increasing pH up to ~pH 9.5. On the basis of this data, we have identified these resonances as the Nɛ2 of His 64.
The 15N spectrum for resting BPN′ at low pH shows that the chemical shift of 190.2 (−187.3) ppm for Nδ1 of active site His 64 is the largest yet reported for a protonated -N+-H (type α+) nitrogen. Interestingly, this is also 4.3 ppm greater than the comparable shift for His57 in α-lytic protease (Bachovchin and Roberts 1978). Increasing chemical shift of 10 ppm for histidine 15N resonances has been attributed to the Asp–His H-bond in α-lytic protease (Bachovchin and Roberts 1978; Bachovchin 1986). The additional increase of 15Nδ1 shift in the subtilisin active site can be attributed to a somewhat stronger Asp 32–His 64 H-bond, although probably <1.4 kcal/mole stronger based on the approximately one unit elevation in pKa compared with α-lytic protease, as will be detailed below. Another factor possibly contributing to 15N chemical shifts in subtilisin is the nearly bifurcated nature of the Asp 32–His 64 H-bond (see Fig. 2A ▶), due to a very different orientation of Asp 32 compared with the Asp 102 in the comparable α-lytic protease structure. Anisotropic shielding by the neighboring Asp 32 group might thus differ by several ppm in subtilisin as compared with that of Asp 102 in α-lytic protease.
Figure 2.
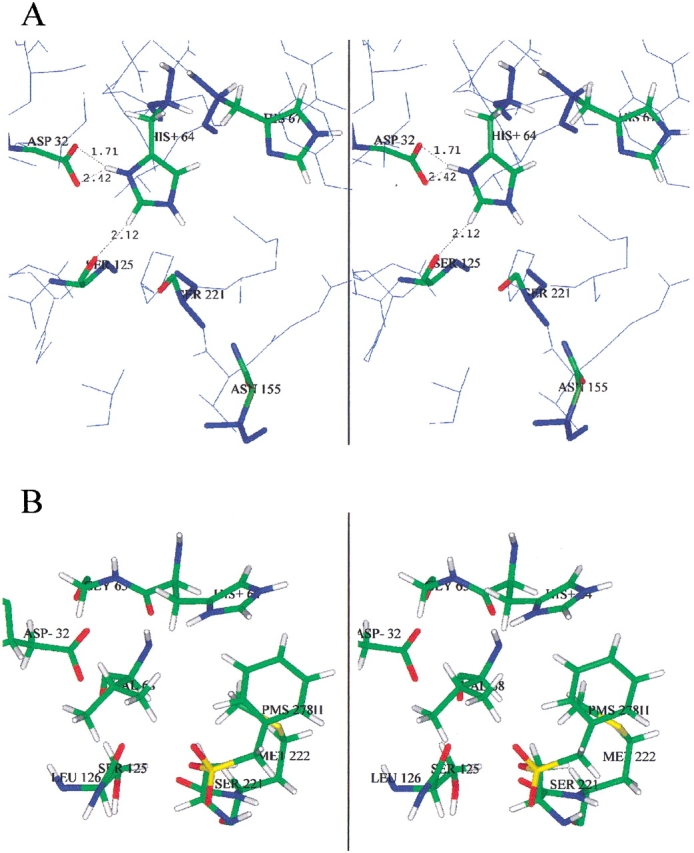
Stereo views of subtilisin BPN′ active site from X-ray crystallography. (A) Resting enzyme (2ST1.pdb; Drenth et al. 1972), showing His 64, His 67, and oxyanion hole mutation site Asn 155. (B) PMSF-inhibited complex (1SUP.pdb; Gallagher et al. 1995).
The 15N chemical shift of Nɛ2 in His 64 in resting subtilisin BPN′ is indicative of type β nitrogen atoms that are not H-bond acceptors. The once-controversial absence of an active site His—Ser H-bond only at low pH in all known serine proteases has an interesting history that was ultimately resolved through 15N chemical shifts (Bachovchin 2001). Now subtilisin can be added to the list of documented examples.
Variation of 15N chemical shift with pH
Variation in pH reveals significant (>2 ppm) movement in only 4 of the 12 15N resonances in Figure 1 ▶. This indicates that only two of the six histidines titrate in the native enzyme, one of which is clearly the active site His 64 (e.g., Fig. 1 ▶, cf. A and C; chemical shifts shown in Table 2). Nδ1 of His 64 does not shift as the pH is raised, but gradually decreases in intensity until disappearing around pH 7.3 (Day 1995). The Nɛ2 resonance, which by analogy with α-lytic protease (Bachovchin and Roberts 1978) has a much larger kinetic window (i.e., a difference of ~80 ppm in the chemical shifts of the acid and base species), has significantly broadened by pH 5.8 and disappeared by pH 6.5, due to intermediate acid-base proton exchange rate. As shown in Figure 3 ▶, the Nɛ2 resonance can be observed at 234 (−143) ppm by solid-state 15N cross polarization magic angle spinning (CP-MAS) NMR in frozen pH 10.5 solution, in which both proton exchange and autolysis can be halted. Comparison of the resting and PMSF complexes by solid-state CP-MAS 15N NMR supports these tentative assignments of the His 64 high pH 15N resonances.
Table 2.
Chemical shift assignments, spin-coupling constants, and p Kavalues of histidines in resting subtilisin BPN′, subtilisin N155A, and autolysis products at 25°C
| Histidine | pH | Cɛ1H (ppm) | Cδ2H (ppm) | Nδ1 (ppm) | Nɛ2 (ppm) | Cɛ1 (ppm) | Cδ2 (ppm) | Nδ1H (ppm) | Nɛ2H (ppm) | 1JCɛ1-H (Hz) |
| 17, pKa < 3 | 5.5 | 7.98 | 7.58 | 251.1 (−126.4)a | 161.0 (−216.5)a | 137.5 | 115.7 | — | 10.83 | 207 |
| 39, pKa < 3 | 5.5 | 7.56 | 7.33 | 249.9 (−127.6)a | 164.7 (−212.8)a | 135.5 | 113.3 | — | 12.10 | 205 |
| 67, pKa < 3 | 5.5 | 7.50 | 6.19 | 259.5 (−118.0)a | 165.6 (−211.9)a | 136.5 | 115.0 | — | 11.00 | 206 |
| 10.5 | 7.47 | 6.10 | 260.7 (−116.7)a | 165.0 (−212.5)a | 136.5 | 114.8 | — | 10.94 | — | |
| 226, pKa < 3 Nδ1H tautomer | 5.5 | 7.50 | 6.44 | 167.6 (−209.9)a | 248.8 (−128.7)a | 139.5 | 125.0 | 11.60 | — | 209 |
| 238 (surface) | 5.5 | 8.74 | 5.92 | 176.6 (−200.9)a | 170.3 (−207.2)a | 133.9 | 116.4 | exch | exch | 217 |
| pKa = 7.30 ± 0.03 | 10.5 | 7.65 | 5.60 | 244.2 (−133.3)a | 163.2 (−214.3)a | 135.0 | 114.4 | — | exch | 208 |
| 64 (active site) | 5.5 | 9.30 | 7.30 | 190.2 (−187.3)a | 173.8 (−203.7)a | 135.5 | 118.0 | ~17.8b | — | — |
| pKa = 7.9 ± 0.3 | 10.5 | 8.51 | 6.71 | 180c (−198)a,c | 235c (−143)a,c | 136.7 | 124.2 | — | — | — |
| J1 (autolysis) | 5.5 | 8.64 | 7.40 | 177.4 (−200.1)a | 172.9 (−204.6)a | 134.0 | 118.3 | exch | exch | — |
| pKa = 6.98 ± 0.05 | 10.5 | 7.72 | 7.10 | — | — | 136.4 | 118.3 | — | exch | — |
| J2 (autolysis) | 5.5 | 8.60 | 7.34 | — | — | 134.5 | 117.5 | exch | exch | — |
| pKa = 6.64 ± 0.05 | 10.5 | 7.76 | 6.98 | — | — | 136.8 | 118.2 | — | exch | — |
a Referenced to 1.0 M HNO3 for historical comparison; values obtained by adding −377.5 ppm.
b From Stratton et al. (2001) at 11°C and 600 MHz.
c Tentative, from 15N CP-MAS solid-state 40 MHz NMR at 200°K (frozen solution; Fig. 3A ▶).
Figure 3.

15N CP-MAS solid-state NMR spectra (40 MHz) of doubly {Nδ1,Nɛ2} 15N histidine-labeled wild-type subtilisin BPN′ (pH 10.5) and 200° K (frozen solution). The spin rate is 4250 Hz. (A) Resting enzyme. (B) PMSF-inhibited complex.
Nδ1 of the second titrating histidine broadens and shifts as the pH is raised, but the resonance is observed at all intermediate pH values. For example, in the PMSF complex of wild-type subtilisin, the Nδ1 of this other titrating histidine broadens as the pH is raised until it sharpens up at 244.4 (−133.1) at high pH, when it reaches the plateau of the titration curve; the broadening of the resonance at intermediate points in the pH titration curve is likely due to intermediate rates of proton exchange. According to the X-ray crystal structure, there are two promising candidates for this second titrating histidine, His 238 and His 17, both of which are near the protein surface. Confirmation that this other titrating group is His 238 arises principally from the Hδ2 chemical shifts, as discussed below.
Note the small variation of 1.2 ppm with changing pH for the extreme low-field 15N resonance (labeled 67-δ1) in Figure 1C ▶. Such a pH-induced drift is orders of magnitude smaller than a pH shift, and is believed to arise as a secondary perturbation of protonating a nearby functional group. The proximity of His 64 and His 67 is evident in Figure 2A ▶. The drift in this 15N resonance, which we attribute to protonation at His 64, together with drifting 1H and 13C resonances, as shown below, is important both in the assignment of His 67 resonances and in determination of the His 64 pKa value.
Assignment of 15N resonances of His 226
From Figure 1B ▶ and a knowledge of 15N chemical shifts, it is evident that the four nontitrating histidines exist in the neutral, unprotonated form at all pH values, and that one of the four occurs as the rare Nδ1-H tautomer 3. That is, we expect the >N-H (type α) resonances at around 168 (−210) ppm and the >N:H (type β) resonances at around 249 (−128) ppm. From X-ray crystallography, it was predicted that His 226 would exist as the rare Nδ1-H tautomer owing to the strong H-bond to the backbone of the neighboring residue Ala 22, thus making it possible to assign 2 more of the 12 15N resonances in Figure 1 ▶. The remaining 15N resonance assignments depend upon NMR data involving additional nuclei.
NOE effects and His motion
The 1H-coupled one-dimensional 15N spectrum of the PMSF complex at low pH (data not shown) reveals an increase of nearly a factor of 2 in the intensity of the His 64 resonances. A significant reduction in peak intensity of the His 64 resonances due to the nuclear Overhauser effect (NOE) in Figure 1D ▶ (PMSF complex) compared with those of Figure 1A ▶ (resting enzyme), all 1H decoupled, is evident. Such a decrease in 15N resonances of His 64 does not occur in the high pH form, indicating that the neutral imidazole in the PMSF complex occupies its normal position in the active site. Using the rule of thumb that the rotational correlation time of a 28-kD protein is about 14 nsec (Malthouse 1986) and the well-known nuclear relaxation equations of Bloembergen et al. (1948), we calculate for these 40 MHz 15N spectra that the correlation time of 15N in His 64 is about 2 nsec. This implies that the active site His has about seven times greater mobility in the low pH PMSF complex than the enzyme as a whole, lending support to catalytic mechanisms involving a moving histidine (Bachovchin 1986; Ash et al. 2000). An X-ray crystal structure (Fig. 2B ▶, 1 ▶SUP.pbd; Gallagher et al. 1995) of subtilisin BPN′ complexed with PMSF also shows the displacement of His 64 in crystalline enzyme from its normal catalytic triad orientation. Mere solvent effects are sufficient to dislodge His 64 from its resting position, as shown by the imidazole ring being flipped 164° about the Cβ-Cγ bond in the X-ray crystal structure of subtilisin BPN′ in 50% DMF (Kidd et al. 1999).
Even though His 238 is in rapid acid-base exchange, and must therefore be fully exposed to solvent, the fact that its NOE behavior is like other immobilized His residues, for example, 17, 39, 226, suggests a lack of mobility of the His 238 side chain. The X-ray crystal structure does show strong H-bonding through it Nɛ2-H proton to Gln 275, and possibly base stacking with the nearby Trp 241.
Two-dimensional 1H/15N correlation NMR spectra
Figure 4 ▶ shows the results of the two-dimensional 1H/15N gradient echo/antiecho heteronuclear muliple quantum coherence (HMQC) experiment, which has the remarkable ability to correlate all 1H and 15N resonances belonging to a particular histidine side chain over both one and two bonds, including all carbon-bound and long-lived nitrogen-bound protons, the latter with negligible loss of signal through saturation transfer. This experiment on a sample of doubly {Nδ1, Nɛ2} 15N-labeled subtilisin N155A complexed to AAPbF ties together the resonances of the four nontitrating histidines 17, 39, 67, and 226. The Z-shaped pattern for His 226 uniquely identifies the rare Nδ1 tautomer and reveals its Hɛ1 and Hδ2 proton chemical shifts. The T patterns labeled His 17, His 39, and His 67 arise from the more common Nɛ2 tautomer. Although many of the cross-peaks in Figure 3 ▶ are of low intensity, they are all confirmed in 13C/1H correlated spectra shown below. His 67, as will be shown, is distinguished both by the drift of its proton resonances with pH and small change upon inhibition by boronic acids, as well as by the magnitude of its 1H chemical shifts calculated from the X-ray crystal structure by the FORTRAN program called SHIFTS available from the Web site of David A. Case (Osapay and Case 1991; Table 3).
Figure 4.

Two-dimensional 1H/15N gradient echo/antiecho HMQC NMR spectrum (600 MHz) for one- and two-bond correlation in doubly {Nδ1,Nɛ2} 15N histidine-labeled subtilisin N155A complexed with AAPbF at pH 6.5 and 30°C. Summation of spectra using primary delays of 12, 17, and 22 msec.
Table 3.
1H chemical shifts of histidines calculated using Case softwareaand coordinates from X-ray crystal structure 2ST1.pdbb
| Histidine | pH | Cɛ1H (ppm)c | Cδ2H (ppm)c | Cɛ1H-Cδ2H (Δ ppm)c |
| His 17 | 5.5 | 7.77 | 7.20 | 0.57 |
| His 39 | 5.5 | 7.61 | 6.87 | 0.74 |
| His 67 | 5.5 | 7.68 | 6.38 | 1.30 |
| His 226 | 5.5 | 7.61 | 6.83 | 0.78 |
| His 238 | 5.5 | 8.37 | 6.38 | 1.99 |
| 10.5 | 7.53 | 6.04 | 1.49 | |
| His 64 (active site) | 5.5 | 8.93 | 7.07 | 1.86 |
| 10.5 | 8.09 | 6.72 | 1.37 |
a FORTRAN program called SHIFTS available from the David A. Case Web site (Osapay and Case 1991).
c Haigh-Mallion ring current model.
The assignment of His 39 versus His 17 or of His 17 versus His 238 rests entirely on the trends exhibited by these 1H chemical shift calculations for both Hɛ1 and Hδ2 in Table 2, which take into account ring current and other shielding effects by neighboring groups in the orientations determined by the crystal structure of BPN′. The low chemical shift Hδ2 resonances of His 238 compared with His 17 in Table 2, and the large spread between Hδ2 and Hɛ1 of His 238 compared with His 17, is largely due to ring current shielding by its neighboring Trp 241. On this basis, we can assign the second titrating histidine (i.e., in addition to the active site His 64) to His 238. It is also clear from Table 2 that the highest chemical shift of the nontitrating Hɛ1 resonances belongs to His 17, and on this basis, we assign His 17 and His 39. Subtilisin Carlsberg has only five histidines, lacking His 17, and can provide future proof of this assignment.
Two-dimensional 1H/13C correlation NMR spectra
Figures 5 ▶ and 6 ▶ show the results of two-dimensional 1H/13C HMQC spectra of uniformly 13C and 15N histidine-labeled subtilisin N155A at pH 5.50. Because of the large 13C chemical shift range covered in these experiments, we have zoomed in on two regions, one showing the one-bond 1H/13Cɛ1 correlations (Fig. 5 ▶), and the other showing 1H/13Cδ2 correlations (Fig. 6 ▶). Changes in peak position as the pH is varied from ~5 to ~11 are then rendered as straight line trajectories in Figures 5 ▶ and 6 ▶, with arrows denoting the direction of increasing pH. The positions of boronic acid-inhibited enzyme, and of resting and PMSF-inhibited enzyme at low and high pH are also indicated on Figures 5 ▶ and 6 ▶. There are two major autolysis products of BPN′, which we have labeled J1 and J2 (Figs. 5 ▶ and 6 ▶, chemical shifts summarized in Table 2). The J1 and J2 resonances grow with time, indicating the extent of sample degradation. We have also found them useful as an internal pH indicator, because during autolysis, the low pH samples tend to rise and the high pH samples tend to fall, all tending toward pH ~8. To counteract pH drift during long NMR accumulations, we have sometimes used 10 mM 2-[N-morpholino]ethanesulfonic acid (MES) (pKa 6.1) for low pH samples, N-[2hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES) (pKa 7.5) at intermediate pH, and 3-[cyclohexylamino]-2-hydroxy-1-propanesulfonic acid (CAPSO) (pKa 9.6) at high pH, all undeuterated buffers.
Figure 5.

Two-dimensional 1H/13Cɛ1 HMQC NMR spectrum (600 MHz) for one-bond correlation in uniformly 13C, 15N histidine-labeled subtilisin N155A, and various complexes at 25°C. Solid-colored lines show trajectories of certain peaks vs. pH, with arrows pointing in the direction of pH increase. Broken colored lines indicate jumps from one, more or less, to the other. Underlying spectra were obtained at pH 5.50. J1 and J2 are the principal autolysis products.
Figure 6.

Two-dimensional 1H/13Cδ2 HMQC NMR spectrum (600 MHz) for one-bond correlation in uniformly 13C, 15N histidine-labeled subtilisin N155A, and various complexes at 25°C. Solid colored lines show trajectories of certain peaks vs. pH, with arrows pointing in the direction of pH increase. Broken colored lines indicate jumps from one, more or less, to the other. Underlying spectra were obtained at pH 5.50. J1 and J2 are the principal autolysis products.
Figure 5 ▶ shows the anomalously large chemical shifts (>9.3 ppm) of the Cɛ1-H proton resonances in resting subtilisin N155A and its complexes with the peptide boronic acid inhibitors AAbF and AAPbF. The significance of these chemical shifts as evidence of H-bonding and the catalytic implications have been presented in the Introduction. The boronic acid complex 1H chemical shifts are pH-independent up to pH ~10. Resting enzyme exhibits a very broad resonance at 9.30 ppm, which does not shift as pH is increased, but simply decreases in intensity, reappearing at 8.51 ppm for high pH values (>10), but still very broad and difficult to observe. By analogy with α-lytic protease, we expected this high pH chemical shift to be closer to 8.25 ppm (Ash et al. 2000), but have observed the 8.51 ppm peak (1H value of two-dimensional peak) on two occasions, using different samples and different NMR spectrometers. The acid-base proton exchange is thus virtually in the slow-exchange limit, as will be shown by computer lineshape simulations below.
The other resonances in Figure 5 ▶, which vary as the pH in increased, His 238, autolysis products J1 and J2, His 64 in the PMSF complex, and the slightly drifting His 67, all move gradually and continuously versus pH, and are thus in the fast-exchange limit. The small drift of His 67 is the only basis for our assignment of the 13Cɛ1 resonances of His 67 versus His 226, which have identical 1Hɛ1 chemical shifts at low pH. The 1Hɛ1 chemical shift data versus pH from several two-dimensional 1H/13C HMQC titrations of PMSF-inhibited subtilisin N155A at 500 MHz (using histidine enriched at Cɛ1 only) are plotted in Figure 7A ▶. Nonlinear least squares fitting to a standard equation for sigmoidal pH titration curves yields pKa values of 7.30 ± 0.03 for His 238 and 7.47 ± 0.02 for His 64 in the PMSF complex at 25°C. Similar data for the major autolysis products J1 and J2 in Figure 7A ▶ produces pKa values of 6.98 ± 0.05 and 6.64 ± 0.05, respectively.
Figure 7.

1H chemical shifts vs. pH for titrating histidines in subtilisin N155A, its PMSF complex, and two major subtilisin autolysis products, J1 and J2. (A) Cɛ1-H 1H NMR data from two-dimensional 1H/13C HMQC NMR spectra of PMSF complex at 500 MHz and of J1 and J2 at 600 MHz. (B) Cδ2-H 1H NMR data from one-dimensional 13C-selective 1H NMR spectra of resting enzyme at 600 MHz.
Repeating the two-dimensional 1H/13C HMQC spectra for the PMSF complex in the absence of 13C decoupling during acquisition, we obtained horizontal pairs of peaks from whose separation we measured the one-bond Cɛ1-H spin-coupling constant 1JCɛ1H, summarized in Tables 1 and 2. Because of the increase in 1JCɛ1H of ~15 Hz, reaching 220 Hz or larger upon protonation of the imidazole ring (Bachovchin et al. 1981), this provides additional confirmation of the imidazole ionization state.
Figure 6 ▶ shows the two-dimensional 1H/13C HMQC correlations for all six His Cδ2-H pairs (underlying spectra at pH 5.50), plus those of autolysis products J1 and J2. The dumbbell-shaped resonances are caused by the large one-bond Cγ-Cδ spin-coupling constants. The magnitudes of these values for 1JCγCδ is generally ~74 Hz, with His 226 exhibiting a somewhat higher value of ~90 Hz (obtained from averaging numerous spectra in addition to the somewhat distorted dumbbell for His 226 shown in Fig. 6 ▶). The 13Cδ2 chemical shifts range from 113 to 118 ppm, except for His 226 and the high pH form of His 64, which give chemical shifts in the 124–125 range, owing to the presence of Nδ1-H tautomer (J.L. Sudmeier, E.M. Bradshaw, K.E. Coffman Haddad, R.M. Day, C.J. Thalhauser, P.A. Bullock, W.W. Bachovchin, unpubl.). Active site His 64 two-dimensional Cδ2-H resonances, both in resting N155A and in its AAbF boronic acid complex, lie very close to those of autolysis products J1 and J2, so that they can be distinguished only in the freshest samples.
When the pH is increased, two-dimensional Cδ2-H resonances move smoothly and continuously for His 238 and His 67 (1H data for both plotted in Fig. 7B ▶), as well as for J1 and J2. In contrast, the Cδ2-H resonance of His 64 disappears at pH >7, re-emerging only at pH values >10 – once again exhibiting slow acid-base exchange behavior. The amount of decreasing drift of His 67 with increasing pH is small, only 0.09 ppm, but certainly measurable. The 1Hδ2 resonance of His 67 also moves (~0.06 ppm decrease; Tables 1 and 2) upon inhibition with boronic acids AAbF and AAPbF, proving its responsiveness to events occurring at the active site nearby. The drift of 1Hδ2 in His 67 occurs at somewhat higher pH than other titrating groups, and least-squares pH titration curve fitting yields a pKa value of 7.9 ± 0.3. The His 238 titration curve in Figure 7B ▶ fit well, using the same pKa value determined from the more accurate data in Figure 7A ▶.
A pKa value for active site histidine of about one unit higher for subtilisin than for α-lytic protease is consistent with the somewhat stronger H-bonding suggested by the greater 15N chemical shift presented above. The amount of increased free energy required in the active site Asp–His H-bond to raise the pKa by one unit is 1.36 kcal/mole. Could the somewhat elevated pKa value of active site His 64 be the result of the N155A mutation, and inapplicable to wild-type subtilitsin BPN′? Three points argue against this idea: (1) the separation of 5.5 Å between active site His 64 and Asn 155 (or Ala 155), (2) the absence of electrostatic charge on Asn and Ala to influence His 64 acidity through space, and (3) the fact that no significant chemical shift difference has been observed between N155A and wild-type subtilisin. Coupled with the well-known unreliability of pKa values obtained by measurement of kinetic constants, we believe that a pKa value of 7.9 ± 0.3 represents the true value for His 64 of wild-type subtilisin BPN′. The fact that the pKa value of the PMSF complex of subtilisin (7.47 ± 0.02) is about one unit higher than that of the PMSF complex of α-lytic protease (6.46 ± 0.02; Farr-Jones 1989), instead of the value for resting α-lytic protease of 7.0 also tends to support the higher pKa value reported here for resting subtilisin BPN′.
One-dimensional 13C-selective 1H NMR spectra
Figure 8 ▶ shows 13C-selective 600 MHz 1H NMR spectra of subtilisin N155A at various pH values, along with calculated His 64 resonance lineshapes. The one-dimensional representations in Figure 8 ▶ contain much the same 1H chemical shift information as that given in Figures 5 ▶ and 6 ▶, but are superior in displaying lineshapes. Figure 8 ▶ also dramatizes the historical problems of degeneracy and peak overlap faced by previous researchers who tried to perform 1H NMR titrations of subtilisin. One can readily confirm the pH independence of His 17, His 39, and His 226. Both proton resonances of His 238 decrease smoothly with increasing pH, its acid and base forms in rapid exchange. The same is true of impurities J1 and J2. The Hδ2 proton of His 67 is noticeably broader than the adjacent resonances in Figure 8 ▶, especially at low pH, and its drifting decrease with increasing pH is unmistakable.
Figure 8.

One-dimensional 13C-selective 1H NMR spectra (600 MHz) of uniformly 13C, 15N histidine-labeled subtilisin N155A at various pH values at 25°C. Asterisks denote principal autolysis products J1 and J2. Smooth lines above each spectrum represent computer-simulated lineshapes for Hɛ1 and Hδ2 of active site His 64 obtained using konOH = 1.0 * 109 M−1s−1 and pKa = 7.9, or the equivalent koffOH = 794 s−1. Linewidths in the absence of chemical exchange of 80 and 160 Hz are assumed for Cɛ1-H acid and base species, respectively, and 50 Hz for all Cδ2-H protons.
In Figure 8B ▶, active site His 64 exhibits a broad resonance at the chemical shift of 9.30 – a value considered anomalously large for any His Hɛ1 in a resting serine protease until quite recently (Ash et al. 2000). In Figure 8A ▶, this peak is even broader and greater in chemical shift, but the enzyme is probably not fully structured at pH <5.5. As the pH is increased, Hɛ1 of His 64 does not gradually begin to decrease as we reported in α-lytic protease (Ash et al. 2000). Instead, it decreases in intensity, remaining visible in some spectra at pH values as high as 6.5, characteristic of slow proton exchange. A similar observation has been made above regarding the 15Nɛ2 resonance of His 64 in resting subtilisin N155A. If the pKa of His 64 were as low as 7.0, these observations would be somewhat surprising. At high pH, presumably due to greater linewidth of the Hɛ1 proton resonance in the neutral His 64 species, we have not yet observed this resonance in the one-dimensional experiment (e.g., Fig. 8G ▶). The Hδ2 proton resonance, however, appears strongly at sufficiently high pH (>10).
Rate and mechanism of His 64 acid-base exchange
To show that the above observations regarding 1H lineshapes are consistent with the active site His 64 chemical shifts and pKa values reported herein, we consider the rate and mechanism of the acid-base exchange. There are two dominant mechanisms for deprotonation of imidazole (Im).
 |
1 |
and
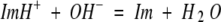 |
2 |
in which the forward and backward reactions for reaction 3 are described by rate constants koffH and konH, respectively, and those for reaction 4 are described by konOH and koffOH, respectively. In dealing with nonexchangeable nuclei on histidine in serine protease active sites, exchange catalyzed by buffers and other mechanisms can be neglected (K.E. Coffman Haddad, J.L. Sudmeier, D.A. Bachovchin, W.W. Bachovchin, unpubl.). That is not the case for histidines in free solution or on protein surfaces in contact with bulk solution.
We can express the chemical lifetimes, τa and τb, of the acid and base forms, respectively, as follows, in which Kw = 1.0 × 10−14, the autoprotolysis constant of water, and Kb, the base dissociation constant is equal to Kw/Ka:
 |
3 |
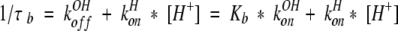 |
4 |
The average or reduced lifetime, τ, entered in certain two-site lineshape calculation algorithms (Nakagawa 1965) is given by
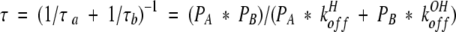 |
5 |
 |
6 |
in which PA and PB are the fractional populations of acid and base species, summing to unity. Equations 7 and 8 are valid for every case from the slow to fast exchange limits, and all exchange rates in between.
From previous NMR studies of 13Cɛ1 lineshapes of active site histidine in α-lytic protease versus pH at 33°C (Bachovchin and Switzman 1983; Ash et al. 2000), we know that the system exhibits fast-exchange broadening and, because the maximum broadening occurs around 0.3 pH units above the pKa value, as predicted by theory (Sudmeier et al. 1980), that water attack, reaction 3 above, controls the lineshape. That is, for α-lytic protease, konH dominates konOH, or
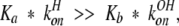 |
7 |
and the equivalent
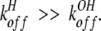 |
8 |
In this case, equations 7 and 8 reduce to
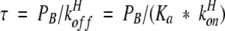 |
9 |
The extra line width, ΔW, caused by fast-exchange broadening is given by
 |
10 |
in which (ΔνAB) is the kinetic window or chemical-shift difference of the acid and base species in Hz. In the case of α-lytic protease, we can combine equations 11 and 12, yielding
 |
11 |
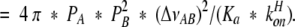 |
12 |
The pKa value of α-lytic protease is 6.87 at 33°C (Ash et al. 2000) and, assuming ΔH for imidazole dissociation of ~11 kcal/mole, closer to 7.1 at 25°C. For α-lytic protease at 33°C, values were determined for koffH = 3.4 * 103 s−1 and konH = 2.5 * 1010 M−1s−1.
For subtilisin, however, it appears by the constancy of the acid species chemical shift with increasing pH that attack by hydroxide ion, reaction 4 above, dominates the 1H lineshape behavior versus pH, and that the opposite of equations 7 and 8 are true:
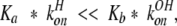 |
13 |
and the equivalent
 |
14 |
Even if konH and kOHon were equal, the almost 10-fold reduction in Ka (i.e., pKa = 7.9 compared with ~7.1 for α-lytic protease), which means an almost 10-fold elevation in Kb, tends to drive equations 15 and 16 nearly one 100-fold to the right, so that for subtilisin, the reduced rate is given approximately by
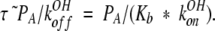 |
15 |
If subtilisin exhibited fast-exchange behavior, then following from equation 17, we would observe maximal line-broadening at pH 7.6 (Sudmeier et al. 1980). However, the Cɛ1-H and Cδ2/-H protons in Figure 8 ▶ are best described by kinetics closer to coalescence or slow exchange. The simulated lineshapes in Figure 8 ▶ are obtained using konOH = 1.0 * 109 M−1s−1 and pKa = 7.9, or the equivalent koffOH = 794 s−1. Linewidths in the absence of chemical exchange of 80 and 160 Hz are assumed for Cɛ1-H acid and base species, respectively, and 50 Hz for all Cδ2-H protons. Values of konH as high as 5.0 * 109 M−1s−1 make negligible change in the calculated subtilisin lineshapes. At konH as high as 1.0 * 1010 the lineshapes look very similar, except that the acid form begins drifting to the right, amounting to about 5% by pH 6.75 (Fig. 8D ▶). Thus, we conclude that although konH may be somewhat slower in subtilisin BPN′ than in α-lytic protease, the elevation in the pKa value of subtilisin is the principal cause of the differences in their lineshape versus pH behavior.
Conclusions
The active site His of subtilisin BPN′ has a pKa value higher than previously reported, 7.9 ± 0.3. This is consistent with 15N NMR evidence for an active site Asp–His H-bond, which is slightly stronger than that of α-lytic protease, although probably less than 1.4 kcal/mole stronger. The higher pKa value also helps account for sluggish proton exchange kinetics, which are dominated by attack of hydroxide on the protonated His 64 imidazolium ion. Subtilisin BPN′ has a strong Cɛ1-H donated H-bond from the active site His 64 presumably due to the backbone carbonyl oxygen of Ser 221, as evidenced by the anomalously large 1H chemical shift of 9.30 for the low pH form. The presence of this H-bond in the evolutionarily distinct subtilisin family lends further support to our proposal, originally based on α-lytic protease, a member of the trypsin family, for a reaction-driven imidazole ring flip mechanism as the key to the catalytic triad function in serine protease catalysis (Ash et al. 2000).
Materials and methods
Mutagenesis of subtilisin BPN′
The pGX5097 vector (gift of Genex Corp., Gaithersburg, MD) encodes the B. amyloliquefaciens BPN′ subtilisin gene with five stabilizing surface mutations (Pantoliano et al. 1989). The BPN′ gene was excised from this Escherichia coli/Bacillus subtilis shuttle vector using BamHI/SalI, and cloned into the same sites of the Bluescript II KS+ (pBKS+) phagemid (Stragagene), which was used for site-directed mutagenesis via the thermal cycling mutagenesis (Ho et al. 1989). The oligonucleotides for the oxyanion hole mutant N155A were as follows: forward primer, 5′-CGACGCACTATAGGGCGAATTGGGTAC-3′; reverse primer, 5′-AC AGCTATGACCATGATTACGATTACGCCAAGC-3′; Ala coding primer, 5′-GGCAGCCGGTGCTGAAGGCACTTCCGGCA-3′; Ala non coding primer, 5′-AAGTGCCTTCAGCACCGGCTGC CGCAACG3′. (Ala mutant codons are underlined.) Two PCR reactions were performed using pBKS+97 vector with (1) the forward primer and the Ala noncoding primer; and (2) the reverse primer and the Ala coding primer. The products of these reactions were then combined with the forward and reverse primers. Reactions contained 0.5 μM oligonucleotides, 10 ng of pBKS+97 DNA, 200 μM nucleotide triphosphates, 0.5 μM oligonucleotides and Vent polymerase (New England Biolabs) with buffers and BSA provided by the manufacturer. Cycling conditions for the first two reactions were as follows: denature 94°C, 45 sec; anneal 52°C, 60 sec; extend 72°C, 90 sec. Cycling conditions for the combined reaction were as above except using 50°C for annealing and 120 sec for extension. The mutated gene was fully sequenced by dideoxynucleotide sequencing and then cloned back into the pGX5097 vector for expression in DB104.
B. subtilis transformation
The DB104 strain of B. subtilis (his-, apr-, npr-) was the gift of R. Doi (Kawamura and Doi 1984). Competent DB104 were produced using the methods R. Doi (Kawamura and Doi 1984). Briefly, DB104 were streaked on 37°C Tryptone Blood Agar Base (TBAB) plates (3.3% TBAB, 0.5% glucose) and grown overnight. Cells scraped from the least dense portion of the plate were used to inoculate SP1 medium (0.2% NH4SO4, 1.4% K2HPO4, 0.6% KH2PO4, 0.1% Na citrate, 0.02% MgSO4, 0.5% glucose, 0.02% casamino acids, 0.1% yeast extract, and 0.01% L-histidine). Cells were grown on a shaker table (200 rpm) at 37°C for ~3.5 h in a Klett flask to 150–200 Klett. The culture was then diluted 1:10 with warm SPII medium (the same as SPI medium with the addition of 0.5 mM CaCl2 and 2.5 mM MgCl2). Cells were grown under the above conditions for an additional 90 min. A 1:100 vol of 100 mM EGTA was added for 10 min before the addition of DNA, which was added at 0.5 to 3.0 μg per 0.5 mL competent cells. The cells were then incubated on a rotary wheel for 90 min at 37°C. Cells were spread on TBAB plates (a solution of nonfat dry milk, autoclaved separately, was added to a concentration of 1%), with 25 μg/mL kanamycin (for pGX5097) or 100 μg/mL chloramphenicol (for pGX2110, gift of Genex Corp).
Subtilisin expression
Subtilisin was grown from B. subtilis DB104 transformed with either the pGX5097 vector (expression a stabilized form of BPN′) or the pGX2110 expression vector (expression of wild-type subtilisin BPN′) (gift of Genex Corp.). Fresh transformations of DB104 were used for each protein growth. On the second day, cells were streaked onto warm TBAB slopes supplemented with 25 μg/mL kanamycin, and the slopes were grown at 37° C for about 24 h. Cells from each slope were used to inoculate 40 mL of medium in a 150-mL baffled flask. Minimal medium for protein production was: 7.23 mM Tris-HCl, 0.384 mM K2PO4 3H2O, 0.449 mM Na citrate, 2.77 mM L-glutamic acid, 0.6 mM Na2SO4, 0.2 g/L tryptone, 1.02 mM NH4Cl, 100 mM CaCl2 2H2O, and 10 mL/L metal solution. Metal solution was composed of 200 mM MgCl2, 70 mM CaCl2 2H2O, 5 mM MnCl2 4H2O, 100 mM ZnCl2, 500 mM FeCl3 6H2O, and 200 mM thiamine HCl. Metal solution was filter sterilized and stored at 4° C. For the natural abundance, protein sample medium was supplemented with 0.5 g/L L-histidine. For the 13C, 15N, and 13C/15N-labeled subtilisin, medium was supplemented with 0.17 g/L 13C, 15N, or 13C/15N isotopically labeled L-histidine (Cambridge Isotope Labs, Andover, MA). Medium contained either 25 μg/mL kanamycin (for pGX5090) or 100 μg/mL chloramphenicol (for pGX2110).
Subtilisin purification
The B. subtilis expression systems result in a secreted protein. Cells were pelleted by centrifugation for 1 h at 10,000 rpm in a JA-10 rotor in a J2-21 centrifuge (Beckman). The supernatant was dialyzed against 25 vol of 1 mM Na phosphate (pH 5.8), overnight, in 12,000–14,000 Da molecular weight cutoff dialysis tubing (Spectrum through Fischer Scientific). The dialyzed protein was passed over a CM-52 column equilibrated in 1 mM Na phosphate (pH 5.8). The column was packed overnight to a volume of 1/10 total sample volume and run at the gravitational flow rate. The pH of the protein was adjusted to ~5.8 prior to loading on to the column. After loading, the column was washed with 1-1/2 column vol of Na phosphate (pH 5.8). Subtilisin protein was eluted from the protein with 2 vol of Na phosphate, 0.1 M NaCl (pH 5.8). Eluent was checked for subtilisin presence by either measuring enzyme activity or SDS-PAGE analysis. Eluted protein was dialyzed overnight in 12–14 kD MWCO dialysis tubing (Spectrum) against 50 vol of 0.1 M NaCl, 0.02 M CaCl2. The protein was subsequently concentrated using a PM10 membrane (Amicon). Chemicals used for purification of subtilisin were reagent grade.
NMR measurements
NMR samples were dialyzed and concentrated in centricon (Amicon) filter concentrators, typically reaching 1.0–1.5 mM in enzyme containing 50–100 mM NaCl, 30 mM CaCl2, 10% D2O for field/frequency lock, and sometimes 5–10 mM MES, HEPES, and/or CAPSO. Some samples that gave evidence of traces of paramagnetic metal impurities were treated by brief exposure to H2S gas, with the insoluble sulfides centrifuged or filtered off, followed by readjusting the pH. The pH was adjusted using small aliquots (~2 μL) with rapid stirring of NaOH and HCl, typically 0.25 M. The pH and enzymatic activity was measured both before and after NMR spectra were recorded, with the average pH being reported. The pH of unbuffered samples drifts toward about pH 8.5. A sample starting at pH 5 can rise as much as one pH unit in 24 h at 25°C due to autolysis, and a sample starting at pH 10.5 can fall one unit. The presence of 10 mM buffer normally keeps the drift to less than 0.1 pH unit. Inhibited samples were prepared by adding threefold excess of PMSF (Sigma) or twofold molar excess of AAPbF or AAbF to the concentrated enzymes. 1H and 13C chemical shifts are reported relative to DSS, and 15N shifts are referenced to liquid NH3, as well as 1 M HNO3.
NMR spectra of liquid samples were obtained using Bruker DRX600, AMX500, and AM400 spectrometers. The DRX600 is equipped with a 5-mm triple resonance inverse probe with triple axis gradients, and standard Avance electronics with digital filtering, digital 2H field/frequency lock, and four pulse transmitter channels. The AMX500 was equipped with a wide-bore magnet and probes, 10-mm selective 15N probe, and an Aspect 3000 computer. The solid-state CP-MAS 15N NMR spectra were run on a Bruker DSX 400 Widebore spectrometer using a spinning speed of 4250 Hz. The one-dimensional 15N NMR spectra were acquired with a 90° pulse (24 μs), spectra width (SW) = 15.1 kHz, total data (TD) = 8 K, recycle time (D1) = 0.8 sec, acquisition time (AQ) = 0.27 sec, and exponential line broadening constant (LB) = 15 Hz. The 600 MHz two-dimensional 1H/15N gradient echo/antiecho HMQC NMR spectra was run using the Bruker pulse program inv4etf3gp from operating system software XWINNMR, version 3.0. This pulse program has the advantage of suppressing water without the need for presaturation or even flip-back pulses, so that minimal losses of N-H protons occurs through saturation transfer. The spectrum in Figure 4 ▶ was obtained using total data of 2K by 256 data points, and is the summation of three spectra using primary delays of 12, 17, and 22 msec, each requiring 21 h. The SW = 9.6 kHz, D1 = 1.2 sec, AQ = 0.11 sec, and the data were processed using the Gaussian-Lorentian window function with LB = −10 Hz in both dimensions and GB2 = 0.012 and GB1 = 0.010. The two-dimensional 1H/13C HMQC NMR spectra were run at both 600 and 500 MHz using standard Bruker pulse programs with water presaturation, typically collecting total data of 2K by 256 data points over a time period of from 8 to 18 h, and using a 1/2J delay of 2.5 msec. We used sweep width SW = 7.2 kHz, D1 = 1.2 sec, AQ = 0.14 sec, and the data were processed using the Gaussian-Lorentian window function with LB2 = −10 Hz and GB2 = 0.010, and shifted sine bell squared using SSB = 2 in the indirect dimension. Heteronuclear single quantum coherence (HSQC) spectra were also used for two-dimensional 1H/13C correlation, giving results of comparable sensitivity with a 1/4J delay of 1.2 msec. The constant-time HSQC variation using a fixed tuning interval of 13.5 msec was also used at times for collapsing the dumbbells caused by carbon–carbon spin-coupling 1JCγCδ. One-dimensional 13C-selective 1H NMR spectra, normally 4K data points, were run at both 600 and 500 MHz, typically from 2 to 4 h using a standard HMQC program stripped from two- to one-dimensional by removal of incremented delays, and using water presaturation and a 1/2J delay of 2.5 msec. We used SW = 7.2 kHz, D1 = 1.2 sec, AQ = 0.29 sec, and exponential multiplication with LB = 6 Hz.
Spectra were processed using the various applicable generations of Bruker software, XWINNMR and UXNMR, and imported into CorelDraw for rendering. The lineshape simulations were carried out on a PC using the shareware program GnuPlot and a home-made implementation of the two-site exchange equations (Nakagawa 1965). The stereo figures of enzymes were rendered using Biosym’s Insight II and imported into CorelDRAW.
Acknowledgments
We thank Dr. Charles Kettner for AAPbF and AAbF boronic acid inhibitors, Dr. J. Wells for expression vectors BPN′ surface mutants, Dr. S. Wong and Dr. R.H. Doi for bacterial strain DB104, and Bruker Instruments for access to 15N solid-state NMR machine. R.M.D. is the recipient of an American Heart Association Scientist Development Grant, National Center, and an American Lung Association Research Grant.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0235203.
References
- Alden, R.A., Birktoft, J.J., Kraut, J., Robertus, J.D., and Wright, C.S. 1971. Atomic coordinates for subtilisin BPN′ (or Novo). Biochem. Biophys. Res. Commun. 4 337–344. [DOI] [PubMed] [Google Scholar]
- Ash, E.L., Sudmeier, J.L., Day, R.M., Vincent, M., Torchilin, E.V., Haddad, K.C., Bradshaw, E.M., Sanford, D.G., and Bachovchin, W.W. 2000. Unusual 1H NMR chemical shifts support (His) C E 1. . .O= =C H-bond: Proposal for reaction-driven ring flip mechanism in serine protease catalysis.Proc. Natl. Acad. Sci. 97 10371–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin, W.W. 1986. 15N NMR spectroscopy of hydrogen-bonding interactions in the active site of serine proteases: Evidence for a moving histidine mechanism. Biochemistry 25 7751–7759. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Contributions of NMR spectroscopy to the study of hydrogen bonds in serine protease active sites. Magn. Reson. Chem. 39 S199–S213. [Google Scholar]
- Bachovchin, W.W. and Roberts, J.D. 1978. Nitrogen-15 nuclear magnetic resonance spectroscopy of the state of histidine in the catalytic triad of α-lytic protease. Implications for the charge-relay mechanism of peptide-bond cleavage by serine proteases. J. Am. Chem. Soc. 100 8041–8047. [Google Scholar]
- Bachovchin, W. and Switzman, S. 1983. Carbon-13 NMR studies of serine protease structure and function. Kinetics and mechanism of proton exchange at the histidyl residue in the catalytic triad of α-lytic protease. Spectros Int. J. 2 219–226. [Google Scholar]
- Bachovchin, W.W., Kaiser, R., Richards, J.H., and Roberts, J.D. 1981. Catalytic mechanism of serine proteases: Reexamination of the pH dependence of the histidyl 1J13C2-H coupling constant in the catalytic triad of α-lytic protease. Proc. Natl. Acad. Sci. 78 7323–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin, W.W., Wong, W.Y., Farr-Jones, S., Shenvi, A.B., and Kettner, C.A. 1988. Nitrogen-15 NMR spectroscopy of the catalytic-triad histidine of a serine protease in peptide boronic acid inhibitor complexes. Biochemistry 27 7689–7697. [DOI] [PubMed] [Google Scholar]
- Bao, D., Cheng, J.T., Kettner, C., and Jordan, F. 1998. Assignment of the N.ɛ.2H and N.δ.1H resonances at the active-center histidine in Chymotrypsin and Subtilisin complexed to peptideboronic acids without specific 15N labeling. J. Am. Chem. Soc. 120 3485–3489. [Google Scholar]
- Bao, D., Huskey, W.P., Kettner, C.A., and Jordan, F. 1999. Hydrogen bonding to active-site histidine in peptidyl boronic acid inhibitor complexes of Chymotrypsin and Subtilisin: Proton magnetic resonance assignments and H/D fractionation. J. Am. Chem. Soc. 121 4684–4689. [Google Scholar]
- Bloembergen, N., Purcell, E.M., and Pound, R.V. 1948. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 73 679–711. [Google Scholar]
- Blomberg, F., Maurer, W., and Ruterjans, H. 1977. Nuclear magnetic resonance investigation of 15N-labeled histidine in aqueous solution. J. Am. Chem. Soc. 99 8149–8159. [DOI] [PubMed] [Google Scholar]
- Bott, R., Ultsch, M., Kossiakoff, A., Graycar, T., Katz, B., and Power, S. 1988. The three-dimensional structure of Bacillus amyloliquefaciens subtilisin at 1.8 Å and an analysis of the structural consequences of peroxide inactivation. J. Biol. Chem. 263 7895–7906. [PubMed] [Google Scholar]
- Bycroft, M. and Fersht, A.R. 1988. Assignment of histidine resonances in the 1H NMR (500 MHz) spectrum of subtilisin BPN′ using site-directed mutagenesis. Biochemistry 27 7390–7394. [DOI] [PubMed] [Google Scholar]
- Cassidy, C.S., Lin, J., and Frey, P.A. 2000. The deuterium isotope effect on the NMR signal of the low-barrier hydrogen bond in a transition-state analog complex of chymotrypsin. Biochem. Biophys. Res. Commun. 273 789–792. [DOI] [PubMed] [Google Scholar]
- Consonni, R., Molinari, H., Greco, F., Zannoni, G., Zetta, L., Carrea, G., and Riva, S. 1992. H-NMR studies of native and fragmented Subtilisin Carlsberg. Biochim. Biophys. Acta 1119 39–44. [DOI] [PubMed] [Google Scholar]
- Day, R.M. 1995. “The relationship between substrate specificity and transition state stabilization in Subtilisin BPN′.” Dissertation, Tufts University, Sackler School of Biomedical Sciences, Department of Biochemistry, Boston, MA.
- Derewenda, Z.S., Derewenda, U., and Kobos, P.M. 1994. (His)C epsilon-H. . .O-C < hydrogen bond in the active sites of serine hydrolases. J. Mol. Biol. 241 83–93. [DOI] [PubMed] [Google Scholar]
- Drenth, J., Hol, W., Jansonius, J., and Koekoek, R. 1972. A comparison of the three-dimensional structures of subtilisin BPN′ and subtilisin novo. Cold Spring Harb. Symp. Quant. Biol. 36 107–116. [DOI] [PubMed] [Google Scholar]
- Farr-Jones, S. 1989. “Serine protease mechanism studied by 15N NMR of the active-site histidine of α-lytic protease.” Dissertation, Tufts University, Sackler School of Biomedical Sciences, Department of Biochemistry, Boston, MA.
- Frey, P.A., Whitt, S.A., and Tobin, J.B. 1994. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science 264 1927–1930. [DOI] [PubMed] [Google Scholar]
- Gallagher, T., Gilliland, G., Wang, L., and Bryan, P. 1995. The prosegment-subtilisin BPN′ complex: Crystal structure of a specific ‘foldase’. Structure 3 907–914. [DOI] [PubMed] [Google Scholar]
- Gerlt, J.A. and Gassman, P.G. 1993. Understanding the rates of certain enzyme-catalyzed reactions: Proton abstraction from carbon acids, acyl-transfer reactions, and displacement reactions of phosphodiesters. Biochemistry 32 11943–11952. [DOI] [PubMed] [Google Scholar]
- Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap-extension using the polymerase chain extension. Gene 77 61–68. [DOI] [PubMed] [Google Scholar]
- House, K.L., Garber, A.R., Dunlap, R.B., Odom, J.D., and Hilvert, D. 1993. 1H NMR spectroscopic studies of selenosubtilisin. Biochemistry 32 3468–3473. [DOI] [PubMed] [Google Scholar]
- Jordan, F. and Polgar, L. 1981. Proton nuclear magnetic resonance evidence for the absence of a stable hydrogen bond between the active site aspartate and histidine residues of native subtilisins and for its presence in thiolsubtilisins. Biochemistry 20 6366–6370. [DOI] [PubMed] [Google Scholar]
- Jordan, F., Polgar, L., and Tous, G. 1985. Proton magnetic resonance studies of the states of ionization of histidines in native and modified subtilisins. Biochemistry 24 7711–7717. [DOI] [PubMed] [Google Scholar]
- Kahyaoglu, A. and Jordan, F. 2002. Direct proton magnetic resonance determination of the pKa of the active center histidine in thiolsubtilisin. Protein Sci. 11 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura, K. and Doi, R.H. 1984. Construction of a Bacillus subtilis double mutant deficient in extracellular alkaline and neutral proteases. J. Bact. 160 442–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd, R.D., Sears, P., Huang, D.H., Witte, K., Wong, C.H., and Farber, G.K. 1999. Breaking the low barrier hydrogen bond in a serine protease. Protein Sci. 8 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, J.R. 1976. The intrinsic pKa-values of functional groups in enzymes: Improper deductions from the pH-dependence of steady-state parameters. CRC Crit. Rev. Biochem. 4 165–173. [DOI] [PubMed] [Google Scholar]
- Krem, M.M., Rose, T., and Di Cera, E. 2000. Sequence determinants of function and evolution in serine proteases. Trends Cardiovasc. Med. 10 171–176. [DOI] [PubMed] [Google Scholar]
- Kretsinger, R.H. 1976a. Calcium-binding proteins. Annu. Rev. Biochem. 45 239–266. [DOI] [PubMed] [Google Scholar]
- ———. 1976b. Evolution and function of calcium-binding proteins. Int. Rev. Cytol. 46 323–393. [DOI] [PubMed] [Google Scholar]
- Lin, J., Westler, W.M., Cleland, W.W., Markley, J.L., and Frey, P.A. 1998. Fractionation factors and activation energies for exchange of the low barrier hydrogen bonding proton in peptidyl trifluoromethyl ketone complexes of chymotrypsin. Proc. Natl. Acad. Sci. 95 14664–14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malthouse, J.G. 1986. 13C NMR of enzymes. Prog. NMR Spectr. 18 1–59. [Google Scholar]
- Nakagawa, T. 1965. The NMR signal shape of identical nuclei exchanging between two different environments. Bull. Chem. Soc. Japan 39 1006–1008. [Google Scholar]
- Neidhart, D., Wei, Y., Cassidy, C., Lin, J., Cleland, W.W., and Frey, P.A. 2001. Correlation of low-barrier hydrogen bonding and oxyanion binding in transition state analogue complexes of chymotrypsin. Biochemistry 40 2439–2447. [DOI] [PubMed] [Google Scholar]
- O’Connell, T.P., Day, R.M., Torchilin, E.V., Bachovchin, W.W., and Malthouse, J.G. 1997. A 13C-NMR study of the role of Asn-155 in stabilizing the oxyanion of a subtilisin tetrahedral adduct. Biochem. J. 326 861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar, S., Brown, M.F., Silver, P., and Schleich, T. 1979. Histidyl and tyrosyl residue ionization studies of subtilisin Novo. Biochim. Biophys. Acta 578 261–268. [DOI] [PubMed] [Google Scholar]
- Osapay, K. and Case, D. 1991. A new analysis of proton chemical shifts in proteins. J. Am. Chem. Soc. 113 9436–9444. [Google Scholar]
- Ottesen, M. and Ralston, G. 1972. The ionization behaviour of subtilisin type novo. Comptes Rendus 38 457–479. [Google Scholar]
- Pantoliano, M.W., Whitlow, M., Wood, J.F., Dodd, S.W., Hardman, K.D., Rollence, M.L., and Bryan, P.N. 1989. Large increases in general stability for subtilisin BPN′ through incremental changes in the free energy of unfolding. Biochemistry 28 7205–7213. [DOI] [PubMed] [Google Scholar]
- Rawlings, N.D. and Barrett, A.J. 1994. Families of serine peptidases. Methods Enzymol. 244 19–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robillard, G. and Shulman, R.G. 1974a. High resolution nuclear magnetic resonance studies of the active site of Chymotrypsin. I. The hydrogen bonded protons of the “charge relay” system. J. Mol. Biol. 86 519–540. [DOI] [PubMed] [Google Scholar]
- ———. 1974b. High resolution nuclear magnetic resonance studies of the active site of Chymotrypsin. II. Polarization of histidine 57 by substrate analogues and competitive inhibitors. J. Mol. Biol. 86 541–558. [DOI] [PubMed] [Google Scholar]
- Russell, A. and Fersht, A. 1986. Commercial samples of subtilisin BPN′. Nature 321 733. [Google Scholar]
- ———. 1987. Rational modification of enzyme catalysis by engineering surface charge. Nature 328 496–500. [DOI] [PubMed] [Google Scholar]
- Stratton, J.R., Pelton, J.G., and Kirsch, J.F. 2001. A novel engineered subtilisin BPN′ lacking a low-barrier hydrogen bond in the catalytic triad. Biochemistry 40 10411–10416. [DOI] [PubMed] [Google Scholar]
- Sudmeier, J.L., Evelhoch, J.L., and Jonsson, N.B.-H. 1980. Dependence of NMR lineshape analysis upon chemical rates and mechanisms: Implications for enzyme histidine titrations. J. Mag. Res. 40 377–390. [Google Scholar]
- Thomas, P.G., Russell, A.J., and Fersht, A.R. 1985. Tailoring the pH dependence of enzyme catalysis using protein engineering. Nature 318 375–376. [Google Scholar]
- Wells, J.A., Cunningham, B.C., Graycar, T.P., and Estell, D.A. 1987a. Recruitment of substrate-specificity properties from one enzyme into a related one by protein engineering. Proc. Natl. Acad. Sci. 84 5167–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells, J.A., Powers, D.B., Bott, R.R., Graycar, T.P., and Estell, D.A. 1987b. Designing substrate specificity by protein engineering of electrostatic interactions. Proc. Natl. Acad. Sci. 84 1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witanowski, M., Stefaniak, L., Januszewski, H., and Grabowski, Z. 1972. Nitrogen-14 nuclear magnetic resonance of azoles and their benzo-derivatives Tetrahedron 28 637–653. [Google Scholar]
- Wright, C.S., Alden, R.A., and Kraut, J. 1969. Structure of subtilisin BPN′ at 2.5 Å resolution. Nature 221 235–242. [DOI] [PubMed] [Google Scholar]