

Abstract
Crry (CR1‐related gene/protein) is a rodent complement regulator that inhibits C3 convertases. CD59 is a conserved protein inhibitor active towards C8 and C9. We have previously produced rat Crry as a recombinant soluble (rs) protein in Pichia pastoris. In this study we produced functionally active rat rsCD59 and a chimeric rsCD59‐Crry protein in P. pastoris. The GPI anchor addition site of rat CD59 (Asn‐79) was replaced either by a stop codon to produce rsCD59, or with the sequence of the first five short consensus repeats of Crry to produce rsCD59‐Crry. Proteins were generated by fermentation and purified by affinity chromatography on an anti‐CD59 column. In a standard classical pathway haemolysis assay, all three rs proteins had inhibitory activity, with 50% inhibition at 0·5 µm (rsCrry and rsCD59‐Crry) and 4·4 µm (rsCD59). In an assay examining inhibition of C5b‐9, in which C5b‐7 was first formed, followed by purified C8 and C9, rsCD59 and rsCD59‐Crry were active with 50% inhibition at 0·8 µm (rsCD59‐Crry) and 1·3 µm (rsCD59). The degree of inhibition was independent of whether the C8 and C9 were of rat or human origin. Therefore, we have produced rsCD59 and rsCD59‐Crry in P. pastoris. The rsCD59 retains its inhibitory activity towards C5b‐9, while rsCD59‐Crry appears to have the combined activities of Crry and CD59. In a haemolytic assay, the inclusion of CD59 to Crry is of no additional benefit to Crry, which may illustrate the overall importance of the C3 convertase step. Yet, inclusion of Crry to CD59 increases the potency of CD59 towards C5b‐9.
Introduction
Activation of the complement cascade can have beneficial effects, such as in direct host defence against pathogens, immune clearance and in the induction and regulation of an immune response. 1 However, it is also clear that complement can lead to tissue damage in a variety of circumstances. These include traditional immune complex and antibody‐mediated inflammation and tissue injury. 2 More recently, it has become apparent that complement activation plays a role in other diverse conditions, such as ischaemia/reperfusion injury, thermal injury and hyperacute xenotransplant rejection.
To prevent injury of self tissue, complement is regulated by both plasma and cell‐associated proteins. The numerous complement regulators at various steps throughout the cascades of the alternative and classical pathways suggest that complement regulation is an important factor in normal human health. A focal point of regulation is at the level of the C3/C5 convertases of both pathways. This occurs in humans via the action of the plasma proteins, factor H and C4‐binding protein and the cell membrane proteins, CR1, decay‐accelerating factor and membrane co‐factor protein. 3 These regulators of complement activation (RCA) proteins inhibit C3/C5 convertases by accelerating their intrinsic decay and/or by acting as a factor I co‐factor for the cleavage and inactivation of C3b and C4b. These all share a common 60–70‐amino acid short consensus repeat (SCR), containing four invariant cysteines that form two intra‐SCR disulphide bonds. 3,4 Another site where complement is regulated is at the level of C5b‐9. CD59, anchored to the cell membrane by a glycosylphosphatidylinositol (GPI) linkage, inhibits the addition of C8 and C9 to C5b‐9, and is therefore an effective inhibitor of the formation of C5b‐9. 5
Because of their utility in studying models of human disease, rats and mice have been used extensively to dissect inflammatory mediator systems. Crry (CR1‐related protein/gene y) is a membrane‐bound intrinsic complement regulatory protein of rodents. 6,7 It has both genetic and functional relatedness to human CR1, yet is much more widely expressed. 8,9 Although decay‐accelerating factor also exists in rats and mice, it has a more limited distribution than Crry (or its human homologue), 10 and thus Crry appears to be the predominant C3 convertase regulator in these animals. In contrast, CD59 is conserved among species and has a widespread distribution as does Crry. 9,11,12
Therefore, the capacity to regulate complement is clearly present in normal animals. Still, in disease states, the tempo of complement activation overwhelms this intrinsic complement regulation. This has led to a surge of enthusiasm to produce complement inhibitors as therapeutics. Most of these are recombinant soluble (rs) proteins of the RCA family, such as rsCR1. 13 A number of other strategies have been employed, such as the use of chimeric recombinant proteins combining the functions of two inhibitors, 14,15 production of transgenic xenografts expressing complement regulators, 16 in vivo transfer of regulatory molecules 17 and ex vivo expression of complement regulators. 18
C5b‐9 generated near the site of C3/C5 convertases can have multiple effects, including cellular activation, injury and death. 19,20 There are a number of disease states in which C5b‐9 is directly pathogenic. 21 Therefore, the capacity to inhibit the production of C5b‐9, either alone or in combination with C3/C5 convertases, may be desirable in such conditions. We have previously produced rsCrry in the methyltrophic yeast, Pichia pastoris. 22 In this study we describe the design, production and functional analysis of rsCD59 and a chimeric rsCD59‐Crry protein.
Materials and methods
Construction of CD59 and CD59‐Crry expression vectors
DNA encoding the 77 N‐terminal amino acids of rat CD59 was obtained by polymerase chain reaction (PCR) from the full‐length cDNA clone of rat CD59 11 using the 5′ primer 5′‐ACCCGGGCTCAGATGCTACAACTGT‐3′ and 3′ primer 5′‐CCTATGGCTTGTCTTCGAAGCT‐3′ (Fig. 1a). This was designed to result in a stop codon prior to Asn‐79, to which the GPI anchor attaches. The resultant 242 base pair (bp) PCR product was sequenced to confirm fidelity and cloned into the pCRII vector (Invitrogen, San Diego, CA). The vector was digested with EcoRI and SmaI and the insert was purified by preparative agarose gel electrophoresis. This was subcloned into the pPIC9 expression vector (Invitrogen) cut with SnaBI and EcoRI. The pPIC9 contains the yeast α‐factor signal sequence, which we have found to be necessary for protein secretion, rather than the native signal sequence of the recombinant. 22
Figure 1.
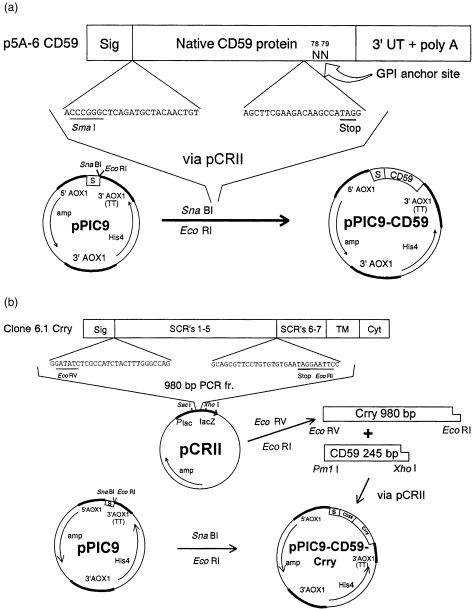
Strategy for integration of rat CD59 (a) and a chimeric rat CD59‐Crry construct (b) into pPIC9. The sequences of the 5′ and 3′ mutagenic primers are shown. The TAG stop codons are underlined. Sig, native signal peptides of CD59 and Crry; TM, transmembrane region of Crry; Cyt, cytoplasmic region of Crry; S, yeast α‐factor signal sequence.
DNA encoding the five N‐terminal SCRs of rat Crry was obtained by PCR from the full‐length cDNA of rat Crry 23 using the 5′ primer 5′‐GGATATCTCGCCATCTACTTTGGGCCAG‐3′ and 3′ primer 5′‐GGAATTCCTATTCACACACAGGAACGCTGC‐3′, and the resultant 980 bp PCR product was sequenced to confirm fidelity and cloned into pCRII (Fig. 1b). To create a CD59‐Crry product, DNA for CD59 was obtained and subcloned into pCRII as above, except that the 3′ primer was 5′‐CCACGTGTGGCTTGTCTTCGAAGCT‐3′, which incorporated a PmlI site after the nucleotides encoding Pro‐77. This was digested with XhoI and PmlI, gel purified and then subcloned into XhoI‐ and EcoRV‐cut pCRII‐Crry. The resultant pCRII‐CD59‐Crry was digested with SmaI and EcoRI, gel purified and ligated into SnaBI‐ and EcoRI‐cut pPIC9. In all cases, after each subcloning step, the orientation and nucleotide sequences were confirmed by restriction enzyme mapping and sequencing across the cloning sites.
Transformation of P. pastoris with CD59 and CD59‐Crry cDNA
The plasmids containing CD59 and CD59‐Crry were linearized with BglII and the P. pastoris strain GS115 (Invitrogen) was transformed by spheroplasting following the specific instructions of the manufacturer. Transformants were distinguished because of disruption of the AOX1 gene, leading to a Mut (methanol utilization slow) phenotype. PCR on genomic DNA was used to confirm that CD59 and CD59‐Crry cDNA were integrated into selected GS115 clones, using the primers listed above as well as primers for the α‐factor and 3′ AOX1.
To verify production of the relevant proteins, initial studies were done with selected P. pastoris clones grown in shake flasks as described previously. 22 By induction of the AOX1 promoter with methanol, recombinant proteins were produced and secreted into the culture supernatant. The appearance of protein products of the predicted size on sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) was used to identify appropriate clones.
Production of rsCD59 and rsCD59‐Crry by fermentation
Both rsCD59 and rsCD59‐Crry were produced by fermentation using a 5‐litre BioFlow 3000 fermenter (New Brunswick Scientific, Edison, NJ). The basic protocol provided by Invitrogen was followed. Briefly, 2·5 l of fermentation basal salts medium, pH 5·0, containing 0·435% PTM1 trace salts and 4% glycerol (v/v), was inoculated with 200 ml of the appropriate GS115 clone previously grown in shaking culture to an optical density at 600 nm (OD600) of > 2·0. The end of this batch glycerol phase was marked by a dissolved oxygen (DO) spike, following which a glycerol‐fed batch phase was begun, in which glycerol feeding was slowly increased to a maximum rate of 50 ml/hr. After the cellular wet weight rose above 350 g/l (typically within 24 hr), glycerol feeding was terminated. Following a DO spike, methanol was used as the carbon source to induce the AOX1 promoter. The rate of methanol feeding was slowly increased to 20 ml/hr. After approximately 96 hr of methanol feeding, the supernatant was harvested.
Purification of recombinant proteins
For affinity purification, 25 mg anti‐rat CD59 monoclonal antibody 6D1 24 was coupled to CNBr‐Sepharose 4B (Pharmacia, Piscataway, NJ) at 1 mg/ml. The harvested supernatant from P. pastoris was brought to pH 7·0, first passed over a 50‐ml column of Sepharose 4B and then over 6D1–Sepharose. The column was washed with 200 ml phosphate‐buffered saline (PBS) followed by 100 ml PBS containing an additional 1 m NaCl. Bound rsCD59 and rsCD59‐Crry were eluted with 20 mm diethylamine, 1 m NaCl, pH 11·5. Protein‐containing fractions were pooled, concentrated and exchanged into PBS in a stirred ultrafiltration cell (Amicon, Bedford, MA) with cut‐offs of 10 000 and 50 000 MW for rsCD59 and rsCD59‐Crry, respectively. As a control for these experiments, rsCrry was produced in P. pastoris and purified by Mono Q chromatography (Pharmacia) as described. 22
Immunoblotting
Serial dilutions of rsCD59, rsCrry and rsCD59‐Crry were applied as 3‐µl spots to nitrocellulose membranes. The membranes were air‐dried, blocked by incubation in PBS containing 5% non‐fat milk powder and 0·1% Tween‐20 (30 min) and rinsed in PBS. The membranes were then incubated (1 hr) in anti‐rat CD59 monoclonal antibody 6D1 or polyclonal rabbit anti‐rsCrry, 25 washed three times in PBS/Tween, then incubated (1 hr) with peroxidase‐conjugated anti‐mouse or anti‐rabbit immunoglobulin G (IgG; Bio‐Rad Laboratories, Hercules, CA). After further washing in PBS/Tween and a final wash in Tris‐buffered saline, the membrane was incubated in ECL reagent according to the manufacturer’s instructions (Pierce Chemical Co, Rockford, IL) and exposed to Kodak film.
N‐terminal amino acid analysis
Purified rsCD59 was separated by SDS–PAGE and transferred to a polyvinyl difluoride (PVDF) membrane (Millipore, Bedford, MA). The protein band was revealed by brief immersion in Coomassie blue R250 (1% in water) and the band was carefully excised for sequencing. N‐terminal amino acid analysis was performed with an Applied Biosystems Procise Sequencer (Foster City, CA).
Complement reagents
Normal rat serum (NRS) diluted in veronal‐buffered saline (VBS) containing 1 mm MgCl2, 0·15 mm CaCl2 and 1% gelatin (GVB++) was used as a source of C1–C9. Normal human serum (NHS) was depleted of C8 by passage over an anti‐C8 immunoaffinity column. 26 C5b‐6 was purified from acute‐phase sera by classical chromatography. 27 Human C7 was purified by affinity chromatography using anti‐C7 monoclonal antibody WU 4‐15 28 (provided by Dr Reinhard Würzner, Institut für Hygiene, University of Innsbruck, Austria). Rat and human C8 and C9 were purified by immunoaffinity or classical chromatography as described previously. 29 In some instances, NRS and NHS diluted in VBS with 1 mm ethylenediaminetetraacetic acid (EDTA), were used as sources of rat and human C8/C9, respectively.
Functional analyses
A standard haemolytic assay was used to examine inhibition of the classical pathway. Recombinant proteins were placed in a doubling dilution series in individual wells of a microtitre plate (10 µl/well) followed by 100 µl antibody‐sensitized sheep E (EA; 2% in GVB++; E, erthyrocytes) and 100 µl NRS in GVB++. The concentration of the latter was chosen to give subtotal haemolysis (90–95%) under standard assay conditions. After 15 min at 37°, the plates were centrifuged, and the supernatants were removed for quantification of haemoglobin release measured at OD412. Percentage haemolysis was calculated by the formula: (experimental OD412 – control OD412 (no NRS))/(100% lysis OD412 (H2O instead of NRS) – control OD412) × 100. Inhibition of haemolysis was expressed as a percentage relative to wells in which buffer alone was added and the concentration at which 50% inhibition was achieved (IC50) was calculated.
Inhibition of C8 and C9 incorporation into C5b‐9 was determined in the following assays. To form EAC5b‐7, EA were incubated with C8‐depleted NHS (1 : 5 in GVB++) for 15 min at 37°. The resultant cells were washed and stored in GVB++. Inhibitors were placed in doubling dilutions in individual wells of a microtitre plate (10 µl/well) followed by EAC5b‐7 (100 µl) and purified rat or human C8 and C9 (100 µl). C9 was added in excess (1 µg/well), while C8 was titrated to give approximately 90% lysis of EAC5b‐7 in the presence of C9 and absence of inhibitors. In other experiments, NHS or NRS diluted in VBS, 1 mm EDTA were used as sources of C8 and C9. Dilutions of sera were chosen to result in 90% haemolysis in the absence of inhibitors.
A reactive lysis assay was also used to evaluate inhibition of C8 and C9 incorporation into C5b‐9. Guinea‐pig E at 2% in 2 ml VBS were incubated with 15 µg C5b6 for 5 min at 37°, followed by 10 µg C7 for 15 min at 37°. The resultant EC5b‐7 were washed with VBS, suspended in 2 ml VBS, and then added in 100 µl aliquots to microtitre plates containing C8 at a dose chosen to result in 90% haemolysis (typically 0·05–0·1 µg/well), C9 (1 µg) and various concentrations of recombinant inhibitors. After incubation for 15 min at 37°, the percentage inhibition of haemolysis was determined as above.
Results
Production and characterization of rsCD59 and rsCD59‐Crry
Figure 1(a) demonstrates the constructed plasmid which contains nucleotides encoding the 77 N‐terminal amino acids of CD59. With inclusion of the α‐factor signal sequence and exclusion of nucleotides for Asn‐79, to which the GPI anchor attaches, this product is predicted to be secreted by P. pastoris. Figure 1(b) illustrates the construct which contains the same 77 N‐terminal amino acids of CD59 but followed by His‐Ile‐Ser‐Pro‐Ser‐Thr‐Leu‐Gly, of which the latter six are the final amino acids of the Crry signal sequence, and then the full five SCRs of Crry. 22 This is predicted to encode a 402 amino acid secreted CD59‐Crry protein.
These constructs were integrated into P. pastoris strain GS115. Two clones of each were identified that produced recombinant protein in shaking culture. Further studies were then performed to generate recombinant proteins by fermentation. The rsCD59 was produced and then purified by affinity chromatography on 6D1–Sepharose. As shown in Fig. 2(a), purified rsCD59 migrated at a MW of 15 000 under non‐reducing conditions. As the MW predicted from the amino acid sequence is 8 600, this indicates glycosylation by P. pastoris. 22 That such glycoslyation occurred was formally confirmed by a deglycosylation mobility shift assay, which showed that rsCD59 decreased in size by approximately 6 000 following treatment with N‐glycanase‐F (not shown). N‐terminal sequencing of rsCD59 confirmed that the expressed protein began with Leu‐1, as in the native sequence, and was identical throughout the sequence obtained (20 residues). 11
Figure 2.
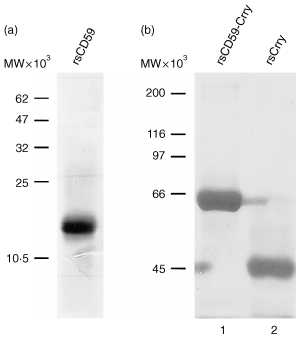
SDS–PAGE analysis of purified recombinant proteins. The rsCD59 was electrophoresed on a 15% acrylamide gel (a), while rsCD59‐Crry and rsCrry were electrophoresed on a 10% acrylamide gel (b). Proteins were stained with Coomassie blue.
The rsCD59‐Crry, which was also produced by fermentation, migrated at 60 000 MW under non‐reducing conditions (Fig. 2b). This is consistent with this chimeric protein containing the 45 000 MW rsCrry (also electrophoresed in Fig. 2b for comparison) 22 and the 15 000 MW rsCD59. As expected, all three recombinant proteins were single chain proteins as shown by SDS–PAGE under reducing conditions (not shown).
Immunoblotting of the three recombinant proteins was performed with anti‐CD59 monoclonal antibody 6D1 and polyclonal rabbit anti‐rat Crry. As expected, the anti‐CD59 and anti‐Crry antibodies both reacted with rsCD59‐Crry; in addition, anti‐Crry identified rsCrry but not rsCD59, and monoclonal antibody 6D1 reacted with rsCD59 but not rsCrry (Fig. 3).
Figure 3.
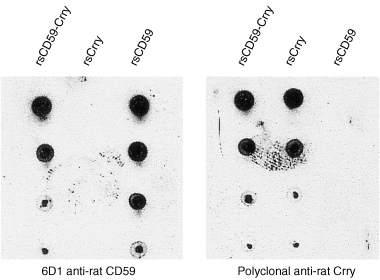
Immunoblot analysis of recombinant proteins. Serial dilutions of rsCD59‐Crry, rsCrry and rsCD59 were applied to nitrocellulose and detected with monoclonal antibody 6D1 anti‐rat CD59 or polyclonal anti‐rat Crry. These results confirm the reactivity of anti‐Crry and anti‐CD59 with the corresponding recombinant protein and that the chimeric rsCD59‐Crry retains reactivity with both antibodies.
Functional studies
The capacity of the different recombinant proteins to inhibit classical pathway‐directed haemolysis was determined using a standard haemolytic assay in the presence of NRS. As shown in Fig. 4, rsCrry and rsCD59‐Crry were equivalent on a molar basis to inhibit complement in this assay (IC50 = 0·5 µm), while rsCD59 was approximately 10‐fold less potent (IC50 = 4·4 µm).
Figure 4.
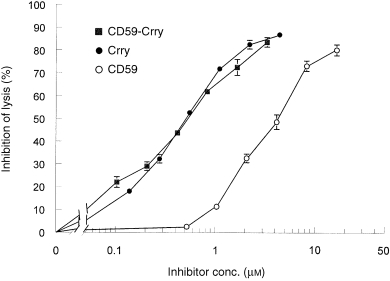
Dose–response relationship between input of recombinant complement inhibitors and inhibition of the classical pathway of C. Data shown are the per cent inhibition of haemolysis relative to no inhibitor and are the mean ± SEM of samples run in triplicate.
In an assay of the terminal complement pathway in which C5b‐9 was formed on EC5b‐7 by adding NRS diluted in EDTA as a source of C8 and C9, rsCD59‐Crry was a more active inhibitor than rsCD59 alone (Fig. 5a), with IC50 values of 1·6 and 4·6 µm, respectively. As expected, rsCrry had no inhibitory activity towards C5b‐9 generation. When purified rat C8 and C9 were added rather than NRS–EDTA, a similar relationship was identified, with rsCD59‐Crry being more potent than rsCD59 (Fig. 5b) (IC50 = 0·8 and 1·3 µm, respectively). In these studies, similar findings were obtained when either NHS–EDTA or human C8 and C9 were used in place of the analogous rat reagents (not shown).
Figure 5.
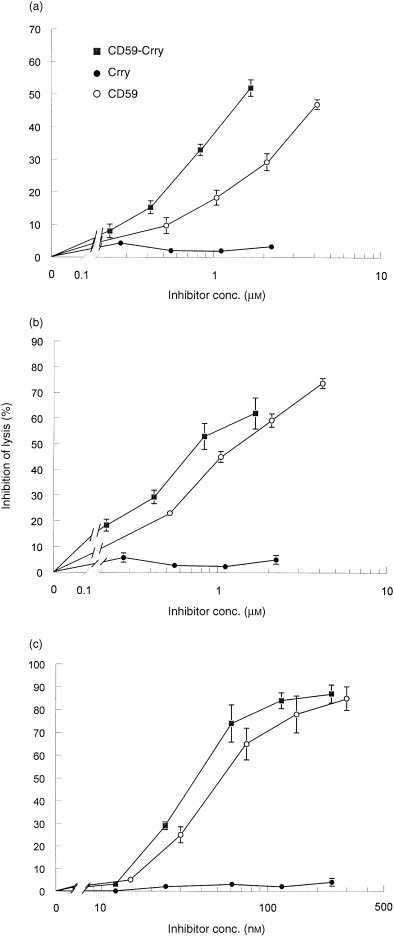
Dose–response relationship between input of recombinant complement inhibitors and inhibition of the terminal complement pathway. EC5b‐7 were formed by incubation of EA with C8‐deficient NHS (a, b) or using purified C5b6 and C7 (c) and completed by addition of NRS in VBS, 1 mm EDTA (a, c) or purified rat C8 and C9 (b) as sources of C8 and C9. Data shown are the per cent inhibition of haemolysis relative to no inhibitor and are the mean ± SEM of samples run in triplicate.
The above studies showed that rsCD59‐Crry was a more potent inhibitor of C5b‐9 than rsCD59. In these assays, EC5b‐7 were generated via classical pathway activation, and therefore antibody and earlier complement components were present on the sheep E surface. As an alternative approach, experiments were performed in which C5b‐7 was deposited using purified C5b6 and C7; C5b‐9 was then completed by addition of purified C8/C9. As shown in Fig. 5(c), rsCD59‐Crry was slightly more potent than rsCD59 in this system, with IC50 values of 37·3 and 53·2 nm, respectively.
Discussion
In this study we have produced recombinant soluble rat CD59. By virtue of its containing amino acids 1–77 of the native protein, 11 and by inclusion of the yeast α‐factor signal sequence in the construct, rCD59 was produced by P. pastoris as a soluble protein. As with the native protein, rsCD59 inhibited the insertion of C8 and C9 into the C5b‐9 membrane attack complex. 24 Comparable to previous studies in which human CD59 was produced as a recombinant soluble protein in Sf9 cells, rat rsCD59 is relatively inefficient in this inhibition. 30 However, as with native rat CD59 and unlike human CD59, rat rsCD59 is not homologously restricted and hence is active towards both rat and human C5b‐9. 12 We have also produced a chimeric rsCD59‐Crry protein containing this same rsCD59 to which the five N‐terminal SCRs of Crry have been added. Thus, rsCD59‐Crry combines the properties of both CD59 and Crry in one molecule. To our knowledge, this is the first time a single molecule containing both C3 convertase inhibition and terminal pathway inhibition has been engineered as a recombinant soluble complement regulator.
The advantages of using P. pastoris to produce recombinant proteins are several. As a eukaryote, appropriate protein processing and folding, and post‐translational modifications occur as with higher eukaryotic systems. Such correct protein processing assures proper function; for example, there are two disulphide bonds within each SCR in RCA proteins. 4 Pichia pastoris can be grown to a high cell density in a defined minimal medium containing methanol to stimulate the AOX1 promoter. Hence, large quantities of recombinant protein can be inexpensively obtained. In our production of rsCD59, rsCD59‐Crry and rsCrry we have achieved typical protein yields from 10 to 100 mg/l. Since P. pastoris secretes little endogenous protein, much of the protein secreted into the medium is the recombinant protein of interest, thereby making purification easier. Hence, large quantities of recombinant protein can be readily produced for use in in vivo studies.
In a standard haemolytic assay which relies upon full activation of the classical pathway from C1 to C9, rsCD59 showed inhibitory activity, consistent with its blocking the formation of C5b‐9 on E. In this assay, rsCrry was nearly 10‐fold more active than rsCD59 on a molar basis, indicating that blocking C3 convertases is a more efficient means of limiting complement activation than inhibition later in the pathway. Interestingly, the curves of rsCrry and rsCD59‐Crry were virtually superimposable, and thus inclusion of CD59 in rsCD59‐Crry provided no greater inhibition in this assay. In contrast, in assays of the terminal complement pathway utilizing EAC5b‐7 cells, rsCD59‐Crry was one and a half to three times more active than rsCD59 at equimolar doses.
We suspected that the enhanced activity of the chimeric protein was due to targeting through Crry to C3 fragments on the E surface, aiding delivery of the CD59 component to the cell. To investigate this finding further we used a purified component system in which no C3 fragment deposition occurs. Here too, the chimeric protein was more active than rsCD59, although the relative difference was small, and thus at least some of the enhanced activity may have been dependent on the presence of early complement components on the target. However, our attempts to identify binding of rsCrry or rsCD59‐Crry to C3b‐coated erythrocytes using flow cytometry have been unsuccessful (data not shown). These negative results are probably owing to the low affinities of these interactions, 31 which do not survive the multiple wash steps required for staining. The small enhancement observed in the purified component assay might indicate that Crry binds directly to the E surface. Alternatively, it may be that inclusion of Crry in rsCD59‐Crry allows CD59 to interact more favourably with membrane‐bound C5b‐9. 14,32 Such an orientation, in which CD59 is placed close to the plasma membrane, has been shown to be necessary for the activity of CD59 present in chimeric proteins containing decay‐accelerating factor 14 or the Fab portion of immunoglobulin. 32 Whatever the mechanism, the data indicate that the chimeric protein is a better inhibitor of the terminal pathway. This unanticipated finding makes rsCD59‐Crry a particularly attractive agent for use in vivo. Whether the human membrane C3 convertase regulators, decay‐accelerating factor and membrane co‐factor protein, have similar ‘targeting’ properties remains to be ascertained.
There are many experimental disease models in rats potentially amenable to treatment with complement inhibitors. In this decade, a great deal of effort has gone into the study of human rsCR1. Given the heterologous nature of rsCR1, these studies have of necessity been in short‐term disease models. 13 Still, they have illustrated the potential value of complement inhibition in diverse models of injury, and have extended the list of diseases that appear to be mediated by complement beyond traditional immune complex diseases to include entities such as ischaemia–reperfusion injury 33 and xenotransplant rejection. 34 The successful use of rsCR1 in animal models has supported its application to analogous acute human diseases and stimulated the production of alternatives to rsCR1 as a means of inhibiting human complement. 14,15,35 Because of the relevance of chronic disease models in rodents to human diseases, we and others have developed homologous complement inhibitors for use in animals. 22,36–38 Here, rsCD59‐Crry was engineered as a potentially valuable agent, since it combines the activities of Crry and CD59.
Armed with these three recombinant complement inhibitors, we will be in a position to shed light on which of them might be the most effective in disease conditions such as immune complex injury of the kidney, 39 or reperfusion injury and xenograft hyperacute rejection which appear to be due to natural antibodies. 40,41 In these various conditions, activation, predominantly through the classical pathway of complement, occurs ultimately leading to the generation of C5b‐9 which can result in cell death, injury and activation events. 19–21 The capacity to inhibit both C3 convertases and C5b‐9 together has theoretical advantages in such disease states. Although our data indicate that inclusion of CD59 to Crry adds little advantage in a classical pathway activation assay, the effects in vivo may be different. Studies of the relative efficacy of rsCrry and rsCD59‐Crry in rodent models of complement‐mediated disease will guide the rational design of complement inhibitors for use in humans.
Acknowledgments
This work was supported by National Institutes of Health grant DK41873; the Wellcome Trust; a chapter grant from the Arthritis Foundation, Greater Chicago Chapter; and, a grant‐in‐aid from the National Kidney Foundation of Illinois. J.J. Alexander was supported by National Institutes of Health training grant DK07510.
Abbreviations
- Crry
CR1‐related protein/gene y
- DO
dissolved oxygen
- GPI
glycosylphosphatidylinositol
- GVB++
VBS containing 1 mm MgCl2, 0·15 mm CaCl2 and 1% gelatin
- IC50
concentration at which 50% inhibition of haemolysis occurs
- NHS
normal human serum
- NRS
normal rat serum
- RCA
regulators of complement activation
- SCR
short consensus repeat
- VBS
veronal‐buffered saline, 145 mm NaCl, 5 mm sodium barbital, pH 7·3
References
- 1.Holers VM. Complement. In: Rich R, editor. Principles and Practices of Clinical Immunology. St. Louis, MO: Mosby; 1995. p. p.363. [Google Scholar]
- 2.Quigg RJ. Glomerular injury induced by antibody and complement. Sem Nephrol. 1991;11:259. [PubMed] [Google Scholar]
- 3.Campbell RD, Law SKA, Reid KBM, Sim RB. Structure, organization, and regulation of the complement genes. Annu Rev Immunol. 1988;6:161. doi: 10.1146/annurev.iy.06.040188.001113. [DOI] [PubMed] [Google Scholar]
- 4.Janatova J, Reid KB, Willis AC. Disulfide bonds are localized within the short consensus repeat units of complement regulatory proteins: C4b‐binding protein. Biochemistry. 1989;28:4754. doi: 10.1021/bi00437a036. [DOI] [PubMed] [Google Scholar]
- 5.Morgan BP, Meri S. Membrane proteins that protect against complement lysis. Springer Sem Immunopathol. 1994;15:369. doi: 10.1007/BF01837366. [DOI] [PubMed] [Google Scholar]
- 6.Wong W, Fearon DT. p65: a C3b‐binding protein on murine cells that shares antigenic determinants with the human C3b receptor (CR1) and is distinct from murine C3b receptor. J Immunol. 1985;134:4048. [PubMed] [Google Scholar]
- 7.Paul MS, Aegerter M, O'brien SE, Kurtz CB, Weis JH. The murine complement receptor gene family. I. Analysis of mCRY gene products and their homology to human CR1. J Immunol. 1989;142:582. [PubMed] [Google Scholar]
- 8.Li B, Sallee C, Dehoff M, Foley S, Molina H, Holers VM. Mouse Crry/p65: Characterization of monoclonal antibodies and the tissue distribution of a functional homologue of human MCP and DAF. J Immunol. 1993;151:4295. [PubMed] [Google Scholar]
- 9.Funabashi K, Okada N, Matsuo S, Yamamoto T, Morgan BP, Okada H. Tissue distribution of complement regulatory membrane proteins in rats. Immunology. 1994;81:444. [PMC free article] [PubMed] [Google Scholar]
- 10.Hinchliffe SJ, Spiller OB, Rushmere NK, Morgan BP. Molecular cloning and functional characterization of the rat analogue of human decay‐accelerating factor. J Immunol. 1998;161:5695. [PubMed] [Google Scholar]
- 11.Rushmere NK, Van Den Berg CW, Harrison RA, Morgan BP. Molecular cloning of the rat analogue of human CD59: structural comparison with human CD59 and identification of a putative active site. Biochem J. 1994;304:595. doi: 10.1042/bj3040595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Den Berg CW, Morgan BP. Complement‐inhibiting activities of human CD59 and analogues from rat, sheep, and pig are not homologously restricted. J Immunol. 1994;152:4095. [PubMed] [Google Scholar]
- 13.Kalli KR, Hsu P, Fearon DT. Therapeutic uses of recombinant complement protein inhibitors. Springer Sem Immunopathol. 1994;15:417. doi: 10.1007/BF01837368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fodor WL, Rollins SA, Guilmette ER, Setter E, Squinto SP. A novel bifunctional chimeric complement inhibitor that regulates C3 convertase and formation of the membrane attack complex. J Immunol. 1995;155:4135. [PubMed] [Google Scholar]
- 15.Higgins PJ, Jone‐long K, Lobell R, Sardonini C, Alessi MK, Yeh CG. A soluble chimeric complement inhibitory protein that possesses both decay‐accelerating and factor I cofactor activities. J Immunol. 1997;158:2872. [PubMed] [Google Scholar]
- 16.McCurry KR, Kooyman DL, Alvarado CG, et al. Human complement regulatory proteins protect swine‐to‐primate cardiac xenografts from humoral injury. Nature Med. 1995;1:423. doi: 10.1038/nm0595-423. [DOI] [PubMed] [Google Scholar]
- 17.Kooyman DL, Byrne GW, McClellan S, et al. In vivo transfer of GPI‐linked complement restriction factors from erythrocytes to the endothelium. Science. 1995;269:89. doi: 10.1126/science.7541557. [DOI] [PubMed] [Google Scholar]
- 18.Nangaku M, Quigg RJ, Shankland SJ, Okada N, Johnson RJ, Couser WG. Overexpression of Crry protects mesangial cells from complement‐ mediated injury. J Am Soc Nephrol. 1997;8:223. doi: 10.1681/ASN.V82223. [DOI] [PubMed] [Google Scholar]
- 19.Morgan BP. Effects of the membrane attack complex of complement on nucleated cells. Curr Top Microbiol Immunol. 1992;178:115. doi: 10.1007/978-3-642-77014-2_8. [DOI] [PubMed] [Google Scholar]
- 20.Nicholson‐weller A, Halperin JA. Membrane signaling by complement C5b‐9, the membrane attack complex. Immunol Res. 1993;12:244. doi: 10.1007/BF02918256. [DOI] [PubMed] [Google Scholar]
- 21.Couser WG. Mediation of immune glomerular injury. J Am Soc Nephrol. 1990;1:13. doi: 10.1681/ASN.V1113. [DOI] [PubMed] [Google Scholar]
- 22.He C, Alexander JJ, Lim A, Quigg RJ. Production of the rat complement regulator, Crry, as an active soluble protein in Pichia pastoris. Arch Biochem Biophys. 1997;341:347. doi: 10.1006/abbi.1997.9989. [DOI] [PubMed] [Google Scholar]
- 23.Quigg RJ, Lo CF, Alexander JJ, Sneed AE, Moxley G. Molecular characterization of rat Crry: widespread distribution of two alternative forms of Crry mRNA. Immunogenetics. 1995;42:362. doi: 10.1007/BF00179397. [DOI] [PubMed] [Google Scholar]
- 24.Hughes TR, Piddlesden SJ, Williams JD, Harrison RA, Morgan BP. Isolation and characterization of a membrane protein from rat erythrocytes which inhibits lysis by the membrane attack complex of rat complement. Biochem J. 1992;284:169. doi: 10.1042/bj2840169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiller B, He C, Salant DJ, Lim A, Alexander JJ, Quigg RJ. Inhibition of complement regulation is key to the pathogenesis of Heymann nephritis. J Exp Med. 1998;188:1353. doi: 10.1084/jem.188.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abraha A, Morgan BP, Luzio JP. The preparation and characterization of monoclonal antibodies to human complement component C8 and their use in purification of C8 and C8 subunits. Biochem J. 1988;251:285. doi: 10.1042/bj2510285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrison RA. In: Handbook of Experimental Immunology. Weir DM, Herzenberg LA, Blackwell C, editors. Cambridge, MA: Blackwell Science; 1996. [Google Scholar]
- 28.Würzner R, Nitze R, Götze O. C7*9, a new frequent C7 allele detected by an allotype‐specific monoclonal antibody. Complement Inflamm. 1990;7:290. doi: 10.1159/000463163. [DOI] [PubMed] [Google Scholar]
- 29.Jones J, Laffafian I, Morgan BP. Purification of C8 and C9 from rat serum. Complement Inflamm. 1990;7:42. doi: 10.1159/000463125. [DOI] [PubMed] [Google Scholar]
- 30.Sugita Y, Ito K, Shiozuka K, et al. Recombinant soluble CD59 inhibits reactive haemolysis with complement. Immunology. 1994;82:34. [PMC free article] [PubMed] [Google Scholar]
- 31.Pangburn MK. Differences between the binding sites of the complement regulatory proteins DAF, CR1, and factor H on C3 convertases. J Immunol. 1986;136:2216. [PubMed] [Google Scholar]
- 32.Zhang H, Yu J, Bajwa E, Morrison SL, Tomlinson S. Targeting of functional antibody‐CD59 fusion proteins to a cell surface. J Clin Invest. 1999;103:55. doi: 10.1172/JCI4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill J, Lindsay TF, Ortiz F, Yeh CG, Hechtman HB, Moore FD., Jr Soluble complement receptor type 1 ameliorates the local and remote organ injury after intestinal ischemia‐reperfusion in the rat. J Immunol. 1992;149:1723. [PubMed] [Google Scholar]
- 34.Xia W, Fearon DT, Moore FD, Jr, Schoen FJ, Ortiz F, Kirkman RL. Prolongation of guinea pig cardiac xenograft survival in rats by soluble human complement receptor type 1. Transplant Proc. 1992;24:479. [PubMed] [Google Scholar]
- 35.Christiansen D, Milland J, Thorley BR, McKenzie IF, Loveland BE. A functional analysis of recombinant soluble CD46 in vivo and a comparison with recombinant soluble forms of CD55 and CD35 in vitro. Eur J Immunol. 1996;26:578. doi: 10.1002/eji.1830260312. [DOI] [PubMed] [Google Scholar]
- 36.Kim Y‐U, Kinoshita T, Molina H, et al. Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay‐accelerating factor and membrane cofactor protein. J Exp Med. 1995;181:151. doi: 10.1084/jem.181.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quigg RJ, Kozono Y, Berthiaume D, et al. Blockade of antibody‐induced glomerulonephritis with Crry‐Ig, a soluble murine complement inhibitor. J Immunol. 1998;160:4553. [PubMed] [Google Scholar]
- 38.Quigg RJ, He C, Lim A, et al. Transgenic mice overexpressing the complement inhibitor Crry as a soluble protein are protected from antibody‐induced glomerular injury. J Exp Med. 1998;188:1321. doi: 10.1084/jem.188.7.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Couser WG, Johnson RJ, Young BA, Yeh CG, Toth CA, Rudolph AR. The effects of soluble recombinant complement receptor 1 on complement‐mediated experimental glomerulonephritis. J Am Soc Nephrol. 1995;5:1888. doi: 10.1681/ASN.V5111888. [DOI] [PubMed] [Google Scholar]
- 40.Parker W, Bruno D, Holzknecht ZE, Platt JL. Characterization and affinity isolation of xenoreactive human natural antibodies. J Immunol. 1994;153:3791. [PubMed] [Google Scholar]
- 41.Weiser MR, Williams JP, Moore FD, Jr, et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183:2343. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]