

Abstract
Acute, low‐dose ultraviolet B radiation (UVR) alters cutaneous immunity at the local site as well as systemically. Within 2–3 days of UVR exposure, recipient mice lose their capacity to develop contact hypersensitivity (CH) when hapten is painted on unexposed skin. This loss correlates temporally with a functional deficit among dendritic antigen‐presenting cells within non‐draining lymph nodes and spleen. In the experiments described, the delayed systemic immune deficiency following acute, low‐dose UVR exposure was found to be eliminated with neutralizing anti‐interleukin‐10 (IL‐10) antibody. Intracutaneous injection of IL‐10 generated a deficiency of systemic immunity as well as a functional deficit among lymph node dendritic cells that was similar to that induced by UVR. The skin itself was found to be the source of the IL‐10 responsible for these defects, and epidermis (presumably keratinocytes) rather than mast cells was found to be the source of IL‐10 within UVR‐exposed skin. The potential relationships are discussed between the delayed systemic immune deficit created by acute, low‐dose UVR, and the systemic immune deficits caused by chronic, high‐dose UVR and by a single, high‐dose UVR exposure.
Introduction
Exposure of skin to ultraviolet B radiation (UVR) has profound effects on the immune system – both the local cutaneous immune response, and the systemic immune response to skin‐derived antigens. Prolonged exposure of mice to high doses of UVR results in a systemic immune deficiency that robs the mice of the capacity to acquire contact hypersensitivity (CH) when hapten is applied epicutaneously, even if the hapten is placed on unexposed skin. 1 Even a single, large exposure of mice to high‐dose UVR (15 000 J/m2) produces a similar systemic immune deficit. 2 Acute, low‐dose UVR exposure (400 J/m2 × 4 days), similar to that originally described by Bergstresser et al., 3 creates an immune deficiency at the exposed site. Application of hapten to the exposed site fails to induce CH in UVB‐susceptible mice. Moreover, hapten applied to skin exposed to the acute, low‐dose UVR protocol results in hapten‐specific systemic tolerance. 4 Of particular interest is the fact that application of hapten to unexposed (dorsal) skin of mice immediately after the animals have completed an acute, low‐dose UVR protocol to ventral skin results in typical CH. 5 This finding indicates that the acute, low‐dose UVR regimen aborts CH induction in a strictly local fashion, i.e. limited to the site of exposure.
However, if hapten is applied to unexposed skin 48 hr or more after acute, low‐dose UVR, a systemic immune deficit is once again observed. 6,7 Mice exposed epicutaneously to hapten via unexposed skin at 48 or 72 hr after acute, low‐dose UVR fail to become sensitized. We have termed this immune aberration the ‘delayed systemic effect’ of acute, low‐dose UVR. We suspect that the delayed systemic effect of acute, low‐dose UVR on systemic immunity is similar mechanically to the immune deficiency caused by prolonged exposure to high doses of UVR, and by a single, large dose of UVR.
Thus, mice can acquire at least three distinct immune abnormalities following cutaneous exposure to UVR: first, an inability to acquire CH when hapten is applied to UVR‐exposed skin; second, acquisition of hapten‐specific tolerance when hapten is applied to UVR‐exposed skin; and third, a systemic immune deficiency that develop 3 or more days after acute, low‐dose, or high‐/chronic‐dose of UVR. There is considerable evidence to indicate that the molecular and cellular pathogeneses of these immune abnormalities are also distinct. For example, the inability of UVB‐susceptible mice to develop CH when hapten is applied epicutaneously immediately after an acute, low‐dose UVR regimen is mediated by excess tumour necrosis factor‐ (TNF‐α) generated within the UVR‐exposed site. Neutralizing anti‐TNF‐α antibodies have been shown to restore the capacity of UVR‐exposed skin to support CH induction in this situation. In the case of hapten‐specific tolerance induced when hapten is applied to skin immediately following acute, low‐dose UVR, the critical mediator has been found to be IL‐10, rather than TNF‐α. 8,9 Neutralizing anti‐IL‐10 antibodies prevent tolerance from developing when hapten is applied to skin immediately after acute, low‐dose UVR. Recent evidence indicates that cutaneous mast cells are the primary immediate source of both TNF‐α and interleukin‐10 (IL‐10) following acute, low‐dose UVR. 10
The immune pathogenesis of the delayed systemic effect following acute, low‐dose UVR remains to be elucidated. We have recently reported that within 72 hr of skin exposure to acute, low‐dose UVR, the lymph nodes and spleens of mice are depleted of dendritic cells that can function as antigen‐presenting cells for CH induction. We have hypothesized therefore that UVR promotes the intracutaneous release of IL‐10, which acts systemically to deplete the secondary lymphoid organs of fully functional APC, and that this deficit is responsible for the failure of UVR‐exposed mice to develop CH when hapten is applied epicutaneously.
Experiments to test this hypothesis form the basis of this report. We found that neutralizing anti‐IL‐10 antibodies were capable of reversing the delayed systemic immune deficit created by acute, low‐dose UVR, that systemic administration of IL‐10 produced a deficiency of APC similar to that of UVR, and that the epidermis, rather than mast cells, of UVR‐exposed skin was the relevant source of IL‐10 that caused the delayed systemic effect.
Materials and methods
Mice
Adult C3H/HeN mice (aged 10–16 weeks) were purchased from Taconic Farms (Germantown, NY). All animal procedures were approved by the institutional Animal Care and Use Committee. Experimental procedures were carried out with the animals under general anaesthesia achieved by intraperitoneal injection of ketamine (12·5 mg/ml; Fort Dodge Lab., Inc., Fort Dodge, IA) and xylazine (0·125 mg/ml; Phoenix Pharmaceutical Inc., St. Joseph, MO). Each experimental protocol was repeated at least twice with similar results.
Reagents
Murine recombinant IL‐10 was purchased from Genzyme (Cambridge, MA). The 2,4‐dinitro‐1‐fluorobenzene (DNFB) was purchased from Sigma Chemical Co. (St. Louis, MO). Neutralizing anti‐IL‐10 antibodies were purified from culture supernatants of hybridoma JES5‐2A5. Rat immunoglobulin G1 (IgG1) antibody (Pharmingen, San Diego, CA) was used as isotype control.
UVR treatment
Dry‐shaved abdominal skin was exposed to UVB light from a bank of four FS‐20 fluorescent lamps with a tube‐to‐target distance of 46 cm, as previously described. 5 These bulbs have a broad emission spectrum (250–400 nm), and high output was primarily in the UVB range (290–320 nm). As measured by an IL 700 radiometer with a SEE 240 UVB photodetector, these lamps delivered an average flux of 1·5 J/m2/second. Mice were exposed to UVB light daily for four consecutive days (400 J/m2/day) on the abdominal cutaneous surface. Three days after the final exposure DNFB (185 µg) in acetone was applied to distant (unexposed) back skin.
Dendritic cell‐enriched suspensions
Lymph node dendritic cells (DCs) were obtained as an enriched suspension according to the method described by Dai and Streilein. 11 Briefly, lymph nodes were removed surgically and minced through a nylon mesh. The cells were then separated on density fractions using low‐density Percoll solution (Pharmacia & Upjohn, Peapack, WN) (1·035 g/dl), layered over a high‐density solution (1·075 g/dl), and centrifuged at 570 g for 20 min. Interface cells were collected and placed in 10‐ml Petri dishes at 37° for 60 min, after which non‐adherent cells were discarded. The adherent cells were then incubated in 10‐ml Petri dishes at 37° overnight. Non‐adherent cells were collected as DC‐enriched suspensions, and these cells displayed a typical dendritic morphology under phase‐contrast microscopy. (data not shown). For hapten‐derivatization, 10 µl of 0·01% DNFB in acetone was added to 1 ml of DC‐enriched cell suspensions (106/ml). Control cells were incubated with acetone only. The suspensions were further incubated for 30 min, then washed and adjusted to 2 × 105 cells/ml in medium. Then, 100 µl of these cell suspensions was injected into the footpads of recipient mice.
Epidermal graft preparations
Ear skin was excised and these cutaneous sheets were immersed in trypsin (0·25%) at 37°. After 50 min of incubation, the epidermis was carefully lifted away from the dermis with fine forceps. These epidermal sheets were then placed, dermal face down, on graft beds prepared on the thoracic wall of recipient mice, as described previously. 12 The graft was held in place with a plaster of Paris bandage.
Induction and assay of contact hypersensitivity
DNFB (185 µg) in acetone was applied (25 µl) to the surface of dry, shaved, body wall skin. Five days later DNFB (15 µg) was painted on the exterior surface of ear pinnae. Ear‐swelling responses were measured with an engineer’s micrometer (Mitsutoyo, Tokyo, Japan) prior to hapten application, and at 24 hr and 48 hr (only 24 hr measurements are presented). Negative control mice received only the pinnae application of DNFB.
Statistical evaluation
Differences between the mean values were determined to be significant using Students’ t‐test.
Results
Neutralizing anti‐IL‐10 antibodies restore the delayed systemic immune deficiency induced by UVR
Neutralizing anti‐IL‐10 antibodies, administered in concert with the four consecutive day regimen of acute, low‐dose UVR, prevent hapten‐specific tolerance when DNFB is painted on the UVR‐exposed site immediately after the last episode of irradiation. 10 This finding offered the possibility that skin‐derived IL‐10 might also be responsible for the systemic deficit in antigen‐presenting cells that emerges 48–72 hr after the UVR regimen. To test this possibility, panels of C3H/HeN mice were subjected to a regimen of UVR consisting of 400 J/m2 delivered to shaved abdominal skin on four consecutive days. Six hr before each exposure, one panel of mice received intraperitoneal injections of 400 µg of neutralizing anti‐IL‐10 antibody. A control panel of mice received the same exposure to UVR plus intraperitoneal injections of 400 µg IgG antibodies. Three days thereafter, both panels of mice, plus a positive control, received an epicutaneous application of DNFB (185 µg) on dorsal (non‐UVR‐exposed) skin. Five days later, the ear pinnae of these mice and of untreated, naive C3H/HeN mice (negative control) were challenged with dilute DNFB (15 µg). The results of ear‐swelling responses assessed 24 and 48 hr later for one representative experiment are displayed in Fig. 1. UVR‐treated mice that received isotype control IgG injections displayed ear‐swelling responses significantly less intense than positive controls, i.e. these mice displayed the delayed systemic effect of acute, low‐dose UVR treatment. By contrast, UVR‐exposed mice that received simultaneously intraperitoneal injections of neutralizing anti‐IL‐10 antibodies mounted ear‐swelling responses comparable to those of positive controls. These results, which indicate that neutralizing anti‐IL‐10 antibody can restore the capacity to acquire CH to mice exposed to an acute, low‐dose UVR regimen, implicate IL‐10 in the pathogenesis of the systemic antigen‐presenting cell defect caused by UVR.
Figure 1.
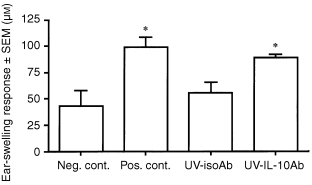
Effect of neutralizing anti‐IL‐10 antibodies on the delayed systemic effect of UVR. Panels of C3H/HeN mice received daily intraperitoneal injections of rat anti‐mouse IL‐10 antibodies (400 µg; UV‐IL‐10Ab) or isotype control (rat IgG1; UV‐isoAb) 6 hr before each exposure of an acute, low‐dose regimen of UVR to shaved abdominal skin (400 J/m2 × 4 consecutive days). Three days after the last exposure, DNFB (185 µg) was painted on unexposed skin (back). Five days later, the ear pinnae of these mice were challenged with 15 µg DNFB, and ear swelling was measured at 24 hr. Bars represent mean ear‐swelling responses ± SEM for groups of five mice each. Asterisks indicate ear‐swelling responses significantly greater than negative control, P < 0·05.
IL‐10 creates a systemic antigen‐presenting deficit similar to that of acute, low‐dose UVR
We have recently reported that DCs from lymph nodes (both draining and non‐draining) are deleteriously affected by acute, low‐dose UVR (Kurimoto and Streilein, manuscript under review). DCs harvested from nodes 72 hr after UVR, and then derivatized in vitro with DNFB, failed to induce CH when injected subcutaneously into naive, syngeneic mice. Our next goal was to determine whether a similar antigen‐presenting cell defect could be produced by administration of IL‐10 to normal mice. Panels of C3H/HeN mice received intradermal injections of IL‐10 (200 ng) into abdominal skin. Control mice received intradermal injections of phosphate‐buffered saline (PBS). Eight hours later, lymph nodes were harvested from sites (cervical, mesenteric) known not to be included in the lymphatic drainage pathways of the injection sites. This strategy insured that any potential effect of IL‐10 would not be due to the direct action of this cytokine on DCs in the nodes immediately upstream from the injection. DC‐enriched cell suspensions were prepared from the harvested nodes, and derivatized with DNFB in vitro. Haptenated DCs were then injected into one hind footpad (2 × 105/100 µl) of naive C3H/HeN mice. The ears of these mice, plus a negative control, were challenged 5 days later with dilute DNFB. The results of a representative experiment are displayed in Fig. 2. As anticipated, recipients of DNFB‐derivatized DCs from donors that received intradermal injections of PBS mounted intense ear‐swelling responses. However, recipients of haptenated DCs from donors whose dermis had received IL‐10 injections failed to acquire CH. Thus, injection of IL‐10 into the dermis at one site in C3H/HeN mice leads to a deficit in DCs harvested from secondary lymphoid organs that are not linked by lymphatic vessels to the injected site. The deficit created within 48–72 hr by exposure of murine skin to an acute, low‐dose UVR regimen resembles the deficit created by intradermal injection of IL‐10.
Figure 2.
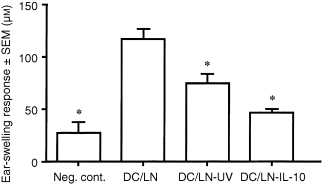
CH‐inducing capacity of dendritic cells obtained from lymph nodes from IL‐10‐treated mice. Panels of mice received IL‐10 (200 ng) injections intradermally into abdominal skin. Eight hours later, lymph nodes not draining the injected skin (all except axillary and inguinal) were removed. Lymphoid cell suspensions were separated on density gradients using Percoll solution 1·075 g/dl, layered over 1·035 g/dl solution and centrifuged. Interface cells were collected and placed in Petri dishes at 37° for 60 min, after which non‐adherent cells were discarded. The adherent cells were then incubated at 37° overnight and non‐adherent cells were collected as cell suspension enriched for DCs. As positive control, cells were prepared from lymph nodes of untreated mice. DC‐enriched suspensions derivatized with DNFB in vitro were injected into footpads of naive syngeneic mice (2 × 104 cells in 100 µl per mouse). Five days later CH was assayed as described in the legend to Fig. 1. Bars represent mean ear‐swelling responses ± SEM for groups of five mice each. Asterisks indicate mean responses significantly less than the positive control (P < 0·0005).
UVR can create a delayed systemic immune deficit in mast cell‐deficient mice
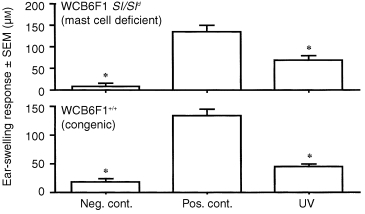
The results of the experiments described above implicate IL‐10 from a cutaneous source as the important stimulus for the emergence of a delayed systemic APC deficit after acute, low‐dose UVR. Niizeki et al. have recently reported that failed CH induced by application of DNFB to exposed skin immediately after the last UVR treatment is dependent upon the acute release of IL‐10 from mast cells at the irradiated site. 13 Therefore, we first attempted to determine whether dermal mast cells were the source of the IL‐10 responsible for the creation of the delayed systemic effect after UVR. Panels of mast cell‐deficient mice (Sl/Sld), and their wild‐type controls, were subjected to the acute, low‐dose UVR protocol on abdominal skin. Three days later, a sensitizing dose of DNFB (185 µg) was applied to dorsal (unexposed) skin. Ear‐swelling responses were elicited by application of dilute DNFB to ear pinnae 5 days later. The results of a representative experiment are presented in Fig. 3. Neither Sl/Sld mice, nor their wild‐type controls, acquired CH when hapten was painted on distant skin 3 days after acute, low‐dose UVR exposure. The ability of UVR to induce a delayed systemic immune deficit in mast cell‐deficient mice renders unlikely the possibility that cutaneous mast cells are the source of the IL‐10 responsible for the delayed deficit.
Figure 3.
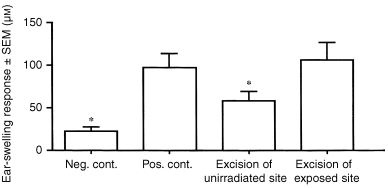
Effect of excision of UVR‐exposed skin on delayed systemic effect of UVR. Panels of C3H/HeN mice received an acute, low‐dose UVR regimen. Immediately after the last exposure, the entire exposed site, or a comparably sized unexposed skin site, was excised. In positive controls, skin comparable in size to the UVR‐exposed site was excised. Wounds were covered with protective dressings. Three days later, DNFB (185 µg) was painted on normal skin (back). Five days later, the ear pinnae of the mice were challenged with 15 µg DNFB, and ear swelling was measured at 24 hr. Asterisks indicate ear‐swelling responses significantly lower than positive control (P < 0·05).
Excision of UVR‐exposed skin prevents the development of a delayed systemic immune deficit
Acute, low‐dose UVR not only causes mast cells in the exposed skin to release IL‐10, but it induces exposed keratinocytes to synthesize and release IL‐10. Whereas the release of IL‐10 from mast cells after UVR is virtually immediate (within 30 min), the production of IL‐10 by keratinocytes is delayed, IL‐10 gene activation becoming apparent 8 or more hours after UVR exposure. 14 Since it takes 48–72 hr for acute, low‐dose UVR to cause a systemic immune deficiency, the possibility exists that UVR‐exposed keratinocytes represent the relevant cutaneous IL‐10 source. To test this possibility, a skin excision experiment was conducted. Panels of C3H/HeN mice received the conventional acute, low‐dose UVR regimen on shaved abdominal wall skin. Immediately after the last exposure, the exposed skin site (epidermis and dermis, but not panniculus carnosis) was removed surgically from one panel of mice. A comparable piece of skin was excised from non‐UVR‐exposed skin of a control panel of mice. DNFB (185 µg) was then applied to unexposed skin of these mice 72 hr later. When the ear pinnae of these mice, plus positive (non‐irradiated) and negative control mice, were challenged with dilute DNFB, the results presented in Fig. 4 were observed. Feeble ear‐swelling responses were detected in mice, from whom skin at an unexposed site had been excised, whereas intense responses (indistinguishable from those of positive controls) were found in mice whose UVR‐exposed skin sites had been removed. This result strongly suggests that UVR‐exposed skin itself produces the factor that is ultimately responsible for the delayed systemic deficit.
Figure 4.
Assay of delayed systemic effect of UVR in mast cell‐deficient mice. Panels of mast cell‐deficient mice (Sl/Sld) and their wild‐type controls received an acute, low‐dose UVR regimen to shaved abdominal skin. Three days after the last exposure, DNFB (185 µg) was painted on unexposed skin (back). Five days later, the ear pinnae of the mice were challenged with 15 µg DNFB, and ear swelling was measured at 24 hr. Asterisks indicate ear‐swelling responses significantly lower than positive control (P < 0·005).
UVR‐exposed epidermis is responsible for creating the delayed systemic immune deficit after UVR
It is not technically possible to excise selectively or remove the epidermis alone from intact skin in vivo, whether the skin is UVR‐exposed or not. Therefore, we approached the possibility that keratinocytes of epidermis are the source of the immunosuppressive factor by adopting a skin‐grafting protocol. More than a decade ago, Streilein et al. reported that grafts prepared from skin to which hapten had been applied epicutaneously were capable of sensitizing naive mice onto which the derivatized grafts were placed orthotopically. 15 If the skin graft was prepared from skin exposed to acute, low‐dose UVR that had been painted with hapten, graft recipients failed to acquire CH. We adapted this model system as follows. C3H/HeN mice received on the dorsal surface of their ear pinnae an acute, low‐dose UVR regimen. Immediately after the last exposure, the pinnae were excised, and the epidermis was carefully removed from the dermis by incubating the tissue first in trypsin. Graft beds were prepared on the thoracic wall of naive C3H/HeN mice, and epidermal sheets were placed on these raw sites. Three days later, recipient mice received a sensitizing dose of DNFB (185 µg) on body wall skin, and their ear pinnae were challenged with dilute DNFB 5 days later. As displayed in Fig. 5, mice that were recipients of epidermal grafts from UVR‐exposed epidermis failed to develop intense ear‐swelling responses, compared to positive controls. We conclude that the stimulus for creation of a systemic immune deficit following acute, low‐dose UVR arises within the exposed epidermis itself.
Figure 5.
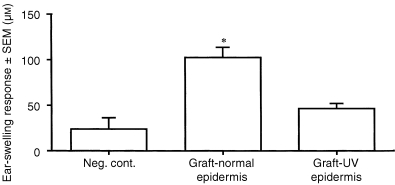
Induction of contact hypersensitivity in mice that received grafts of UVR‐irradiated epidermis. Panels of C3H/HeN mice received an acute, low‐dose regimen of UVR to ear skin. One hour after the last exposure, the ear skin was removed. Cutaneous sheets were peeled away, then immersed in trypsin at 37° for 50 min. Epidermis was separated from dermis and placed as a graft on thoracic wall beds of naive syngeneic mice. Three days later, DNFB (185 µg) was painted on the surface of the right ear pinna. Five days later, the left ear pinnae of these mice were challenged with 15 µg DNFB, and ear swelling was measured at 24 hr. Control epidermal grafts were prepared from pinnae of mice not exposed to UVR. Asterisk indicates ear‐swelling responses significantly greater than negative control (P < 0·05).
Discussion
The discoveries by Kripke and by Daynes that prolonged exposure of mice to high‐dose UVR induced cutaneous tumours and created a systemic immune deficit that proved to be important in tumour development were remarkable and profound. 1,16 The finding that immune deficiency lies at the heart of the pathogenesis of UVR‐induced malignancies revealed the power of the phenomenon of immune surveillance. Although a precise understanding of immune surveillance still eludes us, the importance of a robust immune system in preventing the emergence of cutaneous malignancies cannot be diminished. As a consequence, studies of the effects of UVR on cutaneous immune responses continue to maintain the attention of immunodermatologists.
Over the years, three rather distinct experimental models have been developed to explore the relationship between UVR and the immune response. In addition to the original chronic, high‐dose UVR model, there is a model in which a single, very large dose of UVR (typically 15 000 J/m2) leads to a systemic immune deficiency which is usually tested by attempting to sensitize the recipient with epicutaneous hapten applied 3–5 days later on unexposed skin. 2 The single, high‐dose model bears similarity to the chronic, high‐dose model in that the spleens of both types of UVR‐exposed mice contain suppressor T cells that can inhibit the induction of CH in naive recipients to a range of different haptens. 17 In addition, the sera of both types of mice have been reported to be immunosuppressive. In the single, high‐dose model, IL‐10 has been reported to be the relevant immunosuppressive factor. 18 Moreover, the spleens of mice exposed to chronic UVR, and mice exposed to a single large UVR dose are deficient in antigen‐presenting cells. 19
A third model, developed by Bergstresser and Streilein, exposed mice to an acute, low‐dose UVR regimen. Unlike the two other models, acute, low‐dose UVR creates at the exposed site a profound deficiency of antigen‐presenting cells, and when hapten is applied to that site immediately after the last dose of UVR, contact hypersensitivity fails – at least in a subset of mice (and humans) termed UVB‐susceptible. 20,21 This failure does not result from the generation of regulatory T cells, but from a primary functional deficiency among the cutaneous antigen‐presenting cells necessary for CH induction. The acute, low‐dose UVR model also promotes a form of hapten‐specific tolerance if hapten is painted on the site immediately after the last UVR exposure. This tolerance is mediated primarily by regulatory CD4+ T cells and differs from the systemic immune deficit evoked by either the chronic or single high‐dose UVR regimens because it is hapten‐specific. Several years ago, Shimizu and Streilein 7 reported that mice exposed to the acute, low‐dose UVR regimen also develop a systemic immune deficiency which only becomes apparent 48–72 hr after the last UVR exposure. This delayed systemic immune deficiency has now been shown to correlate with a profound functional defect among dendritic APC in the spleen and in lymph nodes that do not drain the UVR exposure site. In the experiments described in this communication, we explore the basis of this defect in an effort to understand its pathogenesis, and to determine whether this delayed systemic deficiency bears any relationship to the systemic immune defect caused by chronic and acute, high‐dose UVR.
Our results can be summarized as follows: neutralizing anti‐IL‐10 antibodies reversed the systemic immune deficit created by acute, low‐dose UVR and intracutaneous injection of IL‐10 alone caused a similar defect, including a functional deficit among dendritic cells in lymph nodes other than those that drained the injection site. The UVR‐exposed skin itself gave rise to the factor (presumably IL‐10) that caused the systemic deficiency, and the intracutaneous source of that factor(s) was the epidermis itself. We conclude that acute, low‐dose UVR induced a delayed‐in‐onset systemic immune deficiency by provoking epidermal cells (probably keratinocytes) to produce IL‐10 which, by disseminating widely, alters the functional integrity of APC within distant (as well as local) secondary lymphoid organs. The specific defect created among these APC is revealed by their inability to induce hapten‐specific contact hypersensitivity.
We strongly suspect that the intraepidermal source of IL‐10 that leads to the delayed systemic effect after acute, low‐dose UVR is the keratinocyte. This view stems from our knowledge that four consecutive daily exposures of skin to UVR (400 J/m2) results in the virtual obliteration of all Langerhans cells and Thy‐1+ dendritic epidermal T cells. Although the IL‐10 gene is transcriptionally silent in keratinocytes within normal skin, exposure to UVR opens this gene, leading to the production of IL‐10 by these cells. Rivas and Ullrich 22 have reported that supernatants from UVR‐exposed keratinocytes contain IL‐10, and intracutaneous injection of these supernatants 5 days prior to sensitization interferes with the development of delayed hypersensitivity. Similarly, Enk et al. 23 demonstrated that intradermal injection of high doses of IL‐10 (2 µg) followed by application of hapten to the injected site 8 hr later prevented the induction of CH. Experiments with both human and murine keratinocytes from UVR‐exposed skin have revealed that IL‐10 is first produced after a lag time of approximately 6–8 hr, and that peak production is detected several days after UVR exposure. 24,25 These results, taken in conjunction with our observation that a systemic immune deficit is first detectable at 48–72 hr after acute, low‐dose UVR, support the hypothesis that keratinocytes are the principle cells responsible for systemic immune deficiencies detected in mice exposed to single, low‐dose, or acute, low‐dose UVR.
The mechanism by which UVR induces keratinocytes to produce IL‐10 has not been worked out. Shreedhar et al. have recently reported that prostaglandin E2 released from UVR‐exposed keratinocytes induces IL‐10 production via a cascade that involves IL‐4. 26 The time–course of IL‐10 production by UVR‐exposed keratinocytes is compatible with the trigger being a soluble factor. Candidate factors include urocanic acid (UCA), 27 alpha‐melanocyte‐stimulating hormone, 28 and reactive oxygen intermediates 29 – all of which have been implicated in the pathogenesis of UVR‐dependent cutaneous immune deficiencies. Trans‐UCA has been documented as an important photoreceptor in the epidermis, and several laboratories, including ours, have implicated the cis isomer of UCA in the cutaneous immune deficit caused by UVR. 27,30 After acute, low‐dose UVR, cis‐UCA prevents CH induction immediately by promoting the release of TNF‐α. Moreover, cis‐UCA has been found to induce mast cells to release both TNF‐α and IL‐10. However, Grewe et al. 25 have demonstrated that IL‐10 production by UVR‐exposed skin is not interrupted by treatment with anti‐TNF‐α or anti‐IL‐1α antibodies, nor with indomethacin, a prostaglandin synthetase inhibitor. It is possible that activation of the IL‐10 gene within keratinocytes is a direct effect of UVR, perhaps as an immediate early gene response, and perhaps related to the thymine dimers within DNA that characteristically form after UVR treatment. 31 It is relevant that UVR‐dependent DNA damage has been directly implicated in the systemic immune deficiency that occurs within 5 days of a single, large exposure to UVR.
It is worth commenting on the role (or lack thereof) of mast cells in the delayed systemic immune deficit that follows acute, low‐dose UVR. Hart et al. 32 have recently reported that mast cells are essential to the unresponsiveness observed in mice exposed to a single, high dose of UVR. As mentioned previously, our laboratory has reported that the failed CH induced by application of hapten to exposed skin immediately after completion of the acute, low‐dose UVR regimen requires the presence of mast cells. More importantly, the requirement is for IL‐10 released from mast cells in UVR‐exposed skin. Thus, we were surprised to find that acute, low‐dose UVR readily induced a delayed systemic immune deficit in mast cell‐deficient mice. Our discovery that the IL‐10, which is responsible for this deficiency, is derived from epidermis explains why mast cells are unnecessary. These results emphasize the complexity of the pathogenesis of immune deficiencies created by exposing skin to UVR. Moreover, they reveal how much remains to be learned if we are to be able eventually to thwart the development of cutaneous tumours caused by exposure to sunlight.
Acknowledgments
We wish to thank Dr Jacqueline Doherty and Ms Marie Ortega for expert managerial and technical assistance. This work was supported in part by USPHS grant AR‐44130.
Abbreviations
- CH
contact hypersensitivity
- DCs
dendritic cells
- DNFB
dinitrofluorobenzene
- UCA
urocanic acid
- UVR
ultraviolet B radiation
References
- 1.Kripke ML. Immunobiology of photocarcinogenesis. In: Parish JA, editor. The Effect of Ultraviolet Radiation on Immune System. New Brunswick, NJ: Johnson and Johnson Products; 1983. p. 87. [Google Scholar]
- 2.Noonan FP, De Fabo EC. Ultraviolet‐B. dose–response curves for local and systemic immunosuppression are identical. Photochem Photobiol. 1990;52:801. doi: 10.1111/j.1751-1097.1990.tb08685.x. [DOI] [PubMed] [Google Scholar]
- 3.Bergstresser PR, Toews GB, Streilein JW. Natural and perturbed distributions of Langerhans cells: responses to ultraviolet light, heterotopic skin grafting, and dinitrofluorobenzene sensitization. J Invest Dermatol. 1980;75:73. doi: 10.1111/1523-1747.ep12521261. [DOI] [PubMed] [Google Scholar]
- 4.Elmets CA, Bergstresser PR, Tigelaar RE, Wood PJ, Streilein JW. Analysis of the mechanism of unresponsiveness produced by haptens painted on skin exposed to low dose ultraviolet radiation. J Exp Med. 1983;158:781. doi: 10.1084/jem.158.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980;124:445. [PubMed] [Google Scholar]
- 6.De Fabo EC, Noonan FP. Mechanism of immune suppression by ultraviolet irradiation in vivo. I. Evidence for the existence of a unique photoreceptor in skin and its role in photoimmunology. J Exp Med. 1983;157:84. doi: 10.1084/jem.158.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimizu T, Streilein JW. Local and systemic consequences of acute, low‐dose ultraviolet B radiation are mediated by different immune regulatory mechanisms. Eur J Immunol. 1994;24:1765. doi: 10.1002/eji.1830240807. [DOI] [PubMed] [Google Scholar]
- 8.Yoshikawa T, Streilein JW. Genetic basis of the effects of ultraviolet light B on cutaneous immunity. Evidence that polymorphisms at the TNFα and LPS loci govern susceptibility. Immunogenetics. 1990;32:398. doi: 10.1007/BF00241633. [DOI] [PubMed] [Google Scholar]
- 9.Niizeki H, Streilein JW. Hapten‐specific tolerance induced by acute, low‐dose ultraviolet B radiation of skin is mediated via interleukin‐10. J Invest Dermatol. 1997;109:25.. doi: 10.1111/1523-1747.ep12276415. [DOI] [PubMed] [Google Scholar]
- 10.Niizeki H, Alard P, Streilein JW. Calcitonin gene‐related peptide is necessary for ultraviolet B‐impaired induction of contact hypersensitivity. J Immunol. 1997;159:5183. [PubMed] [Google Scholar]
- 11.Dai R, Streilein JW. In vitro culture allows splenic dendritic cells to reach their full potential for T‐cell activation. Reg Immunol. 1993;5:269. [PubMed] [Google Scholar]
- 12.Bacci S, Alard P, Dai R, Nakamura T, Streilein JW. High and low doses of haptens dictate whether dermal or epidermal antigen‐presenting cells promote contact hypersensitivity. Eur J Immunol. 1997;27:442. doi: 10.1002/eji.1830270214. [DOI] [PubMed] [Google Scholar]
- 13.Niizeki H, Alard P, Streilein JW. Calcitonin gene‐related peptide is necessary for ultraviolet B‐impaired induction of contact hypersensitivity. J Immunol. 1997;159:5183. [PubMed] [Google Scholar]
- 14.Ullrich SE. Mechanism involved in the systemic suppression of antigen‐presenting cell function by UV irradiation. keratinocyte‐derived IL‐10 modulates antigen‐presenting cell function of splenic adherent cells. J Immunol. 1994;152:3410. [PubMed] [Google Scholar]
- 15.Streilein JW, Wood PJ, Lonsberry LW, Bergstresser PR. Induction of H‐2 restricted contact hypersensitivity by hapten‐derivatized skin grafts. Transplantation. 1984;37:195. doi: 10.1097/00007890-198402000-00015. [DOI] [PubMed] [Google Scholar]
- 16.Daynes RA, Bernhard EJ, Gurish MF, Lynch DH. Experimental photoimmunology: immunologic ramifications UV‐induced carcinogenesis. J Invest Dermatol. 1981;77:77. doi: 10.1111/1523-1747.ep12479260. [DOI] [PubMed] [Google Scholar]
- 17.Ullrich SE. Suppression of the immune response to allogeneic histocompatibility antigens by a single exposure to ultraviolet radiation. Transplantation. 1986;42:287. doi: 10.1097/00007890-198609000-00012. [DOI] [PubMed] [Google Scholar]
- 18.Swartz RP. Role of UVB‐induced serum factors in suppression of contact hypersensitivity in mice. J Invest Dermatol. 1984;83:305. doi: 10.1111/1523-1747.ep12340434. [DOI] [PubMed] [Google Scholar]
- 19.Kripke ML. Immunological unresponsiveness induced by ultraviolet radiation. Immunol Rev. 1984;80:87. doi: 10.1111/j.1600-065x.1984.tb00496.x. [DOI] [PubMed] [Google Scholar]
- 20.Streilein JW, Bergstresser PR. Genetic basis of ultraviolet‐B effects on contact hypersensitivity. Immunogenetics. 1988;27:252. doi: 10.1007/BF00376119. [DOI] [PubMed] [Google Scholar]
- 21.Yoshikawa T, Rae V, Bruins‐slot W, Van Den Berg JW, Taylor JR, Streilein JW. Analysis of effects of UVB radiation on induction of contact hypersensitivity as a risk factor for skin cancer in humans. J Invest Dermatol. 1990;95:530. doi: 10.1111/1523-1747.ep12504877. [DOI] [PubMed] [Google Scholar]
- 22.Rivas JM, Ullrich SE. Systemic suppression of delayed‐type hypersensitivity by supernatants from UV‐irradiated keratinocytes. J Immunol. 1992;149:3865. [PubMed] [Google Scholar]
- 23.Enk AH, Saloga J, Becker D, Mohamadzadeh M, Knop J. Induction of hapten‐specific tolerance by interleukin 10 in vivo. J Exp Med. 1994;179:1397. doi: 10.1084/jem.179.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enk CD, Sredni D, Blauvelt A, Katz SI. Induction of IL‐10 gene expression in human keratinocytes by UVB exposure in vivo and in vitro. J Immunol. 1995;154:4851. [PubMed] [Google Scholar]
- 25.Grewe M, Gryufko K, Krutmann J. Interleukin‐10 production by cultured human keratinocytes: regulation by ultraviolet B and ultraviolet A1 radiation. J Invest Dermatol. 1995;104:3. doi: 10.1111/1523-1747.ep12613446. [DOI] [PubMed] [Google Scholar]
- 26.Shreedhar V, Giese T, Sung VW, Ullrich SE. A cytokine cascade including prostaglandin E2, IL‐4, and IL‐10 is responsible for UV‐induced systemic immune suppression. J Immunol. 1998;160:3783. [PubMed] [Google Scholar]
- 27.Kurimoto I, Streilein JW. Cis‐urocanic acid suppression of contact hypersensitivity induction is mediated via tumor necrosis factor‐α. J Immunol. 1992;148:3072. [PubMed] [Google Scholar]
- 28.Shimizu T, Streilein JW. Influence of alpha‐melanocyte stimulating on induction of contact hypersensitivity and tolerance. J Dermatol Sci. 1994;8:187. doi: 10.1016/0923-1811(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura T, Pinnell SR, Darr D, et al. Vitamin C abrogates the deleterious effects of UVB radiation on cutaneous immunity by a mechanism that does not depend on TNF‐α. J Invest Dermatol. 1997;109:20. doi: 10.1111/1523-1747.ep12276349. [DOI] [PubMed] [Google Scholar]
- 30.Noonan FP, De Fabo EC. Immunosuppression by ultraviolet B radiation: initiation by urocanic acid. Immunol Today. 1992;13:250. doi: 10.1016/0167-5699(92)90005-R. [DOI] [PubMed] [Google Scholar]
- 31.Kripke ML, Cox PA, Alas G, Yarosh DB. Pyrimidine dimers in DNA initiate systemic immunosuppression in UV‐irradiated mice. Proc Natl Acad Sci USA. 1992;89:7516. doi: 10.1073/pnas.89.16.7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart PH, Grimbaldeston MA, Swift GJ, Jaksic A, Noonan FP, Finlay‐jones JJ. Dermal mast cells determine susceptibility to ultraviolet B‐induced systemic suppression of contact hypersensitivity responses in mice. J Exp Med. 1998;187:2045. doi: 10.1084/jem.187.12.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]