

Abstract
We have found previously that disaccharides (DS) enzymatically generated from heparin or heparan sulphate can modulate tumour necrosis factor‐α (TNF‐α) secretion from immune cells in vitro and cell‐mediated immune reactions in vivo. Here, we show that such DS can modulate the adhesion and migration of human T cells. We found that certain heparin‐ and heparan sulphate‐derived DS induced, in a dose‐dependent manner, the adhesion of human T cells to both extracellular matrix (ECM) and immobilized fibronectin (FN); maximal T‐cell adhesion occurred with 1 ng/ml of DS. The levels of T‐cell adhesion to ECM that were induced by the tested DS molecules resembled those induced by the prototypic chemokine, macrophage inflammatory protein 1β (MIP‐1β). However, the kinetics of DS‐induced T‐cell adhesion to FN resembled that induced by phorbol myristate acetate (PMA), but not that induced by MIP‐1β. This adhesion appeared to involve β1 integrin recognition and activation, and was associated with specific intracellular activation pathways. Although a first exposure of T cells to certain DS molecules appeared to result in cell adhesion, a subsequent exposure of T cells to pro‐adhesive chemokines, such as MIP‐1β or RANTES, but not to other pro‐adhesive stimuli, for example interleukin‐2 or CD3 cross‐linking, resulted in inhibition of T‐cell adhesion to and chemotactic migration through FN. Hence, we propose that the breakdown products of tissues generated by inflammatory enzymes are part of an intrinsic functional programme, and not necessarily molecular waste. Moreover, because the DS molecules exert their modulatory functions within a limited time, it appears that the historical encounters of the tissue‐invading cells with the constituents of inflamed loci may dictate the cells’ behaviour upon subsequent exposure to proinflammatory mediators.
Introduction
The proper functioning of immune cells requires their migration through molecular barriers, such as blood vessel walls and tissues, to inflamed loci. Migration of immune cells across the extracellular matrix (ECM) is necessary for maintaining homeostasis and for performing their designated immunological functions appropriately. 1 The homing and accumulation of T cells at sites of inflammation is regulated by ECM‐degrading enzymes and by cell‐surface expressed adhesion receptors, predominantly those of the β1 integrin [CD49; very late antigens (VLA)] family. 1–3 Ligation by integrins to their glycoprotein ECM ligands, such as fibronectin (FN), laminin and collagens, requires lymphocyte activation and is influenced by diffusible and ECM‐complexed inflammatory cytokines, 2,4 such as tumour necrosis factor‐α (TNF‐α) and interleukin‐2 (IL‐2), 3,5 and by chemokines such as MIP‐1β (macrophage inflammatory protein 1β) and RANTES (regulated on activated T‐cell expressed and secreted). 6 It is becoming increasingly evident that these cytokines and chemokines can control the locomotion of responding lymphocytes by forming chemical gradients, and can influence the consequent sequence of cellular events, including the activation of cell‐adhesion receptors and the detachment of cells from their underlying matrices. 1,3 Besides secreting proinflammatory mediators that guide lymphocyte migration, however, the penetrating lymphocytes also secrete ECM‐degrading enzymes, such as metalloproteases and endoglycosidases, which facilitate the crossing of the ECM by the cells. 7–9
Several years ago, we observed that migrating T cells deploy a heparanase enzyme that degrades the heparan sulphate (HS) scaffold of the ECM. 9 Recently, we discovered that among the degradation products of HS produced by mammalian heparanase are sulphated disaccharide (DS) molecules that can inhibit cell‐mediated immune reactions. 8,10 Sulphated DS molecules that exhibit similar properties can also be generated from heparin by heparinase. 9,11 Therefore, these products of heparin and HS, which are generated and located within inflamed loci, are part of a functional programme, and not just molecular waste. We postulated that these saccharide molecules represent natural feed‐back signals that regulate the intensity of inflammation produced by leucocytes. 8,10 Although the precise mechanism by which the DS molecules manifest their modulatory functions is not yet known, the effect appears to be associated with inhibition of TNF‐α. 8
In this study, we analysed the effects of DS molecules in vitro on the interactions of human T cells with immobilized proteins of the ECM. Specifically, we tested the abilities of DS molecules (i) to induce the adhesion of activated T cells to intact ECM and immobilized FN, and (ii) to regulate the β1 integrin‐dependent cytokine‐ or chemokine‐induced T‐cell adhesion to, and chemotactic migration through, FN.
Materials and methods
Reagents
The following reagents and chemicals were used: recombinant human IL‐2 (specific activity 18×106 U/mg; Chiron BV, Amsterdam, the Netherlands); bovine serum albumen (BSA), FN and collagen IV (CO‐IV) (Sigma Chemical Co., St Louis, MO); HEPES buffer, antibiotics, heat‐inactivated fetal calf serum (FCS), sodium pyruvate and RPMI‐1640 (Beit‐Haemek, Israel); pertussis toxin (List Biological Lab., Campbell, CA); and genistein (Biomol Research, PA); compound GF109203X (bisindolymaleimide I), a protein kinase C (PKC) inhibitor, 12 was kindly donated by Dr Y. Zick (The Weizmann Institute of Science, Rehovot, Israel). Murine monoclonal antibodies (mAb) to α2 (CD49b; IgG1), α4 (CD49d; IgG1), α5 (CD49e; IgG2A) and α6 (CD49f; IgG2A) of human β1 integrins, and the human lymphocyte function‐associated antigen‐1 (LFA‐1) receptor (IgG1), were obtained from Serotec (Oxford, UK). The chemokines stromal cell‐derived (SDF)‐1, MIP‐1β, and RANTES were purchased from PeproTech Inc. (Rocky Hill, NJ). ECM‐coated 24‐well culture plates were purchased from Nova Med (Jerusalem, Israel).
Structures of the DS molecules
The molecular structures of the DS molecules used in this study were as follows. The chemical structures of heparin‐derived DS: (i) α‐UA‐2S‐[1‐ > 4]‐GlcNS, designated DS912, structure O‐(α‐l‐ido‐4‐enepyranosyluronic acid 2‐sulphate)‐(1‐ > 4)‐2‐sulphamine‐2‐deoxy‐d‐glucose; (ii) α‐UA‐2S‐[1‐ > 4]‐GlcN‐6S, designated DS8892 (Sigma), structure O‐(α‐l‐ido‐4‐enepyranosyluronic acid 2‐sulphate)‐(1‐ > 4)‐2‐amino‐2‐deoxy‐d‐glucose 6‐sulphate. HS‐derived DS, designated DS821, is 4‐0‐(2‐deoxy‐6‐0‐sulphoamine‐2‐β‐glucopyranosyl)‐(‐2‐0‐sulpho‐β‐d‐glucopyranoside) uronic acid.
T cells and adhesion assays
Human T cells were obtained from the peripheral blood of healthy human donors and purified as described previously. 5,6 Briefly, leucocytes were isolated on a Ficoll gradient, washed, resuspended in adhesion medium (see below), and incubated (2 hr, 37°, 10% CO2 humidified atmosphere) on Petri dishes. The resulting non‐adherent cells were collected, washed and incubated (2–3 hr at 37° in a 10% CO2 humidified atmosphere) on nylon‐wool columns (Fenwall, IL). Non‐adherent cells were then eluted, washed and counted. The cell population thus obtained contained > 94% CD3+ T cells (usually 44% were CD8+ and 56% were CD4+ T cells), as determined by fluorescein‐activated cell sorter (FACS) analysis. The adhesion of T cells to immobilized protein substrates was examined as described previously. 6 The medium used for adhesion assays (adhesion medium) consisted of RPMI‐1640 supplemented with 0·1% BSA. Activators (various amounts in 50 µl) were used to pretreat the T cells (30 min, tissue culture conditions) and then added to microtitre wells that had been precoated with ECM or ECM‐glycoproteins (1 µg/well), and then blocked with 0·1% BSA in phosphate‐buffered saline (PBS). The moieties involved in the adhesion process were evaluated by adding the following inhibitors to precoated wells along with T cells: GRGDSPK and GRGESP peptides (Sigma) and mAb. Human T cells were labelled with Na251 [Cr]O4 (Amersham, High Wycombe, UK), washed, resuspended in adhesion medium, and seeded in the microtitre plates.
ECM coating of tissue culture plates
35[S]‐labelled bovine corneal endothelial cell ECM‐coated plates were prepared as described previously. 6 Briefly, endothelial cells were dissociated from stock cultures with a trypsin/EDTA solution and plated onto flat‐bottomed 96‐well tissue culture plates at an initial density of 2 × 105 cells/ml. The cells were cultured in Dulbecco’s modified Eagle’s minimal essential medium (DMEM) containing 10% FCS, 0·1% glucose and 4% dextran T‐40. Na2(35S)O4 (540–590 mCi/mmol; 40µCi/ml) was added to the cultures at day 2 and 5 after seeding, with no medium change. Twelve to 14 days after seeding, the subendothelial ECM was exposed by dissolving (5 min, 25°) the cell layer with PBS containing 0·5% Triton‐X‐100 and 20 mm NH4OH, followed by four washes with PBS. The ECM thus exposed remained intact, free of cellular debris, and firmly attached to the entire area of the tissue culture plates.
Chemotaxis assays
Chemotaxis of T cells was assessed as described previously. 5 Briefly, the migration of human T cells (0·5 × 106 cells in adhesion medium/well) was examined in a 48‐well chemotaxis microchamber (Neuro‐Probe Inc., Cabin John, MD). The two compartments of the microchambers were separated by a FN‐coated polycarbonate filter (5 µm pore size; Osmonics Proteins Products, Livemore, CA). Where indicated, MIP‐1β was added to the lower chambers, and T cells were added to the upper chambers together with the DS molecules. After incubation (120 min, 37°, 7·5% CO2 humidified atmosphere), the filters were removed, fixed and stained with a Diff‐Quik staining kit (Dade, Düdingen, Switzerland). The number of migrating T cells, found in the lower side of the filters, was counted in five high‐power fields (500× magnification; WILD Microscope, Heerbrugg, Switzerland). For each group, the results were expressed as the mean number of cells in one high‐power field.
Results
HS‐ and heparin‐derived DS induce T‐cell adhesion to ECM and FN
We analysed the capacities of heparin‐ and HS‐derived DS to affect the adhesion of human T cells to immobilized ECM and its major cell‐adhesive glycoprotein, FN. Freshly purified human T cells were labelled with radioactive chromium and then incubated (30 min) with various DS molecules, derived either from heparin (DS8892 or DS912) or from HS (DS821). The T cells were then placed into microtitre well plates that were coated with either FN or ECM, and their adhesion to these immobilized substrates was measured 30 min later. We found that DS912 and DS821 induced T‐cell adhesion to FN (Fig. 1a) and ECM (Fig. 1b) in a dose‐dependent manner. Maximal T‐cell adhesion occurred with 1 ng/ml of DS821 and 100 ng/ml of DS912. DS8892, in contrast, failed to induce T‐cell adhesion to FN. Interestingly, the levels of T‐cell adhesion to ECM that were induced by DS821 or DS912, which ranged from 25% to 42%, resembled those induced by the prototypic chemokine MIP‐1β (Fig. 1b). MIP‐1β induces T‐cell adhesion to ECM and directional chemotaxis of human leucocytes through FN‐covered layers in vitro. 6 Thus, DS molecules generated from heparin and HS, respectively, by heparinase or heparanase 11 can induce T‐cell adhesion to ECM ligands, similar to the effects of a proinflammatory chemokine, MIP‐1β.
Figure 1.
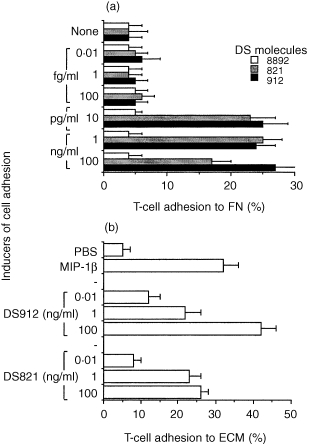
Induction by DS912 and DS821 of T‐cell adhesion to FN (a) and ECM (b). Freshly purified human T cells were labelled with 51Cr, pretreated (30 min, tissue culture conditions) with the indicated DS molecules or MIP‐1β (b; 20 ng/ml), seeded onto either FN‐ or ECM‐coated microtitre wells, and the amount of adherent cells determined 30 min later. Non‐adherent cells were washed away and the remaining bound cells were lysed. Radioactivity of lysates, representing the amount of matrix‐adherent cells, was determined using a γ‐counter. Mean± SD of triplicate wells is depicted. One experiment representative of five.
Kinetics of DS‐induced T‐cell adhesion to FN
T‐lymphocyte adhesion to immobilized ECM ligands requires cell activation, and is transient. In fact T cells must detach their firm association with the molecular ligands of tissues to be able to invade the site of immune reaction. 1 Therefore, we studied the transient nature of T‐cell adhesion to FN induced by DS912, and compared it with T‐cell adhesion induced by phorbol myristate acetate (PMA), a PKC‐activating phorbol ester, or by MIP‐1β. The levels of DS‐induced T‐cell adhesion to FN, which peaked at 45% at 60 min following cell seeding, were higher than those induced by PMA and MIP‐1β (Fig. 2). However, after 60 min the DS‐activated T cells detached rapidly from the matrix. Similar results were obtained with DS821 (data not shown). The kinetics of DS912‐induced T‐cell adhesion to FN resembled that induced by PMA, but not that induced by MIP‐1β. MIP‐1β induced maximal T‐cell adhesion by 90 min after activation, and the subsequent detachment of T cells occurred at 150 min post‐activation. Thus, the pattern of DS‐induced T‐cell adhesion to FN is consistent with the biological time–course of T cell–ECM interactions, which includes a relatively rapid attachment of tissue migrating immunocytes to the substrate followed by their gradual detachment from the ECM moiety.
Figure 2.
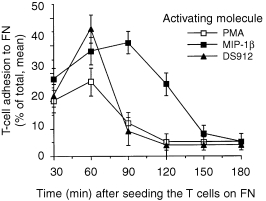
Comparative analysis of the kinetics of PMA‐, MIP‐1β‐ and DS912‐induced T‐cell adhesion to FN. T cells were preincubated (30 min, in tissue culture conditions) with PMA (50 ng/ml), MIP‐1β (20 ng/ml) and DS912 (1 ng/ml), transferred into FN‐coated wells, and the amount of bound cells (percentage of total, mean ± SD) determined at various times. One experiment representative of three.
DS‐induced T‐cell adhesion to FN involves the activation of specific VLA integrins
The recognition and binding of T cells to ECM glycoproteins requires cell activation and is mediated by specific cell‐surface receptors, designated β1 integrins. These integrins, also referred to as VLA, are non‐covalently linked αβ heterodimers, and consist of a large extracellular domain, a single hydrophobic transmembrane α‐helical segment, and a short cytoplasmic domain. 1,4 We therefore undertook to learn whether DS‐induced T‐cell adhesion to FN involves activation‐dependent, specific, intracellular signalling pathways and FN‐specific β1 integrins. Adherence of DS912‐treated T cells to FN was indeed mediated via β1 integrins, as a mAb to CD29 (directed against the extracellular portion of the common β1 chain of integrins) but not to LFA‐1 (a prototypic cell‐to‐cell adhesion receptor) blocked the adhesion (Fig. 3a). Furthermore, DS appeared to activate FN‐specific integrins, as anti‐VLA‐4 and anti‐VLA‐5 mAb, and an Arg‐Gly‐Asp (RGD)‐containing peptide, also inhibited this adhesion. Thus, DS912 and DS821 (data not shown), appear to mediate T‐cell adhesion to FN by activating specific integrins.
Figure 3.
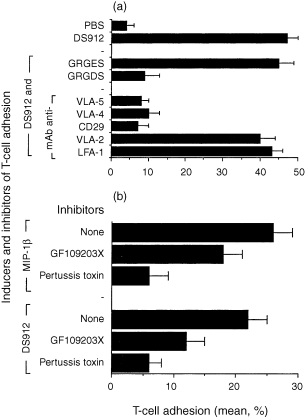
Abrogation of DS‐induced T‐cell adhesion by anti‐integrin antibody, integrin‐ligand motifs peptides (a), and inhibitors of signal transduction pathways (b). T cells were pretreated with the indicated antibodies (1 µg/ml), peptides (200 µg/ml), and intracellular signal transduction inhibitors pertussis toxin (2 µg/ml) and GF109203X (20 nm). The cells were then activated with either DS912 (1 ng/ml) or MIP‐1β (20 ng/ml), seeded onto FN‐coated microtitre wells, and the amount of cell adhesion determined 30 min later. One experiment representative of five.
The involvement of specific intracellular pathways of T‐cell activation induced by DS912 was analysed by using bisindolymaleimide I (GF109203X), a selective cell‐permeable PKC inhibitor, and pertussis toxin, a specific inhibitor of Gα1‐coupled signalling associated with G‐protein coupled receptors, such as chemokine receptors expressed on leucocytes. 6,13 As expected, pertussis toxin markedly inhibited the MIP‐1β‐induced T‐cell adhesion to FN, whereas GF109203X was less inhibitory. The inhibition by GF109203X (used in a concentration shown to inhibit significantly PMA‐induced adhesion of T cells to FN; data not shown) of DS‐induced adhesion was statistically significant (P < 0·05). However, both compounds inhibited DS‐induced T‐cell adhesion, with pertussis toxin causing more inhibition than GF109203X (Fig. 3b). Thus DS‐induced T‐cell adhesiveness to FN appears to involve specific β1 integrins and an active signalling process.
Heparin‐ and HS‐derived DS molecules inhibit T‐cell adhesion induced by chemokines
In vivo migrating T cells probably encounter a combination of pro‐ and anti‐adhesive molecules, including the classical cytokines, along with molecules that are generated by the enzymatic breakdown of tissue. We therefore studied the ability of DS912 and DS8892 to modify the activation‐dependent adhesion of T lymphocytes to FN. T cells were pretreated (30 min; 1 ng/ml) with either DS912 or DS8892, seeded on FN‐coated wells, activated with either phytohaemagglutinin (PHA; a T‐cell mitogen), anti‐human CD3 mAb, IL‐2 or chemokines (MIP‐1β, SDF‐1 and RANTES), and their adhesion to FN determined after 30 min. DS8892 did not inhibit T‐cell adhesion induced by any of the various stimulants (Fig. 4). DS912, in contrast, appeared to be a specific inhibitor of T‐cell adhesion induced by chemokines, as it did not inhibit T‐cell adhesion induced by anti‐CD3 mAb, IL‐2 or PHA. Similar results were obtained with DS821 (data not shown). When T cells were exposed to DS912 or DS821 alone, the cells adhered to the integrin ligand. However, if the DS‐induced cell activation was followed by exposure of the cells to pro‐migratory or pro‐adhesive chemokines, the ability of the treated cells to undergo integrin‐dependent adhesion appeared to be abrogated.
Figure 4.
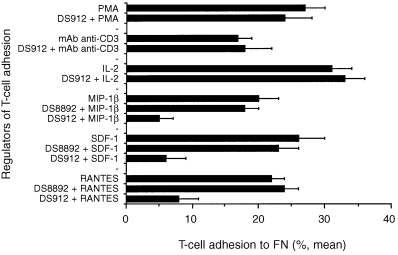
Specific inhibition, by DS912, of chemokine‐induced T‐cell adhesion to FN. T cells pretreated with DS912 (10 ng/ml; 30 min) were added to FN‐coated wells and then activated with PMA (50 ng/ml), anti‐human CD3 (15 µg/ml) mAb, IL‐2 (10 U/ml) or the chemoattractants MIP‐1β, SDF‐1 and RANTES (20 ng/ml). T‐cell adhesion was measured 30 min later. One experiment representative of four.
Effects of DS pretreatment of T cells on chemokine‐induced T‐cell adhesion
We tested whether the order in which T cells were exposed to DS molecules and chemokines affected their adhesion to FN. The results demonstrated that DS921 inhibited T‐cell adhesion induced by RANTES and MIP‐1β only if the cells were pretreated with the DS molecule, or if the DS was added to the cell cultures concomitantly with the chemokines (Fig. 5). Moreover, the DS912 had to removed from the T‐cell cultures prior to their exposure to the chemokines (data not shown). No effects on chemokine‐induced T‐cell adhesion to FN were observed if the cells were first treated with the chemokines (30 min), and only then with DS912. Similar inhibitions of chemokine‐induced T‐cell adhesion were observed using DS821. These results also indicate that the levels of T‐cell adhesion to FN are constant; adhesion was not elevated further when the cells were exposed concomitantly to both DS and chemokines, or to the DS following activation with chemokines. Hence the ability of the DS molecules to inhibit the chemokine‐induced adhesion seems to require a prior exposure of the T cells to the DS.
Figure 5.
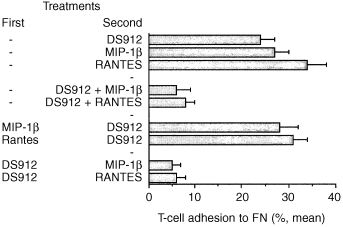
Inhibition of chemokine‐induced T‐cell adhesion to by DS912. T cells were first treated (30 min) with MIP‐1β or RANTES (both at 20 ng/ml), DS912 (10 ng/ml), or buffer alone (none), and were then activated (30 min, tissue culture conditions) with DS912, RANTES and MIP‐1β. The T cells were then added to the FN‐coated microtitre wells. T‐cell adhesion was measured 30 min later. One experiment representative of three.
Inhibition by heparin‐ and HS‐derived DS of MIP‐1β‐induced chemotaxis of T cells
Migration of T cells in vivo is the outcome of a subtle biological equilibrium existing between cellular adhesion and detachment forces. Adhesion and migration of T cells depend on their ability to integrate continuously different pro‐ and anti‐adhesive signals via their various receptors for ECM, chemokines, cytokines and, possibly, antigens. 3,4,14 Therefore, we examined the effects of pretreating (30 min) T cells with DS912, DS821 or DS8892 (0·01–100 ng/ml) on MIP‐1β‐induced T‐cell chemotaxis through FN‐coated polycarbonate membranes. A gradient generated by MIP‐1β, which was placed in the lower compartment of the chemotaxis apparatus, induced a marked (three‐ to fourfold higher than the control) T‐cell migration through FN‐coated membranes. Similar to their anti‐adhesive effects, DS821 and DS912 inhibited, in a dose–response manner, T‐cell chemotaxis towards the MIP‐1β (Fig. 6). Maximum inhibition was observed with 100 ng/ml. DS8892 did not inhibit T‐cell migration, which was reminiscent of the inability of the DS molecule to affect T‐cell adhesion. Thus, exposure to heparin‐ and HS‐derived DS molecules that actively induced T‐cell adhesion to ECM moieties, inhibited both T‐cell adhesion and migration when the cells were subsequently exposed to proinflammatory chemokines.
Figure 6.
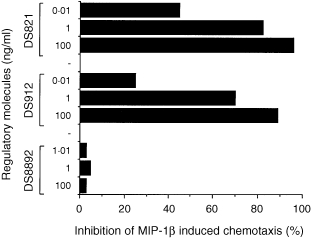
Inhibition by DS of MIP‐1β‐induced chemotaxis of T cells. T cells were pretreated with DS821, DS8892, DS912 (10 ng/ml) or buffer (30 min), placed in the upper wells of a chemotaxis chamber that contained MIP‐1β (20 ng/ml) in the lower compartment, and T‐cell migration towards the chemotactic sources was assessed after 2 hr. Under these conditions, MIP‐1β‐induced T‐cell chemotaxis was always in the range of 25–40% of the cells added in the upper compartments of the chemotaxis chambers. One experiment representative of four.
Analysis of the time required for inhibition by DS912 of MIP‐1β‐induced T‐cell adhesion to FN
The time required for pretreatment of T cells with DS for optimal inhibition of chemokine‐induced T‐cell adhesion to FN was examined. We hoped that such a study would help clarify the possible mechanisms underlying DS‐induced inhibition of T‐cell adhesion, and the relevance of our findings to in vivo situations, in which the migrating T cells may encounter DS and chemokines, not only simultaneously but also at various time‐points. When the T cells were pretreated simultaneously with both MIP‐1β and DS912 (Fig. 7, line A), an inhibition of nearly 95% of T‐cell adhesion to FN was observed. Another group of cells (Fig. 7, line B) was pretreated with DS912 (for 0–150 min), and upon transfer into FN‐coated wells the cells were then exposed to MIP‐1β. The results indicated that DS912 suppresses chemokine‐induced T‐cell adhesion only if the T cells were exposed to the DS for time periods ranging from 0 to 60 min. Interestingly, longer periods of exposure to DS were not effective in inhibiting MIP‐1β‐dependent T‐cell adhesion. A similar pattern of results was obtained using RANTES and DS821 (data not shown). Hence, if the T cells are first exposed to DS and then to chemokines, the DS appear to exert their anti‐adhesive effects in a relatively narrow window of time. However, a complete inhibition of T‐cell adhesion is achieved if the responding T cells are exposed, simultaneously, to both molecules.
Figure 7.
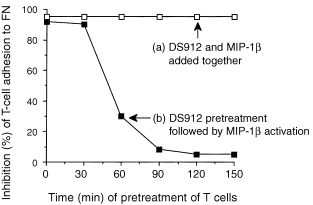
Time required for inhibition, by DS912, of MIP‐1β‐induced T‐cell adhesion to FN. (a) T cells were pretreated, for different time periods, simultaneously with both DS912 (10 ng/ml) and MIP‐1β (20 ng/ml). (b) T cells were first treated with DS912 for the indicated time periods, then with MIP‐1β, and were then transferred into FN‐coated microtitre wells; T‐cell adhesion was measured after 30 min. One experiment representative of three.
Discussion
Intact HS, present in the ECM and on the surface of blood vessel endothelial cells, has been shown to play a major role in leucocyte activation and migration; HS can modify immune cell reactivities, and can bind and present active chemokines to passing immune cells. 4,6,15–19 Previously, we showed that mammalian heparanase and bacterial heparinase can degrade HS and heparin, respectively. 8–10 This degradation specifically generates DS molecules that are capable, at relatively low concentrations, of down‐regulating inflammation in vivo in rodents, and of inhibiting the secretion of the pleiotropic cytokine TNF‐α by immune cells in vitro. 8,10 The dose–response curves of these DS‐mediated inhibitions exhibited bell‐shaped patterns; too little or much of the DS was not inhibitory. In contrast, the inhibitory effects of DS molecules on T‐cell adhesion described here exhibit a plateaued, not a bell‐shaped, dose–response curve. Thus, active DS molecules appear to elicit immunological effects with different dose‐dependencies.
A major source of DS molecules in vivo is probably the HS glycosaminoglycan. Besides providing materials for the assembly of tissues and ECM moieties, HS fulfils an immunological function in situ by presenting pro‐chemotactic chemokines in an immobilized manner to the immune cells that interact with blood vessel walls and ECM. 4,6 Therefore, cleavage of HS by heparanase released from migrating leucocytes can cause not only the disassembly of ECM and tissue barriers, thus enabling leucocyte migration into inflamed tissues, but also the release of HS‐bound mediators and, as proposed here, immunomodulatory molecules to the milieu of tissue degradation and immune reactions.
Our present study is based on the hypothesis that negative feedback mechanisms must be inherent in the inflammatory response. For example, the same heparanase that enables activated T cells to penetrate the tissues seems to produce, from the ECM debris, a DS molecule that provides signals that modify T‐cell adhesion and migration. Interestingly, the heparanase molecule itself can adjust its chemical behaviour to the context: at the mildly acidic pH characteristic of inflammation, heparanase degrades ECM; at a physiological pH the T‐cell heparanase molecule binds HS, but does not degrade it, thereby inducing T‐cell adhesion to the complexed matrix. 3 Context‐dependent alternate functions of heparanase are supported by our recent finding that a cloned T‐cell chemokine, referred to as connective tissue‐activating peptide (CTAP)‐III, can dynamically modify its functions according to the pH in its surrounding environment; depending on the pH of the environment, CTAP‐III can function either as an HS‐degrading enzyme or as a chemokine. 20 Similar findings were reported for biologically purified platelet CTAP‐III. 21 Moreover, the notion that tissue‐degrading enzymes secreted by leucocytes can modify immune cell functions by degrading and producing small signalling molecules is not limited to HS–heparanase interactions. Recently, we found that elastase, an ECM‐degrading proteolytic enzyme, can modify the signalling of IL‐2, a prototypic proinflammatory cytokine. 5 Small peptides generated from IL‐2 by elastase are capable of modulating T‐cell adhesion and chemokine‐induced chemotaxis. Thus T cells that invade into tissues can dynamically regulate their own functions. Both adhesion‐ and migration‐promoting stimuli (for example intact IL‐2) and suppressive by‐products of inflammatory mediators (for example IL‐2 fragments or DS molecules) can be present, although not necessarily simultaneously, within the inflammatory context.
The mechanisms by which heparin‐ and HS‐derived DS exert their adhesion‐ and chemotactic migration‐modifying capacities are not yet fully understood. As shown here, the biological functions of the enzyme‐generated DS molecules are not restricted to their induction of activation of T‐cell adhesion. Certain DS compounds appear to inhibit the T‐cell adhesion and migration that is specifically induced by chemokines, but not other T‐cell activation pathways. One possible mechanism of action of the tested DS may be the regulation of early events implicated in T‐cell activation, such as a rise in intracellular Ca2+ in DS‐treated, responding T cells. Another possible mode of action can be down‐regulation of the function of chemokine receptors on leucocytes, which may result from desensitization of chemokine receptors. This occurs because of phosphorylation of residues in the intraplasmic domain of the receptor. 14 Alternatively, the capacity of DS molecules to inhibit chemokine‐induced T‐cell adhesion and migration may be due to direct chemical interactions between these molecules and chemokines, thereby affecting the chemical integrities of the chemokines and down‐regulating their biological effects. Indeed, a DS structurally similar to DS912 can bind to certain chemokines and alter their chemotactic functions. 22,23 DS binding to chemokines or chemokine receptors, however, cannot account for our findings because relatively low concentrations of DS induced inhibition of T‐cell adhesion, and DS molecules inhibited MIP‐1β‐induced T‐cell adhesion to FN, even if the DS was removed prior to the adhesion assay. These findings support a mechanism by which DS molecules can exert their inhibition of chemokine‐mediated processes via certain active mechanisms, such as desensitization of specific chemokine receptors.
The present results show that DS molecules can induce T‐cell adhesion to ECM and FN in a β1 integrin‐dependent manner; T‐cell adhesion was blocked by inhibitors of β1 integrins and by specific inhibitors (pertussis toxin, and to a lesser effect GF109203X) of signal transduction pathways. Thus, the adhesion‐promoting capacities of DS912 and DS821 appear to involve at least two intracellular activation mechanisms that involve PKC‐ and G protein‐dependent pathways. Pertussis toxin inhibits leucocyte adhesion to ECM and trans‐endothelial migration induced by various chemokines, 6 which implies that the DS molecules exert their T‐cell effects in a manner similar to the heparin‐binding chemoattractants, RANTES and MIP‐1β. Although the existence and mode of action of a putative DS‐binding receptor on T cells have not yet been determined, the discrimination between modulatory (DS821 and DS912) and non‐modulatory (DS8892) DS suggests the involvement of a specific DS‐recognizing moiety on T cells, and thereby a mechanism for intracellular delivery of the versatile immunocyte‐modifying signals of inflammation. Indeed, T cells may possess saccharide‐specific receptors, 24 and receptor cross‐talk in immune cells responding to a chemokine stimulus has been proposed recently. 25 Moreover, it has also recently been found that the interactions between natural killer cells and DS molecules similar to those used here resulted in altered behaviour of the cells. 26
Our findings also imply that the historical encounters of tissue‐invading T lymphocytes with soluble and immobilized constituents of inflamed tissue may dictate the behaviour of the cells upon subsequent exposure to classical proinflammatory mediators. The DS molecules appear to function in a bi‐directional fashion; in one context they induce T‐cell adherence, while in another they inhibit chemokine‐induced adhesion and migration of T cells. The capacities of DS molecules to modulate chemokine‐induced events demonstrates an important biological phenomenon, that immunomodulatory molecules can be more potent when presented within the context of a particular string of signals. 27 Thus, the inflammatory milieu contains not only the classical signals of immune cell activation, but also molecules generated from the tissues or cytokines. These small molecular moieties can then be utilized by the inflammatory cells as signals to cease their own functions, and thus end the inflammatory reaction. The molecular breakdown products described here (DS821 and DS912) may be representatives of a large family of molecules that serve as a feedback regulators of inflammation in situ by reporting the degree of local damage, information that is vital for proper regulation of the ongoing immune reaction.
Acknowledgments
O. Lider is the incumbent of the Weizmann League Career Development Chair in Children’s Diseases. I. R. Cohen is the incumbent of the Mauerberger Chair of Immunology and the Director of the Robert Koch‐Minerva Center for Research in Autoimmune Diseases. Parts of these studies were funded by Peptor Ltd, Nes‐Ziona, Israel.
Abbreviations
- DS
disaccharide
- ECM
extracellular matrix
- FN
fibronectin
- HS
heparan sulphate
- MIP‐1β
macrophage inflammatory protein 1β
- RANTES
regulated on activated T‐cell expressed and secreted
References
- 1.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C, Sporn M. Cytokine in context. J Cell Biol. 1991;113:981. doi: 10.1083/jcb.113.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilat D, Cahalon L, Hershkoviz R, Lider O. Counter‐interactions between tissue‐infiltrating T lymphocytes, pro‐inflammatory mediators, and enzymatically‐modified extracellular matrix. Immunol Today. 1996;17:16. doi: 10.1016/0167-5699(96)80563-9. [DOI] [PubMed] [Google Scholar]
- 4.Adams DH, Lloyd AR. Chemokines: leucocyte recruitment and activation cytokines. Lancet. 1997;349:490. doi: 10.1016/s0140-6736(96)07524-1. [DOI] [PubMed] [Google Scholar]
- 5.Ariel A, Yavin EJ, Hershkoviz R, et al. IL‐2 induces T cell adherence to extracellular matrix: inhibition of adherence and migration by IL‐2 peptides generated by leukocyte elastase. J Immunol. 1998;161:2465. [PubMed] [Google Scholar]
- 6.Gilat D, Hershkoviz R, Mekori YA, Vlodavsky I, Lider O. Regulation of adhesion of CD4+ T lymphocytes to intact or heparinase‐treated subendothelial extracellular matrix by diffusible or anchored MIP‐1β and RANTES. J Immunol. 1994;153:4899. [PubMed] [Google Scholar]
- 7.Owen CA, Campbell EJ. The cell biology of leukocyte‐mediated proteolysis. J Leuk Biol. 1999;65:137. doi: 10.1002/jlb.65.2.137. [DOI] [PubMed] [Google Scholar]
- 8.Lider O, Cahalon L, Gilat D, et al. A disaccharide that inhibits tumor necrosis factor‐α is formed from the extracellular matrix by the enzyme heparanase. Proc Natl Acad Sci USA. 1995;92:5037. doi: 10.1073/pnas.92.11.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fridman R, Lider O, Naparstek Y, Fuks Z, Vlodavsky I, Cohen IR. Soluble antigen induces T lymphocytes to secrete an endoglycosidase that degrades the heparan sulfate moiety of subendothelial extracellular matrix. J Cell Physiol. 1986;130:85. doi: 10.1002/jcp.1041300113. [DOI] [PubMed] [Google Scholar]
- 10.Cahalon L, Lider O, Schor H, et al. Heparin disaccharides inhibit tumor necrosis factor‐α production by macrophages and arrest immune inflammation. Int Immunol. 1997;1997(9):1517. doi: 10.1093/intimm/9.10.1517. [DOI] [PubMed] [Google Scholar]
- 11.Lindahl U, Kjellen L. Heparin and heparan sulfate – what is the difference? Thromb Haemos. 1991;66:44. [PubMed] [Google Scholar]
- 12.Toullec D, Pianetti P, Coste H, Kirilovsky J, et al. The bisindolylmaleimide GF109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:157711. [PubMed] [Google Scholar]
- 13.Grimm MC, Ben‐baruch A, Taub DD, Howard OM, Wang JM, Oppenheim JJ. Opiate inhibition of chemokine‐induced chemotaxis. Ann NY Acad Sci. 1998;840:9. doi: 10.1111/j.1749-6632.1998.tb09544.x. [DOI] [PubMed] [Google Scholar]
- 14.Ben‐baruch A, Grimm M, Bengali K, et al. The differential ability of IL‐8 and neutrophil‐activating peptide‐2 to induce attenuation of chemotaxis is mediated by their divergent capabilities to phosphorylate CXCR2 (IL‐8 receptor B) J Immunol. 1997;158:5927. [PubMed] [Google Scholar]
- 15.Tanaka Y, Kimata K, Wake A, et al. Heparan sulfate proteoglycan on leukemic cells is primarily involved in integrin triggering and its mediated adhesion to endothelial cells. J Exp Med. 1996;184:1987. doi: 10.1084/jem.184.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ihrcke NS, Parker W, Reissner KJ, Platt JL. Regulation of platelet heparanase during inflammation: role of pH and proteinases. J Cell Physiol. 1998;175:255. doi: 10.1002/(SICI)1097-4652(199806)175:3<255::AID-JCP3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 17.Wrenshall LE, Cerra FB, Carlson A, Bach FH, Platt JL. Regulation of murine splenocyte responses by heparan sulfate. J Immunol. 1991;147:455. [PubMed] [Google Scholar]
- 18.Ihrecke NS, Wrenshall LE, Lindman BJ, Platt JL. Role of heparan sulfate in immune system–blood vessel interactions. Immunol Today. 1993;14:500. doi: 10.1016/0167-5699(93)90265-M. [DOI] [PubMed] [Google Scholar]
- 19.Ihrcke NS, Platt JL. Shedding of heparan sulfate proteoglycan by stimulated endothelial cells: evidence for proteolysis of cell‐surface molecules. J Cell Physiol. 1996;168:625. doi: 10.1002/(SICI)1097-4652(199609)168:3<625::AID-JCP15>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 20.Rechter M, Lider O, Cahalon L, et al. A cellulose binding domain‐fused recombinant human T cell chemokine CTAP‐III manifests heparanase activity. Biochem Biophys Res Commun. 1999;255:657. doi: 10.1006/bbrc.1999.0181. [DOI] [PubMed] [Google Scholar]
- 21.Hoogewerf AJ, Leone JW, Reardon IM, et al. CXC chemokines connective tissue activating peptide‐III and neutrophil activating peptide‐2 are heparin/heparan sulfate‐degrading enzymes. J Biol Chem. 1995;270:3268. doi: 10.1074/jbc.270.7.3268. [DOI] [PubMed] [Google Scholar]
- 22.Webb LM, Ehrengruber MU, Clark‐lewis I, Baggiolini M, Rot A. Binding to heparan sulfate or heparin enhances neutrophil responses to interleukin 8. Proc Natl Acad Sci USA. 1993;90:7158. doi: 10.1073/pnas.90.15.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoogewerf AJ, Kuschert GSV, Proudfoot AEI, et al. Glycosaminoglycans mediate cell surface oligomerization of chemokines. J Biol Chem. 1997;36:13 570. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- 24.Bradbury MG, Parish CR. Characterization of lymphocyte receptors for glycosaminoglycans. Immunology. 1991;72:231. [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell JJ, Foxman EF, Butcher EC. Chemoattractant receptor cross talk as a regulatory mechanism in leukocyte adhesion and migration. Eur J Immunol. 1997;27:2571. doi: 10.1002/eji.1830271016. [DOI] [PubMed] [Google Scholar]
- 26.Bezouska K, Yeun C‐T, O'brien J, et al. Oligosaccharide ligands for NKR‐P1 protein activate NK cells and cytotoxicity. Nature. 1994;372:150. doi: 10.1038/372150a0. [DOI] [PubMed] [Google Scholar]
- 27.Atlan H, Cohen IR. Immune information, self‐organization, and meaning. Int Immunol. 1998;10:711. doi: 10.1093/intimm/10.6.711. [DOI] [PubMed] [Google Scholar]