

Abstract
Adenylosuccinate lyase is a homotetramer that catalyzes two discrete reactions in the de novo synthesis of purines: the cleavage of adenylosuccinate and succinylaminoimidazole carboxamide ribotide (SAICAR). Several point mutations in the gene encoding the enzyme have been implicated in human disease. Bacillus subtilis adenylosuccinate lyase was used as a model system in which mutations were constructed corresponding to those mutations associated with severe human adenylosuccinate lyase deficiency. Site-directed mutagenesis was utilized to construct amino acid substitutions in B. subtilis adenylosuccinate lyase; Met10, Ile123, and Thr367 were replaced by Leu, Trp, and Arg, respectively, and the altered enzymes were expressed in Escherichia coli. These purified enzymes containing amino acid substitutions were found to have substantial catalytic activity and exhibit relatively small changes in their kinetic parameters. The major deviations from the wild-type-like behavior were observed upon biophysical characterization. All of these enzymes with amino acid replacements are associated with marked thermal instability. I123W adenylosuccinate lyase exhibits notable changes in the circular dichroism spectra, and a native gel electrophoresis pattern indicative of some protein aggregation. T367R also exhibits alterations at the quarternary level, as reflected in native gel electrophoresis. Experimental results, combined with homology modeling, suggest that the altered enzymes are primarily structurally impaired. The enzyme instability was found to be lessened by subunit complementation with the wild-type enzyme, under mild conditions; these studies may have implications for the in vivo behavior of adenylosuccinate lyase in heterozygous patients. Residues Met10, Ile123, and Thr367 appear to be located in regions of the enzyme important for maintaining the structural integrity required for a stable, functional enzyme.
Keywords: Adenylosuccinate lyase, fumarase superfamily, adenylosuccinate lyase deficiency, site-directed mutagenesis
Adenylosuccinate lyase (E.C. 4.3.2.2.) is an enzyme that catalyzes two steps in the purine biosynthetic pathway, in addition to participating in the purine nucleotide cycle. The two discrete reactions catalyzed are β-eliminations, in which either succinylaminoimidazole carboxamide ribotide (SAICAR) is converted to aminoimidazole carboxamide ribotide (AICAR) and fumarate, or adenylosuccinate (SAMP) is cleaved to form AMP and fumarate (Ratner 1972).
This enzyme is a member of a protein superfamily which includes class II fumarase, δ-crystallin, carboxy-cis,cis-muconate lactonizing enzyme, aspartase, and argininosuccinate lyase. Members of the family share several key characteristics; all catalyze β-elimination reactions in which fumarate is generated as one of the products, each contain the signature sequence “SSxxPxK268xNxxxxE275” (Bacillus subtilis adenylosuccinate lyase numbering), and all share a very high level of structural similarity (Simpson et al. 1994; Weaver et al. 1995; Shi et al. 1997; Turner et al. 1997; Toth and Yeates 2000) despite their low sequence identity plus similarity at the amino acid level. These enzymes are all homotetramers with molecular masses of approximately 200 kD.
Adenylosuccinate lyase is of particular interest biochemically as single-point mutations in the gene encoding the protein give rise to mutated enzymes often associated with severe physical symptoms in humans. Adenylosuccinate lyase deficiency, recognized in 1984 (Jaeken and Van den Berghe 1984), can result in growth and mental retardation, muscle wasting, autistic features, and/or epilepsy. To date, 30 unique point mutations in the gene have been identified, in addition to one deletion (ADSLdb). Normally, in body fluids such as cerebrospinal fluid, blood, and urine, the dephosphorylated derivatives of SAMP and SAICAR, succinyladenosine (S-Ado) and succinylaminoimidazole carboximide riboside (SAICA riboside), respectively, are not detectable. However, these degradation products of substrate become detectable in individuals with adenylosuccinate lyase deficiency. Furthermore, the relative concentrations of S-Ado and SAICA riboside correlate with disease severity (Jaeken et al. 1988): Approximately equal S-Ado and SAICA riboside concentrations are associated with a severe disease state, whereas two to five times greater amounts of S-Ado present are found in patients with milder symptoms. Although this observation is not well understood, the idea that SAICA riboside is the major toxic compound, and that S-Ado acts to protect tissues against its toxicity, has been put forth (Jaeken and Van den Berghe 1984; Jaeken et al. 1988; Stone et al. 1992, 1998). In the literature, it has been hypothesized that a mild disease state arises from a catalytically impaired enzyme, while more severe forms of adenylosuccinate lyase deficiency are caused by structurally impaired enzymes (Race et al. 2000; Ciardo et al. 2001). Only a limited number of biochemical studies have been conducted using the human enzyme (Stone et al. 1992, 1993; Kmoch et al. 2000; Race et al. 2000). A major obstacle has been the instability of the human protein, which led to the enzyme being expressed in fusion with either thioredoxin or maltose binding protein (Kmoch et al. 2000; Race et al. 2000). Even in the thioredoxin–human adenylosuccinate lyase fusion protein, enzymatic activity was lost after freezing and the enzyme had to be purified for each experiment (Race et al. 2000). Because of the instability of human adenylosuccinate lyase (Race et al. 2000), and the highly similar structures of the enzymes in this family, we are using the more stable B. subtilis adenylosuccinate lyase as a model system.
Although more mechanistic details and amino acid participants in the active site of adenylosuccinate lyase are becoming known (Lee et al. 1997, 1998, 1999; Brosius and Colman 2000, 2002), there has still been little consideration of the regions important for tetramer formation and for which amino acids contribute to enzyme stability. Accordingly, we have selected several amino acid substitutions associated with severe forms of human adenylosuccinate lyase deficiency and characterized these enzymes both biochemically and biophysically. Such experiments not only provide structural information, but allow for the examination of the underlying biochemistry associated with several severe forms of adenylosuccinate lyase deficiency.
Adenylosuccinate lyase of Thermotoga maritima has been crystallized and the structure published (Toth and Yeates 2000); the B. subtilis and T. maritima enzymes share 50% identity plus 23% similarity at the amino acid sequence level, while the B. subtilis and human adenylosuccinate lyases share 27% identity plus 17% similarity. Based on this similarity, we previously constructed a homology model of the B. subtilis enzyme using the crystal structure of T. maritima adenylosuccinate lyase (Brosius and Colman 2002); the coincidence of the two structures was remarkable. We have now constructed a homology model of the human enzyme, based on the determined crystal structure of the T. maritima adenylosuccinate lyase. Shown in Figure 1A ▶ is the B. subtilis adenylosuccinate lyase superimposed on the human enzyme model. These homology models assist in relating our experimental results on the B. subtilis model system to the human enzyme.
Figure 1.

(A) An overlay of the B. subtilis adenylosuccinate lyase (cyan) and the human enzyme (white) models. Calculating from the α carbons, the two structures have an RMS value of 0.3 Å. (B) Homology model of B. subtilis adenylosuccinate lyase based upon the Thermotoga maritima crystal structure, PDB 1c3c (Toth and Yeates 2000; Brosius and Colman 2002). On each of the four subunits (labeled I–IV and colored red, cyan, green, and yellow, respectively), the B. subtilis residues Met10, Ile123, and Thr367, corresponding to human enzyme mutations, have been displayed. One of the four active site regions has been encircled in white. There is an active site at each intersection of three subunits.
We herein report the characterization of B. subtilis adenylosuccinate lyases containing amino acid substitutions at positions Met10, Ile123, and Thr367, respectively, which correspond to three substitutions associated with severe adenylosuccinate lyase deficiency in humans. The location of these mutations is shown in Figure 1B ▶.
Results
Purity of the adenylosuccinate lyases with amino acid replacements
In human adenylosuccinate lyase, a severe deficiency arises from the following individual point mutations: M26L, R141W, and S395R (Marie et al. 1999; Race et al. 2000; ADSLdb). Sequence and structural alignments allowed us to determine the equivalent residues between the human and B. subtilis enzymes (see Discussion). The corresponding residues in the B. subtilis enzyme are M10, I123, and T367, respectively. Therefore, we constructed M10L, I123W, and T367R in the more stable B. subtilis enzyme. (The discrepancy in the residue numbers of the human and B. subtilis enzymes arises from the presence of a 16 amino acid N-terminal extension present in the human enzyme [ClustalW alignment, see Fig. 7 ▶]. The function of this extension is not known.) Because the sequences of the bacterial and human enzymes are not identical at positions 123 and 367 (B. subtilis numbering), we constructed as controls I123R and T367S, which change the B. subtilis enzyme’s amino acid to the wild-type human residue at the corresponding position. The purity and subunit molecular weight of the resultant enzymes were assessed by SDS-PAGE gels. Each enzyme is pure and has the correct subunit molecular mass of 50 kD (data not shown).
Figure 7.

A complete sequence alignment of adenylosuccinate lyase from B. subtilis, T. maritima, and H. sapiens generated in Clustal W. These three proteins share 16% identity plus 21.3% high similarity and 15% weak similarity. Conserved residues are shown in white font with a black background, highly similar residues are in white font with a dark gray background, and weakly similar residues are in black front with a light gray background. The alignment numbering corresponds to that of the B. subtilis enzyme. The residues of particular interest, that is, Met10, Ile123, and Thr367 (B. subtilis numbering) are indicated by an asterisk (*) below the alignment.
Kinetic parameters in the direction of AMP formation
The wild-type B. subtilis adenylosuccinate lyase has a Vmax value of 1.56 μmole adenylosuccinate/min/mg at pH 7.0 under standard conditions. Compared to this value, the altered enzymes all retain appreciable amounts of activity (Table 1); the I123W enzyme, though, is reduced more than threefold relative to the wild type in its specific activity, suggesting some catalytic alterations. Interestingly, the M10L enzyme appears to be a more effective enzyme than the wild type, with a Vmax value of 1.90 μmole/min/mg. These enzymes are all uncompromised in their affinity for adenylosuccinate. The Km values determined for adenylosuccinate are virtually identical to that of the wild-type enzyme (3.46 μM). Given the relatively high activity and wild-type-like substrate affinity observed for these altered enzymes, it appears that these residues are not directly involved in catalysis.
Table 1.
The kinetic parameters of the adenylosuccinate lyase enzymes in the direction of AMP formationa
| Enzyme | Vmaxb (μmole/min/mg) | KM (μM) | kcat (s−1) | kcat/KM (s−1M−1) |
| WT | 1.56 ± 0.190 | 3.46 ± 0.44 | 1.30 ± 0.160 | 3.75 × 105 |
| M10L | 1.90 ± 0.129 | 3.80 ± 1.14 | 1.59 ± 0.108 | 4.18 × 105 |
| I123R | 1.24 ± 0.051 | 2.18 ± 0.46 | 1.04 ± 0.042 | 4.76 × 105 |
| I123W | 0.55 ± 0.021 | 3.34 ± 0.80 | 0.46 ± 0.018 | 1.38 × 105 |
| T367R | 1.01 ± 0.036 | 1.63 ± 0.36 | 0.85 ± 0.031 | 5.19 × 105 |
| T367S | 0.96 ± 0.032 | 1.56 ± 0.33 | 0.80 ± 0.042 | 5.14 × 105 |
a Activity assays for adenylosuccinate were carried out spectrophotometrically at 282 nm in 50 mM HEPES, pH 7.0 at 25°C. The calculated standard deviation for each parameter is shown.
bVmax is defined as μmole substrate converted/min/mg enzyme.
pH dependence of Vmax for the wild-type and altered adenylosuccinate lyases
Some differences between the wild-type and disease-associated enzymes are observed in the pH rate profiles. Although all the enzymes yield bell-shaped profiles (Fig. 2 ▶), some differ in their pK values (Table 2). The method for determining values for pK1 and pK2 has been previously described (Brosius and Colman 2000). Among all the enzymes examined, pK1 does not vary appreciably in most cases, nor in the cases of M10L, T367R, and T367S enzymes does pK2. However, pK2 is affected by the mutations at position 123; in both the I123R and I123W enzymes, this value is low relative to the wild-type enzyme.
Figure 2.

pH-Vmax rate profiles for the wild type (filled circles), M10L (open squares), I123R (filled triangles), I123W (open triangles), T367R (open diamonds), and T367S (open circles) adenylosuccinate lyase. All experiments were carried out under saturating amounts of adenylosuccinate (300 μM). The calculated pK values are given in Table 2.
Table 2.
pK values determined from pH-Vmaxrate profiles for wild-type and altered adenylosuccinate lyasesa
| Enzyme | pKa | pK2 |
| WT | 6.78 ± 0.08 | 8.37 ± 0.09 |
| M10L | 6.90 ± 0.05 | 8.44 ± 0.02 |
| I123R | 6.93 ± 0.05 | 7.95 ± 0.06 |
| I123W | 6.96 ± 0.08 | 8.02 ± 0.09 |
| T367R | 7.14 ± 0.07 | 8.17 ± 0.08 |
| T367S | 6.61 ± 0.04 | 8.45 ± 0.04 |
a The calculated standard error for each pK value is shown.
SAICAR/SAMP ratios of the wild-type and disease-associated enzymes
It has been suggested in the literature that, depending on the phenotypic severity of the disease in patients with adenylosuccinate lyase deficiency, the SAICAR and adenylosuccinate activities of the enzyme are differentially altered (Jaeken et al. 1988). In those patients with phenotypically mild adenylosuccinate lyase deficiency, there is a four to five times greater concentration in the body fluids of succinyladenosine than SAICA-riboside; whereas in severe cases, the ratio of the concentrations of the two substrate degradation products is approximately equal. To evaluate the effects of mutations at positions Met10, Ile123, and Thr367, the enzyme activities for both substrates were determined. The ratio of these activities is compared in Table 3. Experiments were carried out in the presence of 90 μM of either substrate, which is saturating relative to the Km (see Table 1 for SAMP values). Km values for SAICAR were determined for the wild type (6.40 ± 0.47 μM) and each altered protein: 10.7 ± 1.03 μM (M10L), 8.37 ± 1.51 μM (I123R), 5.37 ± 0.91 μM (I123W), 13.26 ± 1.47 μM (T367R), and 14.37 ± 1.69 μM (T367S). None of the Km values differs substantially from that of the wild type. The ratio of specific activities of SAICAR/SAMP for the wild type is 1.27. Each altered enzyme has an activity ratio essentially the same as the wild type (~1), indicating that these particular mutations do not impair either activity relative to the other. These results also further support the use of the B. subtilis enzyme as a model for the disease-associated human enzymes, as the altered B. subtilis enzymes yield ratios consistent with that reported in the literature for severe human adenylosuccinate lyase deficiency.
Table 3.
The SAICAR and adenylosuccinate activities of the altered enzymes and wild typea
| Enzyme | SAMP activity μmole/min/mg | SAICAR activity μmole/min/mg | SAICAR/SAMP |
| WT | 1.56 | 1.94 | 1.27 |
| M10L | 1.90 | 1.72 | 0.91 |
| I123R | 1.24 | 1.14 | 0.92 |
| I123W | 0.55 | 0.50 | 0.91 |
| T367R | 1.01 | 1.00 | 0.99 |
| T367S | 0.96 | 0.95 | 0.99 |
a All activities were determined under standard conditions, at pH 7.0, in the presence of either 90 μM adenylosuccinate or SAICAR. The data presented represents an average of at least three activity determinations.
Circular dichroism spectroscopy of the wild-type and altered adenylosuccinate lyases
Because most of the kinetic parameters of these enzymes do not differ appreciably from that of the wild type, circular dichroism was used to probe for differences in secondary structure. The circular dichroism spectra of the altered and wild-type adenylosuccinate lyases at room temperature, each at a concentration of 0.45 mg/mL, are shown in Figure 3 ▶. All of the enzymes exhibit minima at 208 and 222 nm, typical of proteins containing appreciable amounts of α-helix. However, some of these enzymes exhibit differences in their relative intensities. The greatest change is observed in the circular dichroism spectra of I123W adenylosuccinate lyase: In this case, [θ]222 nm is only −9710 deg cm2 dmole−1 compared with −15,200 deg cm2 dmole−1 for the wild type, indicating a decreased amount of helix as a result of the mutation. More subtly altered are the spectra of the I123R and T367S, which appear to actually contain slightly more helix than the wild type. The results suggest that these mutations yield a structurally, rather than a catalytically, impaired enzyme.
Figure 3.

Circular dichroism spectra for the wild type (filled circles), M10L (open squares), I123R (filled triangles), I123W (open triangles), T367R (open diamonds), and T367S (open circles) adenylosuccinate lyase. Each protein is in 20 mM KPO4, containing 20 mM NaCl, pH 7.0, at a final concentration of 0.45 mg/mL.
Thermostability studies of the wild-type and disease-associated adenylosuccinate lyases
Based on both the crystal structure and homology model of the enzyme, the residues we are investigating are not in close proximity to the active site (designated by the white oval in Fig. 1B ▶). Again, it seems likely that these amino acids play an important structural role within the enzyme. To explore this possibility further, thermostability studies of the enzyme at 42.5°C were conducted (Fig. 4 ▶). The temperature, 42.5°C, was selected because it gave the largest differences between the enzymes in a convenient time period. (All other experiments were carried out at 25°C, a temperature at which all the enzymes are stable for hours.) As shown in Figure 4 ▶, the wild-type adenylosuccinate lyase is not completely stable at 42.5°C over the time period studied. However, the altered enzymes, with the exception of T367S, exhibit marked instability relative to the wild type. For example, T367R loses activity with a kobs of 0.16 ± 0.0056 min−1, compared to a wild-type value of 0.0082 ± 0.00050 min−1.
Figure 4.

Rate of inactivation at 42.5°C of the wild-type and altered enzymes. In (A), the experiment was conducted in 20 mM KPO4, 20 mM NaCl, pH 7.0 buffer, whereas in (B) 10% glycerol was also present. Wild type (filled circles), M10L (open squares), I123R (filled triangles), I123W (open triangles), T367R (open diamonds), and T367S (open circles). (Identical symbols are used in [A] and [B] for the enzymes). All enzymes are at a concentration of 1 mg/mL. The enzyme activities are expressed as E/E0 (observed activity/initial activity).
We have shown with the wild-type enzyme, using the technique of light scattering, that glycerol can be used to stabilize the tetrameric form of the enzyme, resulting in greatly enhanced or even complete stability of altered enzymes (J.B. Palenchar and R.F. Colman, unpubl.). Thus, we carried out the thermal stability experiments in 10% glycerol (data not shown). Under these conditions, the wild-type and T367S adenylosuccinate lyases are essentially completely stable over the time period studied. The other altered enzymes are more stable in the presence compared with the absence of glycerol, but still lose some activity at 42.5°C, perhaps indicating the magnitude of structural abnormality arising from these single amino acid substitutions.
Native gel electrophoresis to determine molecular weight and conformation
To explore further the structural impairment of these altered enzymes, native gel electrophoresis was used to examine both molecular weight and enzyme conformation. In Figure 5 ▶, a representative native gel (at 7% polyacrylamide) of the adenylosuccinate lyases is shown. Based on the gels run (from 6%–11% polyacrylamide), the wild-type enzyme has an apparent molecular mass of 209 kD (the tetramer has a theoretical mass of 200 kD). M10L, I123R, and T367S have observed molecular masses similar to the wild type (220 kD, 219 kD, and 202 kD, respectively), and migrate similarly indicating their overall conformation is not substantially altered. However, both the I123W and T367R show differences from the wild-type enzyme. I123W (lane 4) appears to contain large aggregates that did not enter the gel; the concentration of this enzyme added to the gel was identical to that of the wild type, but the concentration in the major stained band is lower than that of the wild type, and notable amounts of enzyme can be seen at the top of the gel. The T367R (lane 5) enzyme exhibited multiple bands on the native gel indicating it, too, forms higher molecular weight species. (Given the distribution of molecular weight species associated with some of these enzymes, techniques such as light scattering and FPLC were not readily interpreted.)
Figure 5.

Native gel electrophoresis (7% polyacrylamide) of the wild-type and altered adenylosuccinate lyases to examine molecular weight and conformation. Only the adenylosuccinate lyases are shown in this figure: (1) wild type, (2) M10L, (3) I123R, (4) I123W, (5) T367R, and (6) T367S. As calculated from the major bands, the molecular masses are 209 kD, 220 kD, 223 kD, 246 kD, and 202 kD, respectively. (Molecular mass standards not shown.)
Enzyme subunit complementation
Previously, for adenylosuccinate lyase, enzyme complementation studies involved the formation of an active hybrid enzyme from two inactive, stable enzymes, each containing a different amino acid substitution; these allowed the identification of the subunit source of residues in the active site (Lee et al. 1999; Brosius and Colman 2002). However, because the altered enzymes described here retain appreciable activity and, because the residues are located far from the active site, such activity-based complementation experiments would be difficult to interpret. Thus, complementation was approached using changes in thermal stability. We hypothesized that a wild type-mutant hybrid enzyme would be more stable than the altered enzyme, and should thus be more resistant to denaturation than the altered enzyme alone. Complementation in terms of enzyme stability has been demonstrated for argininosuccinate lyase (Yu et al. 2001), another member of the fumarase superfamily. Briefly, equal concentrations of one altered enzyme and the wild-type adenylosuccinate lyase were mixed under mild conditions and allowed to incubate at 25°C for 90 min. This time frame and temperature provided maximum reactivation (and, thus, hybridization) with previous enzyme pairs (Lee et al. 1999; Brosius and Colman 2002). After allowing for hybridization, the enzyme pair was transferred to 42.5°C, and the activity was measured as a function of time.
The results of adenylosuccinate lyase complementation between the wild-type and each altered enzyme, as measured by thermal stability, are summarized in Figure 6 ▶. These graphs have been normalized to 100% of the original activity for each enzyme at time zero. (T376S is not shown as this enzyme is similar in stability to the wild-type enzyme.) Using the results for each individual altered and wild-type enzyme, the theoretical line representing no enzyme complementation was calculated (dashed line): In the absence of complementation, the observed activity at each time point should be an average of that for the separate altered and wild-type enzymes. The theoretical lines appear biphasic, with the less stable enzyme rapidly losing all of its activity (i.e., 50% of the total normalized activity), while the more stable enzyme (also representing 50% of the total normalized activity) loses activity more slowly. Deviations from this theoretical line would represent complementation. The experimental data for the mixtures of enzymes appear to follow linear rates of inactivation. For M10L, I123R, I123W, and T367R, the hybrid enzyme deviates from the theoretical line, indicating that complementation is occurring. The exact composition of the hybrids has not been determined, although they can be predicted on the basis of random dissociation and reassociation (Lee et al. 1999). The results demonstrate that stability or instability can be communicated between the altered and wild-type subunits within a hybrid tetramer.
Figure 6.

Thermostability complementation studies at 42.5°C. In all panels, the wild type is represented by filled circles, and the theoretical line representing no complementation is dashed. (A) Wild type, M10L (filled squares), and M10L and WT (open squares) hybrid enzyme. (B) Wild type, I123R (filled squares), and I123R and WT (open squares) hybrid enzyme. (C) Wild type, I123W (filled squares), and I123W and WT (open squares) hybrid enzyme. (D) Wild type, T367R (filled squares) and T367R and WT (open squares) hybrid enzyme. The T367S enzyme is not shown, as it is as stable as the wild-type enzyme. The enzyme activities are expressed as E/E0 (observed activity/initial activity).
Discussion
The importance of three amino acid residues in B. subtilis adenylosuccinate lyase, Met10, Ile123, and Thr367, corresponding to positions in the human enzyme implicated in adenylosuccinate lyase deficiency, have been evaluated in this article by site-directed mutagenesis, and the enzymes with amino acid substitutions were characterized both biochemically and biophysically. These residues were chosen because they are associated with a severe adenylosuccinate lyase deficiency in humans (Race et al. 2000; ADSLdb), and should thus allow a study of the biochemical features of enzymes giving rise to human disease. B. subtilis adenylosuccinate lyase was selected as a model system in which to study the aforementioned mutations. We consider that the use of the B. subtilis enzyme offers a reasonable approach, given the instability of the human enzyme. Additionally, the previous studies with the human recombinant enzyme had to be conducted in the presence of a thioredoxin fusion protein (Race et al. 2000) or maltose binding protein (Kmoch et al. 2000). A sequence alignment of adenylosuccinate lyases from the three species relevant to this work (B. subtilis, Homo sapiens, and T. maritima) is shown in Figure 7 ▶. Although the overall identity plus strong similarity of amino acid sequences of B. subtilis and human adenylosuccinate lyases is 27% plus 17%, respectively, the amino acid sequences are similar in the regions of the three amino acids replaced in this study. Furthermore, the models of the human and B. subtilis adenylosuccinate lyases superimpose well structurally (an RMS value of 0.30 Å was calculated for the overlay of the two models); only minor deviations are noted and these are located away from the three residues being examined (Fig. 1B ▶). This structural agreement between the two enzymes supports our selection of the amino acids in the B. subtilis enzyme, which correspond to the replaced amino acids in human adenylosuccinate lyase.
In human adenylosuccinate lyase deficiency, Met26 is mutated to a leucine. In the sequence alignment (Fig. 7 ▶) Met10 of the B. subtilis enzyme corresponds to Met26 of the human enzyme. Of the more than 40 species from which adenylosuccinate lyase has been sequenced, the majority contain a methionine at this position, with only a few containing conservative substitutions to valine, isoleucine, or leucine (Fig. 7 ▶). Furthermore, the overlay of the structural models of the human and B. subtilis enzymes indicates that the backbones of the two enzymes are totally superimposable in this region of the protein and that the two methionines completely overlap (Fig. 8A ▶1). Thus, M10L B. subtilis adenylosuccinate lyase was constructed and characterized.
Figure 8.
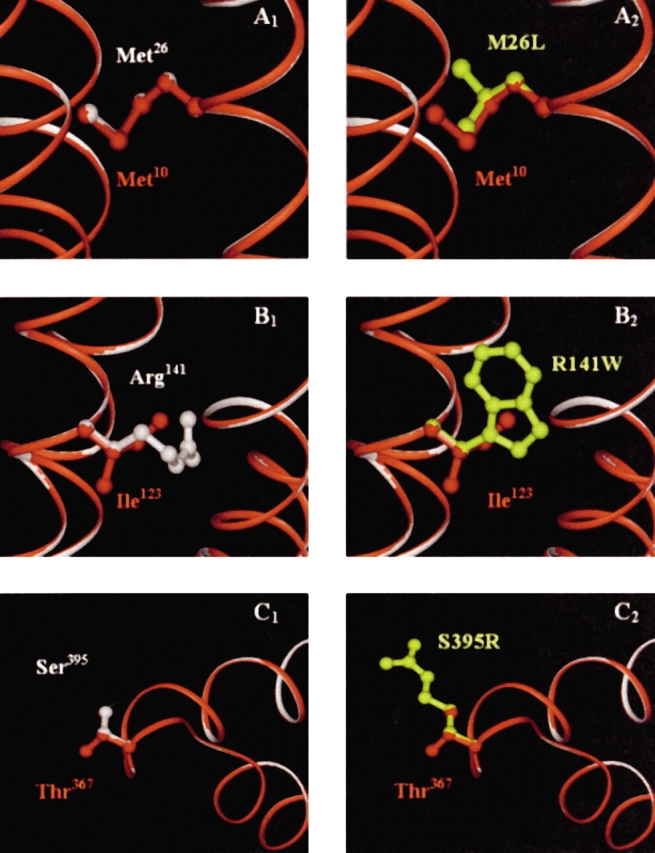
(A–C) An overlay of the human and B. subtilis enzyme homology models. The B. subtilis polypeptide backbone is shown in red, while the human backbone is in white. In (A1), Met10 of B. subtilis (red) and the corresponding wild-type residue in human, Met26 (white) are shown to completely superimpose. (A2) Shows the amino acid substitution to leucine (yellow) in the defective human enzyme. (B1) Shows Ile123 (B. subtilis, red) and Arg141 (wild-type human, white), which overlap through the β-carbons of both arginine and isoleucine. Replacement by a tryptophan (yellow) in the defective human enzyme is shown in (B2). Thr367 (B. subtilis, red) and Ser395 (wild-type human, white) are shown in (C1). These residues completely overlay, except for the additional methyl group of threonine. (C2) Shows the substitution to an arginine (yellow) in the defective human enzyme. The backbones of the two enzymes are completely superimposable in these regions.
Ile123 of the B. subtilis enzyme corresponds to human Arg141 (Fig. 7 ▶), which is mutated to a tryptophan in the deficient enzyme. Again, the human and B. subtilis enzyme models indicate complete superposition of the protein backbone in this region, and isoleucine and arginine overlay through the β-carbon of each (Fig. 8B ▶1). Although this position does not have complete conservation (Fig. 7 ▶), nearly all species have either a positively charged residue (Arg or Lys) or an aliphatic hydrophobic residue (Val, Ile, Ala). Conspicuously absent at this position among the known natural sequences are the larger, aromatic hydrophobic residues such as phenylalanine, tyrosine, or tryptophan. We thus constructed and characterized I123R (to mimic the human wild-type enzyme) and I123W (to mimic the abnormal human enzyme).
Finally, Thr367 of B. subtilis corresponds to the human residue Ser395 (Fig. 7 ▶), which is mutated to an arginine in the deficient state. Sequence conservation is good at this position, with the majority of species containing a threonine, serine, or glycine. The structural superposition of the backbones of the human and B. subtilis enzymes is also excellent in this region (Fig. 8C ▶1); the Thr367 and Ser395 overlap well structurally, except for the extra methyl group of threonine. Therefore, we constructed and characterized both T367S (to mimic the wild-type human enzyme) and T367R (to mimic the abnormal human mutation).
Determination of the kinetic parameters for these enzymes yielded results largely consistent with the hypothesis in the literature that severe deficiency arises from structurally impaired enzymes (Race et al. 2000; Ciardo et al. 2001). The Vmax and Km values of these disease associated enzymes are very similar to those of the wild-type enzyme both for adenylosuccinate (Table 1) and SAICAR (Table 3). The greatest decrease (threefold) in Vmax is for the I123W enzyme, which indicates that there is some perturbation of the catalytic reaction. It should also be noted that the altered proteins intended to mimic the wild-type human enzyme do exhibit activities close to that of the wild type (I123R and T367S). Additionally, the pH-Vmax rate profiles for each enzyme (Fig. 2 ▶) revealed values for pK1 in the altered enzymes that, with one exception, are very similar to that of the wild-type enzyme. The values obtained for pK2 in M10L and T367S are almost the same as that of the wild type, while the remaining enzymes (I123R, I123W, and T367R) have somewhat lower pK2 values. Previous work in our laboratory has identified pK2 as a composite, representing the deprotonation of both His68 and His89, which are important residues within the active site of this enzyme (Lee et al. 1998; Brosius and Colman 2000). These relatively small shifts in pK are probably not the result of a direct influence on the catalytic reaction; rather, they likely reflect secondary effects that can be explained in terms of changes in the enzyme’s structure (see below).
A further indication that amino acid substitutions at Met10, Ile123, and Thr367 do not largely impair the enzyme catalytically is found in the ratio of enzyme activity toward the two substrates, SAMP and SAICAR. Each enzyme has a SAICAR/SAMP activity ratio of approximately 1, indicating that both substrates are handled equally well. Not only is this ratio equal, but all altered enzymes retain substantial amounts of activity toward both substrates relative to the wild-type. Had there been major catalytic impairment, we would have expected more substantial loss of enzyme activity.
In contrast, the biophysical studies of the Met10, Ile123, and Thr367 altered proteins have revealed more notable departures from the wild-type adenylosuccinate lyase behavior than did the kinetic characterization. If Met10 of B. subtilis adenylosuccinate lyase is replaced by a leucine, corresponding with adenylosuccinate lyase deficiency, the largest deviations from the wild-type enzyme are found in the thermostability studies. M10L is markedly unstable, and rapidly loses activity at 42.5°C. This instability is not completely eliminated by the addition of glycerol (which is known to stabilize the enzyme tetramer), suggesting there is some alteration in the enzyme’s structure. However, this enzyme exhibits essentially the same electrophoretic mobility on native gels as does the wild type, showing that it can form a tetrameric species. Additionally, the circular dichroism spectrum of M10L adenylosuccinate lyase is superimposable upon that of the wild-type enzyme, indicating the type and amount of secondary structure is not substantially altered due to the mutation. These structural changes must be more subtle than detectable by this spectroscopic method.
Adenylosuccinate lyase, and other members of the superfamily, contains a core 20-helix bundle in the center of the protein (Fig. 1 ▶). Met10 is located in the N-terminal region of the enzyme, and is oriented inward towards an adjacent helix that is part of the core bundle. Replacing Met10 with a leucine does change the hydrophobicity; however, recent reports in the literature suggest that this replacement has yet a larger impact. The nonbonded interaction between the sulfur of the methionine side chain and the oxygen of the carbonyl backbone on the adjacent helix may play a role in protein stability (Iwaoka et al. 2001, 2002a, 2002b). Such nonclassical interactions are thought to weakly stabilize proteins through the π(C=O)→σ*(S) orbital overlap. This type of interaction is thought to be as strong as 2.5 kcal/mole. If the various rotamers of Met10 are examined in both the homology model and the T. maritima crystal structure, there is a close interaction between two such groups; the thioether of the Met10 is ~3.2 Å from the carbonyl oxygen of Thr317 on the neighboring helix. Indeed, we observed significant protein instability upon substituting Met10 with a leucine, suggesting the sulfur of this methionine does play a role in stabilizing in the protein through a nonbonded interaction with the adjacent helix of the core bundle.
Of the three amino acids examined, the isoleucine to tryptophan mutation (the disease-associated mutation) results in the most drastic structural aberrations in the enzyme. These changes were observed by circular dichroism, by native gel electrophoresis, and by thermostability measurements. Although I123W adenylosuccinate lyase retains a spectrum typical of a helical protein, its molar ellipticity at 222 nm is only about 60% that of the wild-type enzyme, signifying the amount of helix has been reduced. Native gel electrophoresis indicates that, although the major band for the I123W enzyme corresponds to the molecular weight of tetramer, higher molecular weight species corresponding to aggregated protein are also observed. In addition, the I123W enzyme is inactivated at 42.5°C at a rate about seven times greater than the wild type, indicative of its thermal instability both in the presence and absence of glycerol. (This mutation to a tryptophan in the human recombinant enzyme was described by Race et al. [2000] to yield thermostable enzyme. The studies of Race et al. used adenylosuccinate lyase fused to thioredoxin, and it is likely that the relative stability was imparted by the attached thioredoxin protein. It should be noted, however, that all of these fusion proteins lost activity upon storage and fresh enzyme had to be purified for each experiment.) Furthermore, enzyme structural impairment is the most likely explanation for the altered pK2 values observed in the pH rate profiles for the enzymes with substitutions for Ile123. Adenylosuccinate lyase has a multisubunit active site composed of three subunits (Brosius and Colman 2002). If the positioning of the monomers within the tetramer is even slightly altered, the enzyme groups reflected in the pH rate profiles could be perturbed, resulting in small changes in the pK values.
The I123R enzyme, which corresponds to the wild-type human enzyme, is much more similar to the wild type by circular dichroism. Additionally, native gel electrophoresis reveals this enzyme exists solely as a tetramer, and does not have the aggregation problems associated with mutation to tryptophan. Unexpectedly, I123R adenylosuccinate lyase is less thermally stable than the wild type. The presence of the arginine at this position in the wild-type human enzyme may, in fact, contribute to the seemingly greater instability inherent in the wild-type human enzyme. Perhaps the introduction of an Arg at position 123 of the B. subtilis enzyme causes the instability associated with the human wild-type protein. In the bacterial enzyme, this instability may arise from subtle structural perturbations, which are too minor to observe by either circular dichroism or native gel electrophoresis.
Ile123 of adenylosuccinate lyase is located within the core helix bundle of the enzyme. Mutating Ile123 to a large tryptophan is akin to introducing a “molecular wedge” within the helix bundle; there is little room to accommodate such a large amino acid (Fig. 8B ▶2). The unfavorable interactions associated with all tryptophan rotamers at position 123 will probably disrupt the helix bundle, leading to structural alterations at the secondary, tertiary, and quaternary levels, as evidenced by changes in circular dichroism, native gels, and thermostability.
An amino acid replacement of Thr367 by an arginine (corresponding to the human disease) also gives rise to a structurally impaired adenylosuccinate lyase. Although the circular dichroism spectrum of T367R adenylosuccinate lyase is similar to the wild type, native gel electrophoresis indicates that this enzyme aggregates to multiple higher molecular weight species. The T367R enzyme, also exhibits striking thermal instability, which is only partially alleviated by glycerol. (This mutation to an arginine in the human recombinant enzyme was also classified by Race et al. as thermally stable, although it lost activity at 53°C more rapidly than the wild-type enzyme. Again, it is likely that the relative stability observed was due to the attached fusion protein.) This structural aberration is probably responsible for the small shifts in pK values observed for the T367R enzyme.
The wild-type human enzyme contains a serine at the position equivalent to 367. The T367S B. subtilis enzyme we constructed behaves similarly to the wild type in every aspect we examined. The contrasting properties of the T367S and T367R enzymes suggest that the introduction of a positively charged residue at position 367 is not well tolerated. Thr367 is located on the protein surface, away from helix bundle (Figs. 1 ▶, 8 ▶C1). The larger arginine introduced at this position could hinder proper enzyme function by folding in towards the enzyme’s interior and engaging in electrostatic interactions that would normally not be present, thus introducing rigidity into a region that may require flexibility. Alternatively, the additional positive surface charge could cause aggregation between adenylosuccinate lyase molecules because of new electrostatic attractions. In vivo, a similar electrostatic effect could result in unfavorable interactions with other proteins.
Our experimental results, combined with the homology modeling and sequence alignments, support the idea that complete amino acid conservation at these positions is not essential; rather, certain types of amino acids (e.g., large hydrophobic or positively charged residues) are not tolerated in these regions. Residues Met10, Ile123, and Thr367, and the surrounding local area, are important for maintaining the structure required for a stable, functional enzyme. Unfavorable substitutions at these positions give rise to structurally altered adenylosuccinate lyases.
Finally, we have observed that enzyme instability associated with these amino acid substitutions can, in some cases, appear to be decreased by subunit complementation experiments using the wild-type enzyme. If there is dissociation of both the wild type and an amino acid-substituted enzyme, followed by random reassociation, hybrid enzymes are generated containing varying numbers of each type of subunit. (For all possible hybrid combinations, see Lee et al. 1999.) Enzyme subunit complementation does occur in these cases, as monitored by thermal stability; each pair behaves differently from that expected in the absence of complementation. That the different pairs examined exhibited varying levels of stability may be reflective of incomplete randomization, or may indicate that particular mutations destabilize the hybrid enzyme more than others. These complementation experiments may have implications for the in vivo behavior of adenylosuccinate lyase. The experiments were performed under extremely mild conditions (i.e., subunit randomization took place at 25°C in phosphate buffer, pH 7.0), suggesting that complementation could occur within the body. Most patients with adenylosuccinate lyase deficiency are not homozygous for the mutations; rather, the majority are heterozygous and have either two enzymes with two different amino acid substitutions, or both wild-type and an enzyme with an amino acid replacement (ADSLdb). Enzyme subunit complementation in vivo may be responsible for lessening the degree of disease severity.
Through a set of mutations at positions Met10, Ile123, and Thr367 of B. subtilis adenylosuccinate lyase, we have constructed amino acid substitutions that are equivalent to those associated with severe human adenylosuccinate lyase deficiency. These mutations have provided insights into the biophysical basis of three forms of the disease. Our results demonstrate that these residues are at critical positions within the tetramer, and can unfavorably influence enzyme structure at both the secondary and quaternary levels, ultimately resulting in enzyme instability.
Materials and methods
Materials
From Sigma, adenylosuccinate (SAMP), aminoimidazole carboxamide ribotide (AICAR), adenosine 5′-monophosphate, fumarate, MES1, HEPES, TAPS, and imidazole were obtained. The protein assay concentrate was from BioRad. Oligonucleotides for site-directed mutagenesis and sequencing (nonfluorescent) were obtained from BioSynthesis. Fluorescent oligonucleotides for sequencing were from Li-Cor. All other chemicals were of reagent grade.
Site-directed mutagenesis
The Stratagene QuikChange mutagenesis kit was used to introduce single amino acid substitutions to the pBHis plasmid, which encodes Bacillus subtilis adenylosuccinate lyase as previously described (Redinbo et al. 1996; Lee et al. 1997). The following oligonucleotides and their complements were used: Met10: CAA CAG CTG AAC TGT CCG CGA TTT GG (Leu); Ile123: CAG ATT TGT TGA CTG GAT AAA AGA AAA AGC (Trp); CAG ATT TGT TGA CCG TAT AAA AGA AAA AGC G (Arg); Thr367: GAC ACA GGC CTG CGT CGT GAA GAA GC (Arg); GAC ACA GGC CTG TCT CGT GAA GAA GC (Ser). DNA sequencing was performed at either the University of Delaware Cell Biology Core Facility using a Long-Readir 4200 DNA sequencer (Li-Cor) or at the Delaware Biotechnology Institute, and University of Delaware Center for Agricultural Biotechnology using an ABI Prism model 377 DNA sequencer (PE Biosystems).
The protein contains a six-His tag at the N terminus, was overexpressed in E. coli strain BL21(DE3), and was purified using Qiagen Ni-NTA agarose as previously described (Redinbo et al. 1996; Lee et al. 1997). The purity of the resultant enzymes was assessed by 12% polyacrylamide gels containing 0.1% sodium dodecyl sulfate (Laemmli 1970). After purification, the enzyme was aliquoted, rapidly frozen, and stored at −80°C in 20 mM sodium phosphate containing 20 mM sodium chloride and 0.05% sodium azide, pH 7.0. Full enzyme activity was retained for at least 6 months after the purification. Protein concentrations were determined either by the method of Bradford (1976) using the wild-type adenylosuccinate lyase as the standard, or by absorbance at 280 nm using E2801% = 10.6 (Lee et al. 1997).
SAICAR synthesis
SAICAR was synthesized enzymatically starting from AICAR (10 mM) and fumarate (150 mM), using adenylosuccinate lyase (0.4 mg/mL) in 50 mM HEPES, pH 7.0 (1 mL total volume). Reaction progress was monitored by PEI cellulose plates using 1 M ammonium acetate, pH 6.40, as solvent (Stone et al. 1993). After approximately 2 h, the enzyme was removed from the reaction mixture using a Centricon-10 filtration device (Millipore). The substrates were then applied to a DEAE-cellulose column (40 mL resin) equilibrated with 10 mM ammonium bicarbonate, and eluted using a 10 mM (1 L) to 300 mM (1 L) ammonium bicarbonate linear gradient. To desalt the product, a Dowex 50 column (BioRad AG 50W-X4, 100–200 mesh, hydrogen form, 40 mL) was used. SAICAR was eluted using water. The final product was obtained in 13% yield.
Kinetics of B. subtilis adenylosuccinate lyases
Prior to activity determination, each enzyme was incubated at a minimum concentration of 0.4 mg/mL in 20 mM potassium phosphate, containing 20 mM sodium chloride, pH 7.0, for 30 min at 25°C. Adenylosuccinate activity is monitored by the time-dependent decrease in absorbance at 282 nm as adenylosuccinate is converted to AMP, using the difference extinction coefficient of 10,000 M−1cm−1 (Tornheim and Lowenstein 1972). As SAICAR is converted to AICAR, an increase in absorbance may be followed at 267 nm using the difference extinction coefficient of 700 M−1cm−1 (Woodward and Braymer 1966). Standard assay conditions consist of 50 mM HEPES, pH 7.0, at 25°C, with 60 μM adenylosuccinate. Specific activity is defined as μmole substrate converted per minute per milligram enzyme when either substrate is utilized.
The kcat and Km values were determined for both SAICAR and adenylosuccinate by varying the substrate concentration. The Michaelis-Menten plots were analyzed, and error estimates calculated, using SigmaPlot software (SPSS Inc.). For the determination of the ratios of SAMP and SAICAR activity, 90 μM of either substrate was used to ensure saturating conditions. At adenylosuccinate concentrations greater than 150 μM in the assay mixture, substrate conversion was monitored at 290 nm using a difference extinction coefficient of 4050 M−1cm−1, due to high absorbance at 282 nm.
The pH dependence of Vmax was measured for the wild-type and altered enzymes, using the buffers MES (pH 6.3–6.9), HEPES (pH 6.8–8.0), and TAPS (pH 7.9–8.8), each constant in the basic species of the buffer (0.03 M). Adenylosuccinate, at a saturating concentration of 300 μM, was present in the assay mixture. The pH of each assay solution was measured after the rate determination. Data was analyzed using SigmaPlot software.
For the thermostability studies, each enzyme was preincubated for 30 min at 25°C in 20 mM potassium phosphate pH 7.0, containing 20 mM sodium chloride. After this incubation, the enzyme was transferred to a 42.5°C water bath, and activity was assayed under standard conditions as a function of time. This temperature was chosen because it gives the largest difference in behavior between the wild-type and altered enzymes in the shortest time period.
Circular dichroism spectroscopy
Experiments were performed using a Jasco J-710 spectropolarimeter. The samples were scanned five times, averaged, and the background buffer spectrum subtracted out. The mean molar ellipticity was then calculated from the following equation: [θ] = θ/10 nCl, where [θ] is the measured ellipticity (millidegrees), n is the number of amino acids in one subunit (437, including the six His tag), C is the molar concentration of enzyme subunits, and l is the pathlength in centimeters (0.1 cm).
Molecular weight determination
The molecular weights of both the wild-type and altered proteins were determined by native gel electrophoresis (Hedrick and Smith 1968; Brosius and Colman 2002). Gels were composed of 20 mM potassium phosphate, 0.115% (v/v) N,N,N′, N′-tetramethylethylenediamine, 0.2 mg/mL ammonium persulfate, and appropriate volumes of H2O and acrylamide:bis-acrylamide to yield gels with final acrylamide concentrations ranging from 6%–11%. Each gel had a final pH of 7.0. Adenylosuccinate lyase has a theoretical pI of 6.13 (including the six-histidine tag); thus, the gels were run with the anode at the bottom of the gel. The electrophoresis buffer was 20 mM potassium phosphate, pH 7.0. Bromophenol blue served as the tracking dye. The gels were run at 25°C, at 25 mA, until the tracking dye almost reached the bottom of the gel (approximately 2 h). The molecular masses of all enzymes were determined by the method of Hedrick and Smith. Ovalbumin (45 kD), bovine serum albumin (67 kD), alcohol dehydrogenase (141 kD), and ferritin (450 kD) were used as the molecular weight standards. All samples and standards were prepared in 20 mM potassium phosphate, pH 7.0, containing 10% glycerol.
Enzyme subunit complementation as measured by thermostability
The wild type paired with each individual altered enzyme was tested for complementation by monitoring thermostability. For comparison, the thermostability of each individual enzyme was monitored in parallel with the mixture of the wild-type and altered protein. Samples had a final concentration of 1.4 mg/mL (0.7 mg/mL wild type plus 0.7 mg/mL altered enzyme for the mixture), and were incubated for 90 min at 25°C in 20 mM potassium phosphate, containing 20 mM sodium chloride, pH 7.0. The incubated enzyme(s) was then transferred to a 42.5°C water bath, and activity was monitored as a function of time under standard conditions.
Homology modeling of human adenylosuccinate lyase
Using the crystal structure of T. maritima adenylosuccinate lyase (Toth and Yeates 2000) as a template (PDB 1c3c), a homology model of human adenylosuccinate lyase was created. Homology modeling methods for B. subtilis adenylosuccinate lyase have been described elsewhere (Brosius and Colman 2002). With the exception that the 16 additional N-terminal amino acids of the human enzyme were truncated prior to modeling, this method was unaltered.
Acknowledgments
This work has been supported by NIH 1-R01-DK 60504 (R.F.C.), NIH Training Grant T32 GM-08550 (for J.B.P.), and the University of Delaware HHMI Undergraduate Biological Sciences Education Program (for J.M.C.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AICAR, aminoimidazole carboxamide ribotide
SAICAR, succinylaminoimidazole carboxamide ribotide
SAMP, adenylosuccinate
MES, 2-(N-morpholino) ethanesulfonic acid
HEPES, N-(2-hydroxyethyl) piperazine-N′-2-ethanesulfonic acid
TAPS, (N-tris)-[hydroxymethyl] methyl-3-aminopropane sulfonic acid
ADSLdb, Adenylosuccinate Lyase Mutations Database Homepage
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0303903.
References
- ADSLdb: Adenylosuccinate lyase Mutations Database Home Page (http://www.icp.ucl.ac.be/adsldb/mutations.html
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brosius, J.L. and Colman, R.F. 2000. A key role in catalysis for His89 of adenylosuccinate lyase of Bacillus subtilis. Biochemistry 39 13336–13343. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Three subunits contribute amino acids to the active site of tetrameric adenylosuccinate lyase: Lys268 and Glu275 are required. Biochemistry 41 2217–2226. [DOI] [PubMed] [Google Scholar]
- Ciardo, F., Salerno, C., and Curatolo, P. 2001. Neurologic aspects of adenylosuccinate lyase deficiency. J. Child Neurol. 16 301–308. [DOI] [PubMed] [Google Scholar]
- Hedrick, J.C. and Smith, A.J. 1968. Size and charge isomer separation and estimation of molecular weights of proteins by disc gel electrophoresis. Arch. Biochem. Biophys. 126155–164. [DOI] [PubMed] [Google Scholar]
- Iwaoka, M., Takemoto, S., Okada, M., and Tomoda, S. 2001. Statistical characterization of nonbonded S—O interactions in proteins. Chem. Lett. 132–133.
- ———. 2002a. Weak nonbonded S—X (X=O, N, and S) interactions in proteins. Statistical and theoretical studies. Bull. Chem. Soc. Jpn. 75 1611–1625. [Google Scholar]
- Iwaoka, M., Takemoto, S., and Tomoda, S. 2002b. Statistical and theoretical investigations on the directionality of nonbonded S—O interactions. Implications for molecular design and protein engineering. J. Am. Chem. Soc. 124 10613–10620. [DOI] [PubMed] [Google Scholar]
- Jaeken, J. and Van den Berghe, G. 1984. An infantile autistic syndrome characterized by the presence of succinylpurines in body fluids. Lancet 2 1058–1061. [PubMed] [Google Scholar]
- Jaeken, J., Wadman, S.K., Duran, M., van Sprang, F.J., Beemer, F.A., Holl, R.A., Theunissen, P.M., de Cock, P., Van den Berghe, F., Vincent, M.-F., et al. 1988. Adenylosuccinase deficiency: An inborn error of purine nucleotide synthesis. Eur. J. Pediatr. 148 125–131. [DOI] [PubMed] [Google Scholar]
- Kmoch, S., Hartmannova, H., Stiburkova, B., Krijt, J., Zikanova, M., and Sebesta, I. 2000. Human adenylosuccinate lyase (ADSL), cloning and characterization of full-length cDNA and its isoform, gene structure and molecular basis for ADSL deficiency in six patients. Hum. Mol. Genet. 9 1501–1513. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lee, T.T., Worby, C., Dixon, J.E., and Colman, R.F. 1997. Identification of His141 in the active site of Bacillus subtilis adenylosuccinate lyase by affinity labeling with 6-(4-bromo-2, 3-dioxobutyl)thioadenosine 5′-monophosphate. J. Biol. Chem. 272 458–465. [PubMed] [Google Scholar]
- Lee, T.T., Worby, C., Bao, Z.Q., Dixon, J.E., and Colman, R.F. 1998. Implication of His68 in the substrate site of Bacillus subtilis adenylosuccinate lyase by mutagenesis and affinity labeling with 2-[(4-bromo-2, 3-dioxobutyl)thio]adenosine 5′-monophosphate. Biochemistry 37 8481–8489. [DOI] [PubMed] [Google Scholar]
- Lee, T.T., Worby, C., Bao, Z.Q., Dixon, J.E., and Colman, R.F. 1999. His68 and His141 are critical contributors to the intersubunit catalytic site of adenylosuccinate lyase of Bacillus subtilis. Biochemistry 38 22–32. [DOI] [PubMed] [Google Scholar]
- Marie, S., Cuppens, H., Heuterspreute, M., Jaspers, M., Tola, E.Z., Gu, X.X., Legius, E., Vincent, M.-F., Jaeken, J., Cassiman, J.-J., et al. 1999. Mutation analysis in adenylosuccinate lyase deficiency: Eight novel mutations in the re-evaluated full ADSL coding sequence. Hum. Mutat. 13 197–202. [DOI] [PubMed] [Google Scholar]
- Race, V., Marie, S., Vincent, M.-F., and Van den Berghe, G. 2000. Clincal, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 9 2159–2165. [DOI] [PubMed] [Google Scholar]
- Ratner, S. 1972. Argininosuccinases and adenylosuccinases. In The enzymes (ed. P.D. Boyer), 3rd ed., Vol. 7, pp. 167–197. Academic Press, New York.
- Redinbo, M.R., Eide, S.M., Stone, R.L., Dixon, J.E., and Yeates, T.O. 1996. Crystallization and preliminary structural analysis of Bacillus subtilis adenylosuccinate lyase, an enzyme implicated in infantile autism. Protein Sci. 5 786–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, W., Dunbar, J., Jayasekera, M.M.K., Viola, R.E., and Farber, G.K. 1997. The structure of l-aspartate ammonia-lyase from Escherichia coli. Biochemistry 36 9136–9144. [DOI] [PubMed] [Google Scholar]
- Simpson, A., Bateman, O., Driessen, H., Lindley, P., Moss, D., Mylvaganam S., Narbor, E., and Slingsby, C. 1994. The structure of avian eye lens d-crystallin reveals a new fold for a superfamily of oligomeric enzymes. Nat. Struct. Biol. 1 724–733. [DOI] [PubMed] [Google Scholar]
- Stone, R.L., Aimi, J., Barshop, B.A., Jaeken, J., Van den Berghe, G., Zalkin, H., and Dixon, J.E. 1992. A mutation in adenylosuccinate lyase associated with mental retardation and autistic features. Nat. Genet. 1 59–63. [DOI] [PubMed] [Google Scholar]
- Stone, R.L., Zalkin, H., and Dixon, J.E. 1993. Expression, purification, and kinetic characterization of recombinant human adenylosuccinate lyase. J. Biol. Chem. 286 19710–19716. [PubMed] [Google Scholar]
- Stone, T.W., Roberts, L.A., Morris, B.J., Jones, P.A., Ogilvy, H.A., Behan, W.M.H., Duley, J.A., Simmonds, H.A., Vincent, M.F., and van den Berghe, G. 1998. Succinylpurines induce neuronal damage in the rat brain. Adv. Exp. Med. Biol. 431 185–189. [DOI] [PubMed] [Google Scholar]
- Toth, E.A. and Yeates, T.O. 2000. The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway. Structure 8 163–174. [DOI] [PubMed] [Google Scholar]
- Tornheim, K. and Lowenstein, J.M. 1972. The purine nucleotide cycle: The production of ammonia from aspartate by extracts of rat skeletal muscle. J. Biol. Chem. 247 162–169. [PubMed] [Google Scholar]
- Turner, M.A., Simpson, A., McInnes, R.R., and Howell, P.L. 1997. Human argininosuccinate lyase: A structural basis for intragenic complementation. Proc. Natl. Acad. Sci. 94 9063–9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, T.M., Levitt, D.G., Donnelly, M.I., Wilkens Stevens, P.P., and Banaszak, L.J. 1995. The multisubunit active site of fumarase C from Escherichia coli. Nat. Struct. Biol. 2 654–662. [DOI] [PubMed] [Google Scholar]
- Woodward, D.O. and Braymer, H.D. 1966. Purification and properties of Neurospora adenylosuccinase. J. Biol. Chem. 241 580–586. [PubMed] [Google Scholar]
- Yu, B., Thompson, G.D., Yip, P., Howell, P.L., and Davidson, A.R. 2001. Mechanisms for intragenic complementation at the human argininosuccinate lyase locus. Biochemistry 40 15581–15590. [DOI] [PubMed] [Google Scholar]