

Abstract
Osmolytes increase the thermodynamic conformational stability of proteins, shifting the equilibrium between native and denatured states to favor the native state. However, their effects on conformational equilibria within native-state ensembles of proteins remain controversial. We investigated the effects of sucrose, a model osmolyte, on conformational equilibria and fluctuations within the native-state ensembles of bovine pancreatic ribonuclease A and S and horse heart cytochrome c. In the presence of sucrose, the far- and near-UV circular dichroism spectra of all three native proteins were slightly altered and indicated that the sugar shifted the native-state ensemble toward species with more ordered, compact conformations, without detectable changes in secondary structural contents. Thermodynamic stability of the proteins, as measured by guanidine HCl-induced unfolding, increased in proportion to sucrose concentration. Native-state hydrogen exchange (HX) studies monitored by infrared spectroscopy showed that addition of 1 M sucrose reduced average HX rate constants at all degrees of exchange of the proteins, for which comparison could be made in the presence and absence of sucrose. Sucrose also increased the exchange-resistant core regions of the proteins. A coupling factor analysis relating the free energy of HX to the free energy of unfolding showed that sucrose had greater effects on large-scale than on small-scale fluctuations. These results indicate that the presence of sucrose shifts the conformational equilibria toward the most compact protein species within native-state ensembles, which can be explained by preferential exclusion of sucrose from the protein surface.
Keywords: Osmolytes, hydrogen, deuterium exchange, conformational dynamics, thermodynamic stability, preferential exclusion
Osmolytes are small organic solutes such as sugars, methylamines, and amino acids that are accumulated in certain osmotically stressed and freeze-resistant organisms, as well as in the medulla of the mammalian kidney (Yancey et al. 1982; Garcia-Perez and Burg 1991). These compounds are known to inhibit the large-scale conformational changes associated with protein denaturation (Yancey et al. 1982; Garcia-Perez and Burg 1991). Osmolytes stabilize the native state because they are preferentially excluded from the protein’s surface (Lee and Timasheff 1981; Timasheff 1998). Preferential exclusion increases the chemical potential of the protein, proportionally to its solvent-exposed surface area. Thus, by LeChatelier’s principle, osmolytes favor the more compact native state over the structurally expanded denatured state.
Preferentially excluded solutes should also inhibit conformational fluctuations within the native state because such movements are concomitant with increases in the solvent-exposed surface area of the protein (Kendrick et al. 1997; DePaz et al. 2000; Kim et al. 2000). Even under conditions thermodynamically favoring the native state, protein structures are not static, but fluctuate on a rugged energy landscape that exhibits many local energy minima (Karplus and McCammon 1983; Frauenfelder et al. 1991; Onuchic et al. 1997). Protein structural dynamics studied by native-state hydrogen exchange (HX) have revealed that the residue-specific HX rates show large regional differences (Mayo and Baldwin 1993; Bai et al. 1995; Miller and Dill 1995). The heterogeneity in HX rates indicates that the native state of proteins is an ensemble of conformational substates that rapidly interconvert through various scales of unfolding and folding (Mayo and Baldwin 1993; Bai et al. 1995; Miller and Dill 1995). Furthermore, conformational flexibility is known to be essential to protein functions such as substrate and ligand binding (Frauenfelder et al. 1991), properties that have been shown to be altered greatly by osmolytes (e.g., Fields et al. 2001).
The biologically important, fundamental effects of osmolytes on conformational fluctuations within native states of proteins remain controversial, despite numerous investigations using various techniques. Native-state HX studies of several proteins with infrared spectroscopy have shown that sucrose, a model osmolyte, reduces both fast and slow HX rates, implying that it affects not only unfolding, but also interconversions between species within the native-state ensemble (Kendrick et al. 1997; DePaz et al. 2000; Kim et al. 2000). Sucrose also inhibits the formation of aggregation-competent transition states, which exhibit slight surface area increases (10%–20%) relative to the native-state protein (Kendrick et al. 1998; Webb et al. 2001; Kim et al. 2002; Krishnan et al. 2002). In contrast, other native-state HX studies performed with NMR spectroscopy have shown that osmolytes inhibit global unfolding of proteins, with no detectable effects on small-scale conformational fluctuations (Wang et al. 1995; Foord and Leatherbarrow 1998).
It is difficult to reconcile the apparently contradictory results of earlier studies because they used different proteins, spectroscopic methods, and sample preparation techniques. The aim of the present study is to examine the same proteins that were used in the NMR spectroscopic HX studies, with the sample preparation and analysis methods used in the IR spectroscopic studies. We used sucrose as a model osmolyte and ribonuclease (RNase) A, its subtilisin-cleaved form, RNase S, and cytochrome c (Cyt c) as model proteins. In addition to quantifying the effects of sucrose on HX with IR spectroscopy, we determined the effects of this osmolyte on the proteins’ secondary and tertiary structures with far- and near-UV circular dichroism (CD) spectroscopy. Finally, we measured the thermodynamics of unfolding for each of the proteins as a function of sucrose concentration.
Results
Effects of sucrose on the secondary and tertiary structures of the proteins
The effects of sucrose on the secondary and tertiary structures of three model proteins were examined under native conditions (25°C, pH 7.4 in H2O buffer), using far- and near-UV CD and IR spectroscopy. Far-UV CD spectra showed that the overall molar ellipticity of each protein was slightly increased in the presence of 1 M sucrose, without shifts in band positions (Fig. 1 ▶). A similar observation had been reported previously for RNase A in the presence of 1 M sucrose (Lee and Timasheff 1981). However, spectral deconvolution documented that there were no significant changes in the secondary structural compositions of the three proteins in the presence of sucrose, which was also confirmed by the second-derivative IR spectra of the amide I region (data not shown). Because the far-UV CD spectrum of a protein arises primarily from the spatial arrangement of amide groups, a small increase in molar ellipticity implies that the native-state ensemble is shifted to favor species with enhanced order of secondary structures (Johnson Jr. 1986; Sreerama and Woody 1993).
Figure 1.

The far- (A–C) and near-UV (D–F) CD spectra of RNase A (A,D), RNase S (B,E), and Cyt c (C,F) in the absence (solid lines) and presence (dashed-dotted lines) of 1 M sucrose at 25°C.
The near-UV CD spectra of RNase A and S displayed a trough in the 300–250-nm region (Fig. 1D,E), which mainly results from Tyr side chains (Strickland 1972; Goux and Hooker 1980). The addition of sucrose made the trough more negative around 275 nm, without shifts in band positions. This spectral change indicates that the microenvironment of at least one of the Tyr residues becomes more hydrophobic in the average conformation of the native-state ensemble (Strickland 1972; Goux and Hooker 1980; Johnson Jr. 1986). For Cyt c, however, the near-UV CD spectra of the protein in absence and presence of 1 M sucrose were indistinguishable.
Effect of sucrose on the thermodynamic stability of the proteins
All three proteins exhibited increased thermodynamic stability of the native state in proportion to sucrose concentration, as evidenced by larger values of Cm (the GdnHCl concentration at the midpoint of the unfolding transition region) and Tm (the midpoint of the thermal unfolding transition region), and higher ΔGu (the free energy of unfolding in H2O;Figs. 2, 3 ▶ ▶; Table 1). A complete thermal unfolding curve of Cyt c could not be obtained because of the protein’s high specific heat capacity around neutral pH (Privalov and Makhatadze 1990), resulting in the coincidence of the posttransition region with the boiling point of water at mile-high elevation (95°C; Fig. 3C ▶). However, sucrose increased the onset temperature of thermal denaturation compared with that for the control sample in buffer alone (Table 1).
Figure 2.
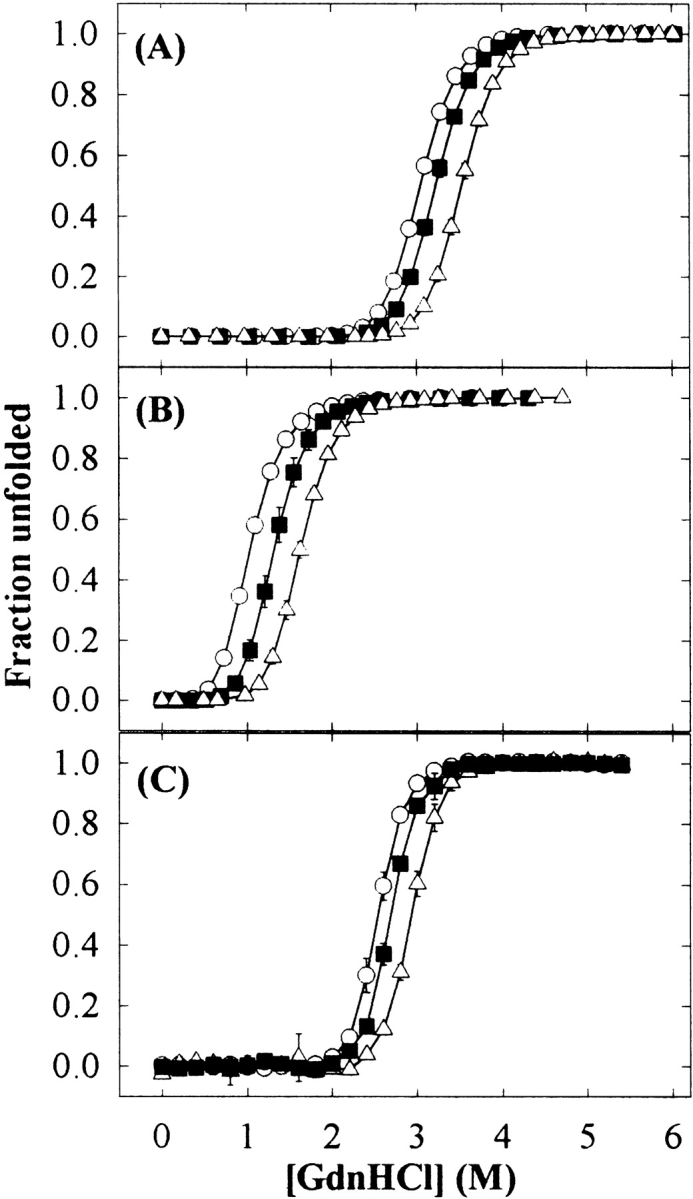
GdnHCl-induced unfolding curves for RNase A (A), RNase S (B), and Cyt c (C) in 0 (○), 0.5 (▪), and 1 M (▵) sucrose concentrations. Error bars represent the standard deviation for triplicate samples.
Figure 3.
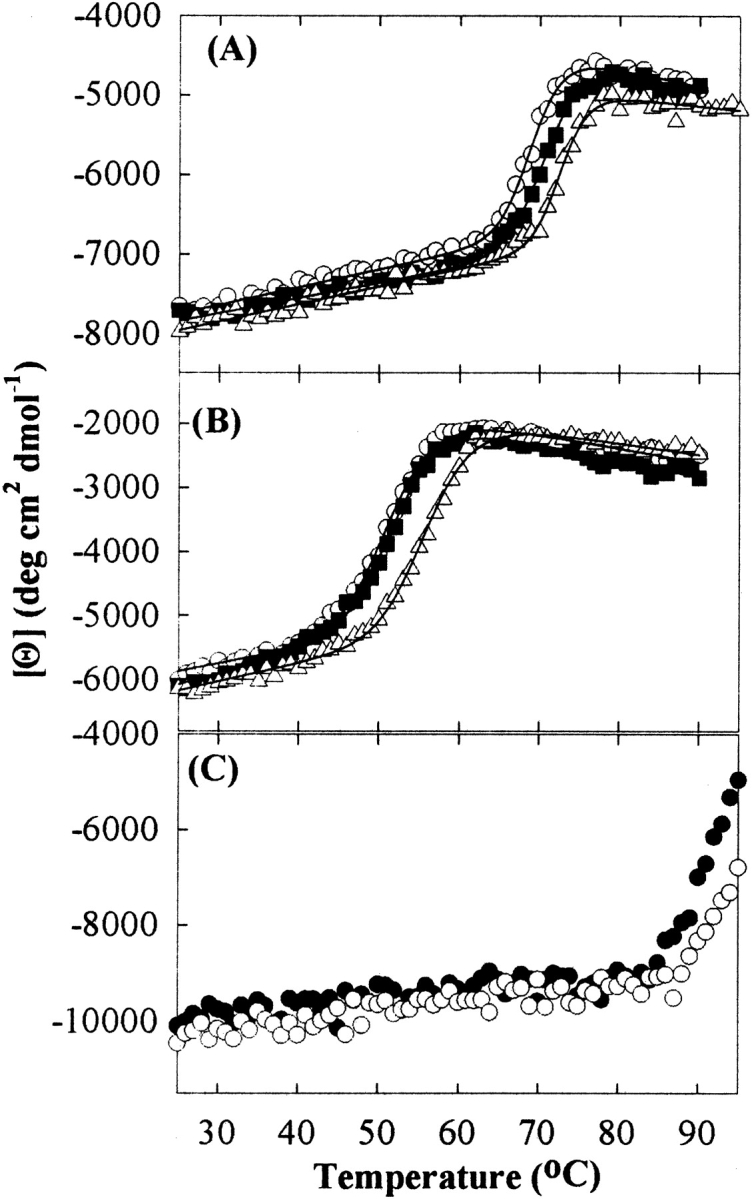
Thermal-induced unfolding curves of RNase A (A), RNase S (B), and Cyt c (C) as a function of sucrose concentration; for RNase A and S, 0 (○), 0.5 (▪), and 1 M (▵) sucrose concentrations; for Cyt c, 0 (•) and 0.75 M (○) sucrose concentrations. The solid lines in A and B represent the fitted curve by the complex, nonlinear least-squares equation to the raw data.
Table 1.
Thermodynamic parameters of RNase A and S, and Cyt c as a function of sucrose concentration determined by GdnHCl- and thermal-induced unfolding a
| Proteins | [Sucrose] (M) | Cm (M) | −m (kcal/mole/M) | ΔGu (kcal/mole) | ΔΔGu (kcal/mole) | Tm (°C) |
| RNase A | 0 | 3.04 ± 0.01 | 2.77 ± 0.01 | 8.46 ± 0.02 | — | 69.1 |
| 0.5 | 3.22 ± 0.02 | 2.74 ± 0.03 | 8.87 ± 0.10 | 0.41 ± 0.10 | 71.0 | |
| 1 | 3.53 ± 0.02 | 2.78 ± 0.10 | 9.84 ± 0.39 | 1.38 ± 0.39 | 72.7 | |
| RNase S | 0 | 1.03 ± 0.02 | 3.30 ± 0.08 | 3.56 ± 0.04 | — | 51.4 |
| 0.5 | 1.31 ± 0.04 | 3.20 ± 0.31 | 4.31 ± 0.43 | 0.75 ± 0.43 | 52.1 | |
| 1 | 1.63 ± 0.02 | 3.04 ± 0.06 | 5.00 ± 0.07 | 1.44 ± 0.08 | 56.2 | |
| Cyt c | 0 | 2.54 ± 0.03 | 3.70 ± 0.19 | 9.43 ± 0.43 | — | 85b |
| 0.5 | 2.69 ± 0.02 | 3.66 ± 0.12 | 9.76 ± 0.25 | 0.33 ± 0.50 | ||
| 1 | 2.93 ± 0.03 | 3.48 ± 0.23 | 10.22 ± 0.56 | 0.79 ± 0.71 | 89c |
Values represent the mean ± standard deviation for triplicate samples.
a ΔΔGu values were the differences in ΔGu values relative to that in 0 M sucrose, respectively.
b Onset temperature of thermal unfolding in 0 M sucrose.
c Onset temperature of thermal unfolding in 0.75 M sucrose.
The m values with or without 1 M sucrose for all three proteins were indistinguishable within the experimental error (Table 1). The m value is critical to measure ΔGu in the linear extrapolation method as shown in equation 7 below (Pace 1986). It was observed that if the equilibration time in the presence of sucrose was not long enough (<24 h), the m value could be underestimated, leading to underestimation of ΔGu (data not shown).
Effect of sucrose on native-state HX of the proteins
The effects of sucrose on conformational fluctuations within native states for the model proteins were investigated by HX studies with IR spectroscopy. IR spectroscopy provides an average HX rate over all amide hydrogens in a protein (Hvidt and Nielsen 1966; Barksdale and Rosenberg 1983). As HX progressed, there were marked changes in the shape and frequency of the amide I band (1600–1700 cm−1) as well as gradual decreases in the intensity of the amide II band (centered around 1550 cm−1), accompanied by an increase in the intensity of the amide II′ band (centered around 1450 cm−1; Fig. 4 ▶; Hvidt and Nielsen 1966; Barksdale and Rosenberg 1983; Haris et al. 1986; Dong et al. 1996).
Figure 4.
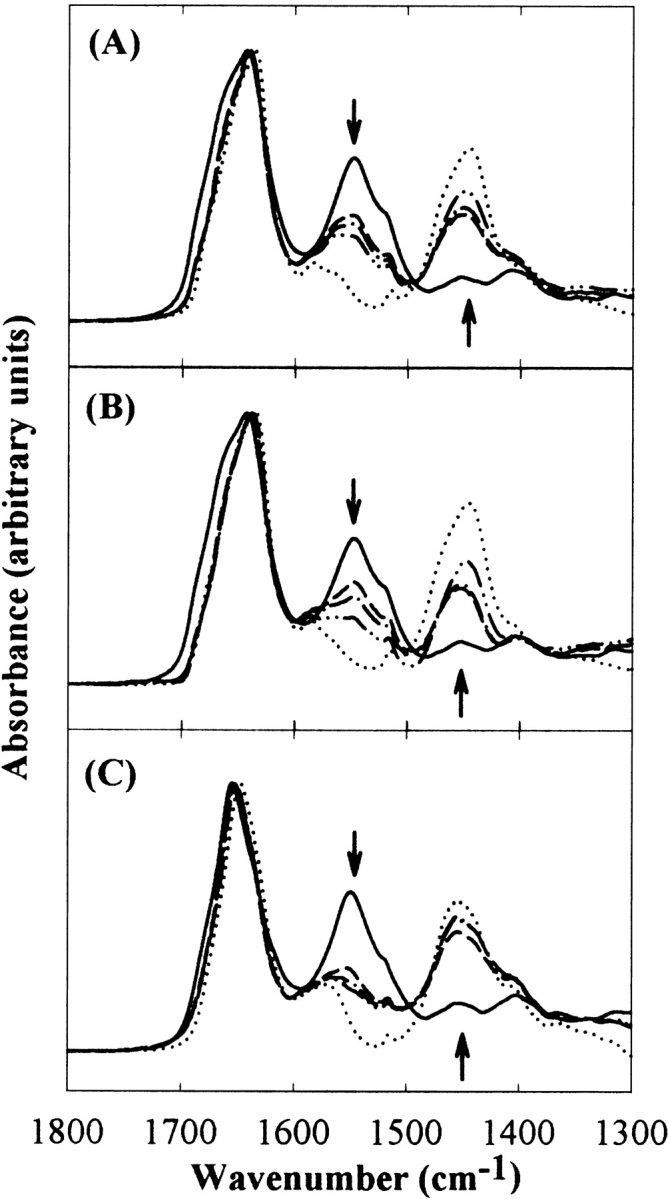
Representative IR amide absorbance spectra of HX for RNase A (A), RNase S (B), and Cyt c (C), showing the amide I, II, and II′ peaks. The solid and dotted lines represent the spectra of nondeuterated proteins in H2O buffer and the completely deuterated proteins in 100% D2O buffer, respectively. Other lines are representative spectra of proteins exposed to 75% D2O buffer. Arrows indicate the direction of spectral changes with the progress of HX.
Figure 5 ▶ shows the fraction of unexchanged amide hydrogens (X) of the three proteins as a function of time of incubation in 75% D2O buffer, in the absence or presence of 1 M sucrose. About 45% of amide hydrogens in RNase A, 49% in RNase S, and 68% in Cyt c were already exchanged, within the time between sample preparation and acquisition of the first IR spectrum (90 sec). This rapid initial exchange is mainly due to solvent-exposed amide hydrogens (Hvidt and Nielsen 1966; Barksdale and Rosenberg 1983; Haris et al. 1986; Dong et al. 1996). Sucrose slightly decreased the initial fraction of exchanged hydrogens for all three proteins (Fig. 5 ▶). Furthermore, for the three proteins, sucrose substantially increased their HX-resistant core regions. After 4 d, ∼36% and ∼42% of the amide hydrogens of RNase A, ∼22% and ∼28% of the amide hydrogens of RNase S, and ∼19% and ∼28% of the amide hydrogens of Cyt c were not exchanged in the absence and presence of 1 M sucrose, respectively.
Figure 5.
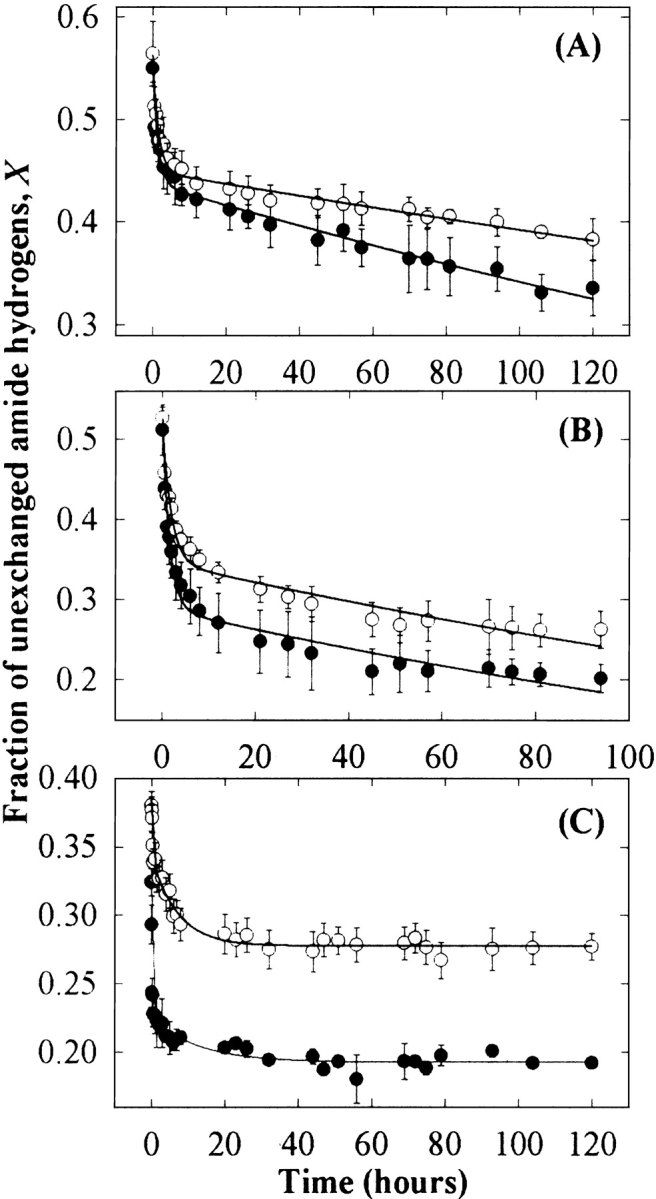
The fraction of unexchanged amide hydrogens of RNase A (A), RNase S (B), and Cyt c (C) in the presence (○) and absence (•) of 1 M sucrose. The solid lines represent the fitting curve with equation 3. Error bars indicate the standard deviation for triplicate samples of RNase A and S and for duplicate samples of Cyt c.
HX showed two phases: very rapid within the first 6 h and much slower over the next several days (Fig. 5 ▶). These HX profiles are consistent with previous observations for these proteins monitored by FT-IR or mass spectrometry (Nonnenmacher et al. 1971; Nabedryk-Viala et al. 1976; Haris et al. 1986; de Jongh et al. 1995; Dong et al. 1996). The HX rate constants were obtained by two exponential decay functions with equation 3 below for fast (<6 h) and slow (>6 h) phases, based on the previous classifications for RNase A and S (Nonnenmacher et al. 1971; Wang et al. 1995; Chakshusmathi et al. 1999) and Cyt c (Foord and Leatherbarrow 1998). For RNase A and S, sucrose reduced very slightly both fast and slow HX (Table 2). For Cyt c, the effects of sucrose on HX rates were more significant for the fast HX than the slow HX (Table 2).
Table 2.
HX parameters of RNase A and S, and Cyt c with or without 1 M sucrose
| HX rate constantsa (h−1) | |||||
| Proteins | HX rates | 0 M sucrose | 1 M sucrose | HX retardation factorb | ΔΔGHXc (kcal/mole) |
| RNase A | Fast | 0.70 | 0.63 | 1.11 | 0.06 |
| Slow | 0.0025 | 0.0013 | 1.92 | 0.39 | |
| RNase S | Fast | 0.64 | 0.53 | 1.21 | 0.11 |
| Slow | 0.0047 | 0.0038 | 1.24 | 0.13 | |
| Cyt c | Fast | 2.13 | 0.60 | 3.55 | 0.75 |
| Slow | 0.00036 | 0.00034 | 1.06 | 0.35 | |
a Values were obtained by fitting with equation 3 for the triplicate samples for RNase A and S and duplicate samples for Cyt c.
b Calculated by k0/ks, where k0 and ks are HX rate constant with and without 1 M sucrose, respectively.
c Calculated by the ΔΔGHX = −RT ln(k0/ks).
However, the above approach of fitting an arbitrary number of exponentials to the data (e.g., a “slow” and “fast” exponential decay) to provide an estimate of exchange rate constants is subjective, and is difficult to interpret in cases in which solution conditions can influence exchange rates. Therefore, an alternative approach was adopted to provide an objective, quantitative comparison of HX rates as a function of X in the presence and absence of sucrose, as described in Materials and Methods in detail. Assuming that sucrose does not change the order in which individual hydrogens exchange for deuterium, the HX rate constant at given fractions of exchange can be compared in the presence and absence of sucrose (Fig. 6 ▶). For all proteins, sucrose reduced the average rate constant at all degrees of exchange where comparisons could be made.
Figure 6.
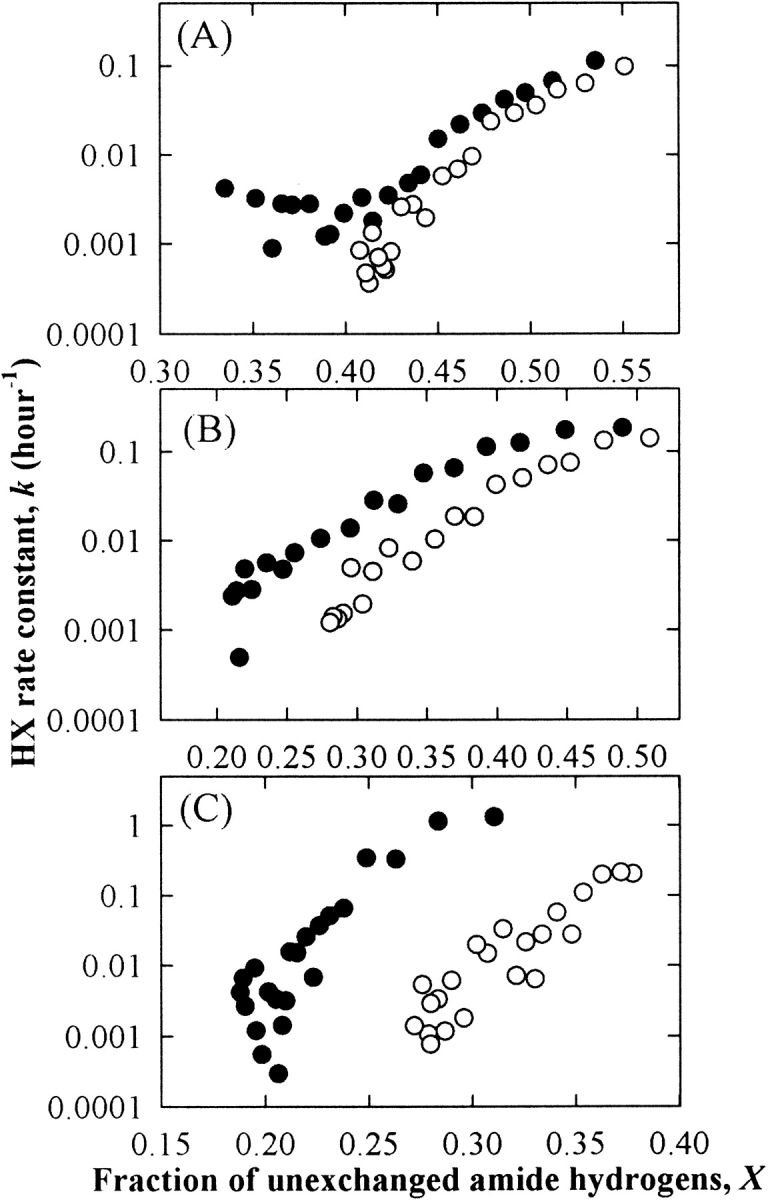
The HX rate constant as a function of X for RNase A (A), RNase S (B), and Cyt c (C) in the presence (○) and absence (•) of 1 M sucrose.
Discussion
Native-state HX is a useful tool for monitoring various scales of conformational fluctuations of proteins because conformational interconversions can be manifested as closed (HX-resistant) and open (HX-susceptible) conformations (Karplus and McCammon 1983; Mayo and Baldwin 1993; Bai et al. 1995; Miller and Dill 1995; Onuchic et al. 1997). Such conformational fluctuations within the native-state ensemble of proteins may range from small and frequent to large and rare, and are reflected in relatively fast and slow HX rates, respectively (Karplus and McCammon 1983; Mayo and Baldwin 1993; Bai et al. 1995; Miller and Dill 1995; Onuchic et al. 1997). Residues that are on the surface of the protein can exchange without any conformational fluctuations, whereas those that are buried in the interior will require varying degrees of protein unfolding to allow exchange (Mayo and Baldwin 1993; Bai et al. 1995; Miller and Dill 1995; Onuchic et al. 1997). We expect then that sucrose, which should inhibit any degree of protein unfolding, should have the largest effect on the rates of HX for those residues requiring the largest conformational change for exposure to solvent, and only minimal or no effect on surface-exposed residues. Our results for all three proteins are consistent with this hypothesis and document that sucrose can inhibit conformational fluctuations within the native state of proteins as well as increase the free energy of unfolding.
It is important to note that the large effects of sucrose on protein HX noted in this study cannot be ascribed to an intrinsic effect of sucrose on amide hydrogen exchange. Previously, Wang et al. (1995) showed by NMR spectroscopy that 1 M sucrose has no significant effects on the intrinsic chemical HX rate constants for the model peptide, poly-DL-alanine. Furthermore, a previous HX study for subtilisin as a function of water activity showed that HX rates were not affected by the water activity, indicating that D2O molecules at lower activity are still able to access exchangeable hydrogens (Hutcheon et al. 2000). Thus, sucrose-induced changes in water activity do not affect intrinsic HX rates.
Further insight into these effects of sucrose can be gained from examination of the sugar’s impact on the apparent free energy of HX, ΔGHX, which can be determined from the observed HX rate constant (Kim et al. 1993; Mayo and Baldwin 1993). ΔGHX values determined for relatively small-scale fluctuations are much less than those for complete unfolding, whereas those for large-scale fluctuations are similar to those for unfolding (Kim et al. 1993; Mayo and Baldwin 1993; Bai et al. 1995; Huyghues-Despointes et al. 1999). We expect that the effect of sucrose on ΔGHX will be small for residues that exchange via relatively small-scale conformational fluctuations. In contrast, for more protected, slowly exchanging residues, the effect of sucrose on ΔGHX should be similar to that on the free energy of unfolding ΔΔGu (Kim et al. 1993; Huyghues-Despointes et al. 1999). The change in ΔGHX in the presence of sucrose, ΔΔGHX, can be determined by ΔΔGHX = −RT ln(ks/k0; Foord and Leatherbarrow 1998; Neira et al. 1999), where R is the gas constant, T is temperature, k0 is the HX rate constant in the absence of sucrose, and ks is the HX rate constant in the presence of 1 M sucrose. ΔΔGHX calculated with HX rate constants from the fitting with two exponential decays using equation 3 below (see Table 2) gave much smaller values relative to ΔΔGu, even for the slowly exchanging hydrogens.
Therefore, we calculated ΔΔGHX based on the HX rate constants at different fractions of HX, obtained by equation 5 below (Fig. 6 ▶), and used these values in the determination of a coupling factor between effects of sucrose on HX and complete unfolding, which is defined as:
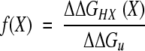 |
1 |
If HX occurs through global unfolding, the value of f(X) will be equal to 1 (Kim et al. 1993; Huyghues-Despointes et al. 1999). In contrast, f(X) for surface residues should be nearly 0, and that for partially buried residues should range from 0 to 1. For RNase A and S, f(X) values were gradually increased from ∼0.2 for up to ∼50% exchange (i.e., fast-exchanging hydrogens) to ∼1 at 60% exchange for RNase A and 75% for RNase S (i.e., slowly exchanging hydrogens; Fig. 7 ▶). This analysis further supports the conclusion that sucrose restricts both small- and large-scale conformational fluctuations in proteins, with greater effects on the latter. For Cyt c, f(X) could not be reliably computed because the fraction of exchange in the presence and absence of sucrose only overlapped for two data points, at the onset on the HX experiment.
Figure 7.

The coupling factor, f(X), between effects of sucrose on HX and complete unfolding as a function of the fraction of unexchanged amide hydrogens, X, for RNase A (•) and RNase S (○).
Sucrose also increased the size of the HX-resistant core regions of the proteins (Fig. 5 ▶), consistent with the apparent increase in secondary structural ordering (Fig. 1 ▶). Similar effects of sucrose on HX were found in earlier studies with interleukin-1 receptor antagonist (Kendrick et al. 1997), immunoglobulin light chains (Kim et al. 2000), and subtilisin (DePaz et al. 2000). Thus, sucrose shifts the conformational equilibria within the native-state ensemble toward more compact, structurally ordered species with lower surface area.
These effects of sucrose can be explained by its preferential exclusion from the protein surface, in which sucrose thermodynamically favors minimization of protein surface area (Lee and Timasheff 1981; Kendrick et al. 1997, 1998; Timasheff 1998). Native-state HX studies have shown that large-scale fluctuations (e.g., global unfolding) involve relatively large increases in solvent-accessible surface area, whereas small-scale fluctuations may occur with minor changes in solvent-accessible surface area between the closed and open conformations (Mayo and Baldwin 1993; Bai et al. 1995). On the basis of the statistical mechanical analysis of HX for Staphylococcal nuclease, Hilser and colleagues (Wooll et al. 2000) have recently suggested that small-scale fluctuations are associated with surface area changes of ∼10%. We have previously shown that sucrose can inhibit conformational changes associated with similar surface area increases (10%–20%) that occur upon formation of aggregation-competent transition states within the native-state ensembles of interferon-γ (Kendrick et al. 1998; Webb et al. 2001), an immunoglobulin light chain variable domain (Kim et al. 2002), and granulocyte colony stimulating factor (Krishnan et al. 2002).
Further evidence for osmolytes affecting conformational changes in the native state comes from the modulation of enzymatic functions by osmolytes (Burg et al. 1996, 1999; Fields et al. 2001). Enzymatic functions of proteins, such as substrate binding, transition state formation, and product release, require conformational shifts from one substate to another, which will carry minor degrees of surface area change compared with global unfolding (Frauenfelder et al. 1991; Fields et al. 2001). Yet osmolytes can greatly alter these functions (e.g., Fields et al. 2001).
Many experimental conditions, such as pH, temperature, buffer salts, added solutes, and protein concentration, can affect HX rates and profiles (Hvidt and Nielsen 1966; Barksdale and Rosenberg 1983; Mayo and Baldwin 1993; Bai et al. 1995; Miller and Dill 1995; Chakshusmathi et al. 1999; Kim et al. 2000). In addition, sample preparation methods can greatly alter the HX rates. We note that, unlike the sample preparation used in this study, many other HX studies have used direct dissolution of lyophilized proteins in 100% D2O buffer to initiate HX (Mayo and Baldwin 1993; Bai et al. 1995; Wang et al. 1995; Dong et al. 1996; Foord and Leatherbarrow 1998; Chakshusmathi et al. 1999). The direct dissolution method may distort HX kinetics because of the presence of a significant portion of unfolded protein molecules in the dried solid (Prestrelski et al. 1993; Dong et al. 1995). As a result, the residues that are normally deeply buried in the native conformation are exposed to solvent during rehydration (Dong et al. 1995), leading to much faster HX than that expected for a fully folded protein.
Furthermore, the effects of osmolytes on conformational dynamics of the native state may not be resolved if the starting condition for HX is dissolving unfolded protein molecules in 100% D2O buffer. For example, we have observed that when Cyt c was dissolved in 100% D2O, >95% of the amide hydrogens were already exchanged before the first spectrum was acquired (90 sec), and there were no significant differences in the HX rates with or without up to 1.5 M sucrose (data not shown). The previous HX data for RNase A and S dissolved in 100% D2O showed much lower fraction values of the initial unexchanged hydrogens than those we reported here (Nonnenmacher et al. 1971; Dong et al. 1996; Chakshusmathi et al. 1999). Our approach of diluting native protein in H2O buffer into D2O buffer to initiate HX provides the resolution needed to detect the effects of osmolytes on conformational fluctuations in the native state.
In conclusion, sucrose shifts the conformational equilibria within native-state ensembles of the proteins toward more ordered, compact conformations. From a protein stability standpoint, the shift away from more expanded species accounts for the capacity of sucrose to inhibit physical and chemical degradation of proteins during long-term storage in aqueous solution (Kendrick et al. 1997; DePaz et al. 2000; Kim et al. 2000). Similarly, favoring the most compact substates could explain how osmolytes can alter the catalytic function of enzymes (Fields et al. 2001). Thus, it appears that in addition to inhibiting protein denaturation, a fundamental property of osmolytes is to influence the conformational dynamics of the native state.
Materials and methods
Materials
Lyophilized (from water) bovine pancreatic RNase A (highly purified, CalBiochem), RNase S (Type XII-S, Sigma), and horse heart Cyt c (Type VI, Sigma) were used without further purification. Protein concentrations were determined spectrophotometrically using extinction coefficients of 9.8 × 103 M cm−1 at 278 nm for RNase A and S (Lee and Timasheff 1981; Catanzano et al. 1996), and 1.06 × 105 M cm−1 at 409 nm for Cyt c (Elöve et al. 1994). D2O (99.9 atom % D) from Aldrich-Sigma, guanidine HCl (GdnHCl) from Sigma, and highly purified sucrose from Pfanstiehl Laboratories were used. Other chemicals were of reagent grade or higher quality. Throughout the experiments, 10 mM potassium phosphate buffer (pH 7.4; “buffer”) was used. Values of pH for D2O buffer represent apparent values of pH, without correction for isotope effects. Deuterated sucrose was prepared as previously described (Kendrick et al. 1997; Kim et al. 2000), which was used to prepare 100% D2O buffer containing 1.33 M sucrose for HX studies.
HX with IR spectroscopy
HX was determined in real time by recording IR spectra with a Bomem MB-104 IR spectrometer. The spectrometer was continuously purged with dry air from a Balston dryer to eliminate water vapor in the IR radiation path. The protein stock solutions (40 mg/mL) were first prepared in H2O buffer. The HX reaction was initiated by mixing the protein stock solution with appropriate D2O buffer to give a final protein concentration of 10 mg/mL in 75% D2O buffer, with or without 1 M sucrose. All samples also contained 0.1% (w/v) sodium azide to prevent microbial growth. The sample was injected immediately in a liquid IR cell (P/N 20500, Specac) with CaF2 windows separated by a 6-μm Mylar spacer (Chemplex). A time-dependent series of spectra ranging from 500 to 4000 cm−1 was recorded in single beam mode with a 4-cm−1 resolution. The time from the sample preparation to acquisition of the first spectrum was ∼90 sec. During the first 60 min, 32- or 64-scan interferograms were recorded, and subsequently 128-scan interferograms were collected. Reference spectra of the appropriate buffer blank were recorded under identical scan conditions. Protein spectra were obtained according to the previously established criteria (Dong et al. 1990) and normalized to the same intensity of amide I peak after the baseline regions of 1800–1750 cm−1 were normalized to zero (Dong et al. 1996; Kendrick et al. 1997; Kim et al. 2000).
To determine the fraction of unexchanged amide hydrogens, spectra for nondeuterated (in H2O buffer) and deuterated (in 100% D2O buffer) proteins were obtained under the same scanning conditions. To fully deuterate the proteins, the lyophilized proteins were dissolved in D2O (10 mg/mL), incubated at elevated temperatures (below the proteins’ thermal unfolding transitions, i.e., 55°C for RNase A, 50°C for RNase S, and 60°C for Cyt c) for various times, and then cooled to room temperature for spectral acquisition. As a function of time of incubation, there was a red shift of the amide I band and a reduction in intensity of amide II caused by deuteration of the protein backbone (data not shown). After ∼1 h for RNase A and S and ∼10 h for Cyt c, these spectral parameters no longer changed with further incubation, and the spectra were assumed to be representative of the fully deuterated proteins (Haris et al. 1986; Dong et al. 1996).
The fraction of unexchanged amide hydrogens, X, was calculated using the equation (Barksdale and Rosenberg 1983):
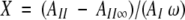 |
2 |
where AI and AII are the intensity of the amide I and II bands of proteins as a function of exposure time to 75% D2O buffer, respectively. AII∞ is the intensity of amide II band of the fully deuterated protein, and ω is the ratio of AII0/AI0, with AII0 and AI0 being the respective intensity of amide II and I bands of the proteins in H2O buffer. AII was measured at the wavenumber for maximum intensity of amide II in the spectrum of the nondeuterated protein, that is, at 1547.3 cm−1 for RNase A and RNase S, and 1549.9 cm−1 for Cyt c.
To obtain the HX rate constants, the fraction of unexchanged amide hydrogens was fitted to a double exponential decay curve with SigmaPlot5 software (SigmaPlot) after the initial value was normalized to 1,
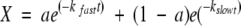 |
3 |
where a represents the fractional contribution, kfast and kslow are fast and slow HX rate constants, respectively, and t is time in hours.
Alternatively, X was analyzed to compare HX rate constants at the same amide hydrogen fractions of exchange in the presence and absence of 1 M sucrose as described below. HX of the model proteins under native conditions has been interpreted in terms of an EX2 mechanism (Mayo and Baldwin 1993; Bai et al. 1995; Foord and Leatherbarrow 1998; Neira et al. 1999), in which the structural reclosing is much faster than the intrinsic chemical HX rate of unprotected peptide groups (Hvidt and Nielsen 1966; Barksdale and Rosenberg 1983). Then, X can be assumed to be the sum of the first-order reactions of HX for all hydrogens (N) and can be expressed as (Hvidt and Nielsen 1966; Barksdale and Rosenberg 1983):
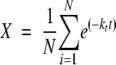 |
4 |
where ki is the HX rate constant for the i-th amide hydrogen. The exchange rate data obtained from IR experiments does not allow for assignment of kis to individual amide hydrogens. Instead, we approximate the extent of exchange by:
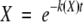 |
5 |
where k(X) is the observed, instantaneous HX rate constant averaged over all exchanging amide hydrogens. k(X) values were estimated from the slopes of semilogarithmic plots of X versus time, after the raw data for X versus time were smoothed using an exponential smoothing function (damping factor = 0.5) on Excel software (Microsoft). The k(X) values were designated as k0 in buffer and ks in 1 M sucrose, respectively.
CD spectroscopy
CD spectroscopy was performed with an Aviv 62DS spectrometer (Aviv) equipped with a Peltier temperature control unit. Far-UV (200–260 nm) and near-UV (250–340 nm) spectra were recorded for samples in 0.1- and 1-cm path length quartz cuvettes, respectively, with a step size of 0.5 nm. The signal was averaged for 5 sec at each wavelength. The protein concentrations were 0.3 and 1 mg/mL for far- and near-UV CD spectra, respectively. The protein samples in H2O buffer with or without 1 M sucrose were equilibrated at room temperature overnight before measurements. For each spectrum, two or three scans were averaged. Raw spectra were corrected for the appropriate buffer blank and converted to mean residue ellipticity, [ϑ], in degree centimeter-squared per decimole (Johnson Jr. 1986),
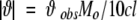 |
6 |
where ϑobs is the observed ellipticity in millidegrees, M0 is the mean residue molecular weight, c is the protein concentration in milligrams per milliliter, and l is the path length of the cell in centimeters. For the estimation of secondary structural composition of proteins, the CD spectra were deconvoluted using SELCON (Sreerama and Woody 1993).
GdnHCl- and thermally induced denaturation
GdnHCl-induced unfolding curves were obtained by using the CD spectrometer to determine ellipticity at 222 nm for the protein (0.3 mg/mL) at 25°C as a function of GdnHCl concentration in H2O buffer. Samples were equilibrated in GdnHCl solutions at room temperature for 24–48 h prior to spectral acquisition. For thermally induced unfolding, ellipticity at 222 nm for proteins (0.3 mg/mL) in H2O buffer was recorded during heating from 25° to 95°C at 1°C intervals and 1°C/min. The average recording time was 10 sec at each temperature.
For GdnHCl-induced unfolding, the linear extrapolation method was used to calculate the fraction of native protein (Pace 1986; Kim et al. 2000, 2001). The free energy of unfolding in H2O (ΔGu) was calculated with the following equation (Pace 1986):
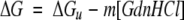 |
7 |
where ΔG is the free energy of unfolding as a function of GdnHCl concentration and m (≡δΔG/δ[GdnHCl]) is the linearized dependence of ΔG on the GdnHCl concentration in the transition region. The GdnHCl concentration at the midpoint of the unfolding transition region, Cm, was calculated at ΔG = 0. Data for thermally induced unfolding were analyzed by complex sigmoid, nonlinear least-squares fitting to determine the midpoint of the unfolding transition region, Tm (Kim et al. 2000, 2001).
Acknowledgments
Supported by National Science Foundation grant BES-0138595.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
HX, hydrogen–deuterium exchange
IR, infrared
CD, circular dichroism
RNase, ribonuclease
Cyt c, cytochrome c
GdnHCl, guanidine HCl
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0242603.
References
- Bai, Y., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995. Protein folding intermediates: Native-state hydrogen exchange. Science 269 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barksdale, A.D. and Rosenberg, A. 1983. Acquisition and interpretation of hydrogen exchange data from peptides, polymers, and proteins. Methods Biochem. Anal. 28 1–111. [DOI] [PubMed] [Google Scholar]
- Burg, M.B., Kwon, E.D., and Peters, E.M. 1996. Glycerophosphocholine and betaine counteract the effect of urea on pyruvate kinase. Kidney Int. Suppl. 57 S100–S104. [PubMed] [Google Scholar]
- Burg, M.B., Peters, E.M., Bohren, K.M., and Gabbay, K.H. 1999. Factors affecting counteraction by methylamines of urea effects on aldose reductase. Proc. Natl. Acad. Sci. 96 6517–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanzano, F., Giancola, C., Graziano, G., and Barone, G. 1996. Temperature-induced denaturation of ribonuclease S: A thermodynamic study. Biochemistry 35 13378–13385. [DOI] [PubMed] [Google Scholar]
- Chakshusmathi, G., Ratnaparkhi, G.S., Madhu, P.K., and Varadarajan, R. 1999. Native-state hydrogen-exchange studies of a fragment complex can provide structural information about the isolated fragments. Proc. Natl. Acad. Sci. 96 7899–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jongh, H.H., Goormaghtigh, E., and Ruysschaert, J.M. 1995. Tertiary stability of native and methionine-80 modified cytochrome c detected by proton-deuterium exchange using on-line Fourier transform infrared spectroscopy. Biochemistry 34 172–179. [DOI] [PubMed] [Google Scholar]
- DePaz, R.A., Barnett, C.C., Dale, D.A., Carpenter, J.F., Gaertner, A.L., and Randolph, T.W. 2000. The excluding effects of sucrose on a protein chemical degradation pathway: Methionine oxidation in subtilisin. Arch. Biochem. Biophys. 384 123–132. [DOI] [PubMed] [Google Scholar]
- Dong, A., Huang, P., and Caughey, W.S. 1990. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry 29 3303–3308. [DOI] [PubMed] [Google Scholar]
- Dong, A., Prestrelski, S.J., Allison, S.D., and Carpenter, J.F. 1995. Infrared spectroscopic studies of lyophilization- and temperature-induced protein aggregation. J. Pharm. Sci. 84 415–424. [DOI] [PubMed] [Google Scholar]
- Dong, A., Hyslop, R.M., and Pringle, D.L. 1996. Differences in conformational dynamics of ribonucleases A and S as observed by infrared spectroscopy and hydrogen–deuterium exchange. Arch. Biochem. Biophys. 333 275–281. [DOI] [PubMed] [Google Scholar]
- Elöve, G.A., Bhuyan, A.K., and Roder, H. 1994. Kinetic mechanism of cytochrome c folding: Involvement of the heme and its ligands. Biochemistry 33 6925–6935. [DOI] [PubMed] [Google Scholar]
- Fields, P.A., Wahlstrand, B.D., and Somero, G.N. 2001. Intrinsic versus extrinsic stabilization of enzymes: The influence of solutes and temperature on A4-lactate dehydrogenase orthologs from warm-adapted and cold-adapted marine fishes. Eur. J. Biochem. 268 4497–4505. [DOI] [PubMed] [Google Scholar]
- Foord, R.L. and Leatherbarrow, R.J. 1998. Effect of osmolytes on the exchange rates of backbone amide hydrogens in proteins. Biochemistry 37 2969–2978. [DOI] [PubMed] [Google Scholar]
- Frauenfelder, H., Sligar, S.G., and Wolynes, P.G. 1991. The energy landscapes and motions of proteins. Science 254 1598–1603. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez, A. and Burg, M.B. 1991. Renal medullary organic osmolytes. Physiol. Rev. 71 1081–1115. [DOI] [PubMed] [Google Scholar]
- Goux, W.J. and Hooker, T.M. 1980. Chirooptical properties of proteins. 1. Near-ultraviolet circular dichroism of ribonuclease-S. J. Am. Chem. Soc. 102 7080–7087. [Google Scholar]
- Haris, P.I., Lee, D.C., and Chapman, D. 1986. A Fourier transform infrared investigation of the structural differences between ribonuclease A and ribonuclease S. Biochim. Biophys. Acta 874 255–265. [DOI] [PubMed] [Google Scholar]
- Hutcheon, G.A., Parker, M.C., and Moore, B.D. 2000. Measuring enzyme motility in organic media using novel H–D exchange methodology. Biotechnol. Bioeng. 70 262–269. [PubMed] [Google Scholar]
- Huyghues-Despointes, B.M., Scholtz, J.M., and Pace, C.N. 1999. Protein conformational stabilities can be determined from hydrogen exchange rates. Nat. Struct. Biol. 6 910–912. [DOI] [PubMed] [Google Scholar]
- Hvidt, A. and Nielsen, S.O. 1966. Hydrogen exchange in proteins. Adv. Protein Chem. 21 287–386. [DOI] [PubMed] [Google Scholar]
- Johnson Jr., W.C. 1986. Circular dichroism and its empirical application to biopolymers. Methods Biochem. Anal. 31 61–163. [DOI] [PubMed] [Google Scholar]
- Karplus, M. and McCammon, J.A. 1983. Dynamics of proteins: Elements and function. Annu. Rev. Biochem. 52 263–300. [DOI] [PubMed] [Google Scholar]
- Kendrick, B.S., Chang, B.S., Arakawa, T., Peterson, B., Randolph, T.W., Manning, M.C., and Carpenter, J.F. 1997. Preferential exclusion of sucrose from recombinant interleukin-1 receptor antagonist: Role in restricted conformational mobility and compaction of native state. Proc. Natl. Acad. Sci. 94 11917–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick, B.S., Carpenter, J.F., Cleland, J.L., and Randolph, T.W. 1998. A transient expansion of the native state precedes aggregation of recombinant human interferon-γ. Proc. Natl. Acad. Sci. 95 14142–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.-S., Fuchs, J.A., and Woodward, C.K. 1993. Hydrogen exchange identifies native-state motional domains important in protein folding. Biochemistry 32 9600–9608. [DOI] [PubMed] [Google Scholar]
- Kim, Y.-S., Wall, J.S., Meyer, J., Murphy, C., Randolph, T.W., Manning, M.C., Solomon, A., and Carpenter, J.F. 2000. Thermodynamic modulation of light chain amyloid fibril formation. J. Biol. Chem. 275 1570–1574. [DOI] [PubMed] [Google Scholar]
- Kim, Y.-S., Cape, S.P., Chi, E., Raffen, R., Wilkins-Stevens, P., Stevens, F.J., Manning, M.C., Randolph, T.W., Solomon, A., and Carpenter, J.F. 2001. Counteracting effects of renal solutes on amyloid fibril formation by immunoglobulin light chains. J. Biol. Chem. 276 1626–1633. [DOI] [PubMed] [Google Scholar]
- Kim, Y.-S., Randolph, T.W., Stevens, F.J., and Carpenter, J.F. 2002. Kinetics and energetics of assembly, nucleation, and growth of aggregates and fibrils for an amyloidogenic protein: Insights into transition states from pressure, temperature, and co-solute studies. J. Biol. Chem. 277 27240–27246. [DOI] [PubMed] [Google Scholar]
- Krishnan, S., Chi, E.Y., Webb, J.N., Chang, B.S., Shan, D., Goldenberg, M., Manning, M.C., Randolph, T.W., and Carpenter, J.F. 2002. Aggregation of granulocyte colony stimulating factor under physiological conditions: Characterization and thermodynamic inhibition. Biochemistry 41 6422–6431. [DOI] [PubMed] [Google Scholar]
- Lee, J.C. and Timasheff, S.N. 1981. The stabilization of proteins by sucrose. J. Biol. Chem. 256 7193–7201. [PubMed] [Google Scholar]
- Mayo, S.L. and Baldwin, R.L. 1993. Guanidium chloride induction of partial unfolding in amide proton exchange in RNase A. Science 262 873–876. [DOI] [PubMed] [Google Scholar]
- Miller, D.W. and Dill, K.A. 1995. A statistical mechanical model for hydrogen exchange in globular proteins. Protein Sci. 4 1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabedryk-Viala, E., Thiery, C., Calvet, P., and Thiery, J.M. 1976. Hydrogen-isotope exchange of oxidized and reduced cytochrome c: A comparison of mass spectrometry and infrared methods. Eur. J. Biochem. 61 253–258. [DOI] [PubMed] [Google Scholar]
- Neira, J.L., Sevilla, P., Menéndez, M., Bruix, M., and Rico, M. 1999. Hydrogen exchange in ribonuclease A and ribonuclease S: Evidence for residual structure in the unfolded state under native conditions. J. Mol. Biol. 285 627–643. [DOI] [PubMed] [Google Scholar]
- Nonnenmacher, G., Viala, E., Thiery, J.M., and Calvet, P. 1971. A deuterium–hydrogen exchange study of inhibitor-induced conformational changes in ribonuclease A. Eur. J. Biochem. 21 393–399. [DOI] [PubMed] [Google Scholar]
- Onuchic, J.N., Luthey-Schulten, Z., and Wolynes, P.G. 1997. Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem. 48 545–600. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Prestrelski, S.J., Tedeschi, N., Arakawa, T., and Carpenter, J.F. 1993. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophys. J. 65 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privalov, P.L. and Makhatadze, G.I. 1990. Heat capacity of proteins. II. Partial molar heat capacity of the unfolded polypeptide chain of proteins: Protein unfolding effects. J. Mol. Biol. 213 385–391. [DOI] [PubMed] [Google Scholar]
- Sreerama, N. and Woody, R.W. 1993. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209 32–44. [DOI] [PubMed] [Google Scholar]
- Strickland, E.H. 1972. Interactions contributing to the tyrosyl circular dichroism bands of ribonuclease S and A. Biochemistry 11 3465–3474. [DOI] [PubMed] [Google Scholar]
- Timasheff, S.N. 1998. Control of protein stability and reactions by weakly interacting cosolvents: The simplicity of the complicated. Adv. Protein Chem. 51 355–432. [DOI] [PubMed] [Google Scholar]
- Wang, A., Robertson, A.D., and Bolen, D.W. 1995. Effects of a naturally occurring compatible osmolyte on the internal dynamics of ribonuclease A. Biochemistry 34 15096–15104. [DOI] [PubMed] [Google Scholar]
- Webb, J.N., Webb, S.D., Cleland, J.L., Carpenter, J.F., and Randolph, T.W. 2001. Partial molar volume, surface area, and hydration changes for equilibrium unfolding and formation of aggregation transition state: High pressure and cosolute studies on recombinant human IFN-γ. Proc. Natl. Acad. Sci. 98 7259–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooll, J.O., Wrabl, J.O., and Hilser, V.J. 2000. Ensemble modulation as an origin of denaturant-independent hydrogen exchange in proteins. J. Mol. Biol. 301 247–256. [DOI] [PubMed] [Google Scholar]
- Yancey, P.H., Clark, M.E., Hand, S.C., Bowlus, R.D., and Somero, G.N. 1982. Living with water-stress: Evolution of osmolyte systems. Science 217 1214–1222. [DOI] [PubMed] [Google Scholar]