

Abstract
We have investigated whether CD95–CD95 ligand interactions are important in anterior chamber-associated immune deviation (ACAID) induced by soluble protein antigen, and if so, to identify the participating cells on which these molecules are expressed. Peritoneal exudate cells as antigen-presenting cells (APC) obtained from B6.lpr/lpr, B6.gld/gld and C57BL/6 mice were cultured with ovalbumin (OVA) and transforming growth factor-β2 (TGF-β2) overnight, then injected intravenously into C57BL/6 or B6.lpr/lpr recipients. Some B6.lpr/lpr mice were reconstituted with naive T cells from wild-type C57BL/6 donors. In other experiments, B6.lpr/lpr and B6.gld/gld mice received an anterior chamber injection of OVA followed 7 days later by subcutaneous immunization with OVA plus adjuvant. Delayed hypersensitivity (DH) was assessed with an ear swelling assay. T cells activated in vitro with OVA-pulsed, TGF-β-treated APC were tested in vivo for their capacity to suppress DH expression in a local adoptive transfer assay. The results indicate that when ACAID was induced by in-vitro generated ACAID-inducing cells, the APC expressed CD95L, and recipient T cells expressed CD95. The capacity of in vitro generated regulatory T cells to suppress DH expression to OVA in vivo was not governed by CD95–CD95L interactions. When OVA was injected into the anterior chamber of naive mice, CD95 expression was required for ACAID induction, although ACAID was readily induced in CD95L-deficient mice. We conclude that CD95–CD95L interactions are required in ACAID for the initial stage of APC presentation of eye-derived antigens to T cells, and that CD95–CD95L interactions participate at one or more additional step in the process by which ACAID is induced by soluble protein antigens.
Introduction
Antigens injected into the anterior chamber of the eye induce a stereotypic deviant systemic immune response termed anterior chamber-associated immune deviation (ACAID).1 The key antigen-specific characteristics of this response are (i) failed induction and expression of delayed hypersensitivity (DH) and (ii) intact, and sometimes even enhanced, production of antibodies.2,3 In immunologically naive mice, it is believed that indigenous intraocular antigen-presenting cells (APC) capture injected antigen, migrate across the trabecular meshwork to escape from the eye, and traffic via the blood to the spleen.4–6
Previous studies have shown that interactions of CD95 and its ligand (Fas and Fas ligand) are required if ACAID is to be induced by intracameral injection of haptenated splenocytes or virus antigens of herpes simplex virus-1 (HSV-1).7 These investigators speculated that induction of ACAID in the first instance relies upon CD95 expression by TNP-splenocytes, and upon CD95L expression by recipient cells (undefined). In the case of HSV-1 antigens, they speculated that recipient T cells must express both CD95 and CD95L. Studies reported from another laboratory have shown that interaction of CD95–CD95L is required for ACAID induction by injection of allogeneic splenocytes into the anterior chamber.8 In this example, the authors proposed that ACAID-inducing allogeneic splenocytes must express CD95, not its ligand, and that recipient cells (again undefined) must express CD95L. Although CD95–CD95L expression in all of these studies have identified the reactant expressed on one cell type during ACAID induction, but the expression of the counter-receptor on the responding cell type has been inferred, rather than demonstrated.
In the present study, we determined whether CD95–CD95L interactions are important when ACAID is induced by a soluble protein antigen. Our results indicate that the ability of in-vitro generated, ovalbumin (OVA)-pulsed, transforming growth factor-β2 (TGF-β2)-treated APC to induce ACAID in vivo requires expression of CD95L on the APC, and CD95 expression by recipient T cells. However, injection of OVA into the anterior chamber induced ACAID in CD95L deficient, but not CD95 deficient mice, implying that CD95–CD95L interactions are involved in more than one step of ACAID induction and expression.
Materials and methods
Animals
C57BL/6 mice at 6–8 weeks of age were purchased from Taconic Farms (Taconic, NY). The FasL-mutant, B6.gld/gld. and the Fas-negative mutant, B6.lpr/lpr were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were maintained and used experimentally by following the guidelines described by the Association for Research in Vision and Ophthalmology (ARVO, Bethesda, MD) resolution for use animals in research. All treatments were conducted under either ketamine HCl and xylazine anaesthesia.
Antigen
OVA (Sigma, St. Louis, MO) was used as the soluble protein antigen.
Preparation of T-cell reconstituted mice
B6.lpr/lpr mice were reconstituted by a modification of an original method previously described.9,10 In brief, recipient mice received 4 × 107 naive C57BL/6 T cells intravenously 24 hr prior to use. The injected T cells were isolated from spleens and lymph nodes of donors and enriched through T-cell enrichment columns (Biotecx Laboratories, Inc, Houston, TX).
Incubation of peritoneal exudate cells (PEC) with antigen and TGF-β
PEC taken from C57BL/6 mice, B6.gld/gld mice and B6.lpr/lpr mice, were collected 3 days after injection of 2·5 ml of thioglycolate (Sigma) intraperitoneally. PEC were treated as described previously.11 Briefly, 8 × 105 PEC/well were incubated overnight in 24-well plates in the presence of 5 mg/ml of OVA and 5 ng/ml of TGF-β2 (R and D Systems, Minneapolis, MN). Cells were incubated in serum-free medium composed of RPMI-1640, 10 mm HEPES, 0·1 mm nonessential amino acids, 1 mm sodium pyruvate, 100 U/ml penicillin, 100 µg/ml streptomycin (all from Biowhitaker, Walksville, MD), 1 × 10−5 m 2-mercaptoethanol (2-ME; Sigma Chemical Co., St. Louis, MO), supplemented with 0·1% bovine serum albumin (BSA; Sigma Chemical Co.), ITS+ culture supplement (1 µg/ml iron-free transferrin, 10 ng/ml linoleic acid, 0·3 ng/ml Na2Se, and 0·2 µg/ml Fe(NO3)3) (Collaborative Biochemical Products, Bedford, MA). On the following day, non-adherent cells were decanted, and the tightly adherent PEC that remained were harvested from the plates by vigorous pipetting, then were washed three times in Hank’s balanced salt solution (HBSS). When analyzed by flow cytometry for expression of F4/80, > 99% of these cells were positive (data not shown). A total of 100 µl of HBSS containing 2 × 105 PEC/ml was injected into the tail vein of naive syngeneic mice or B6.lpr/lpr mice.
Administration of antigen
For intraocular inoculation, mice received an injection of OVA (50 µg/2 µl HBSS) into the anterior chamber (AC) of the right eye as previously described.4 For conventional sensitization, 100 µl of an emulsion produced from a 1:1 mixture of OVA (2 mg/ml) and complete Freund’s adjuvant (CFA; Life Technologies, Grand Island, NY) was injected s.c. into the nape of the neck.
Assay of DH
Mice received an intradermal injection of 200 µg/10 µl HBSS of OVA into the right ear pinnae. After 24 and 48 hr, the ear swelling was measured by micrometer (Mitutoyo, MTI Corporation, Paramus, NJ).
Local adoptive transfer assay (LAT)
The LAT assay has been described in detail previously, and has been used for the detection of regulatory cells that impair the expression of DH.12 Naive mice received an anterior chamber injection of OVA (50 µg/2 µl); 7 days later, their spleens were removed, and rendered into single-cell suspensions. These splenocytes which are known to contain a population of regulatory cells were used as regulators, and were mixed with OVA-pulsed APC and responder (DH-mediating effector cells) cells obtained from various strains of mice immunized to OVA plus CFA. This cell mixture (10 µl) was injected into the right ear pinnae of naive syngeneic mice, and ear swelling was measured 24 and 48 hr later. Reduced ear swelling responses compared with positive controls indicate that regulatory cells have impaired the expression of DH (efferent phase). Splenocytes were harvested and depleted of red blood cells (RBC) by treatment of with Tris–NH4Cl. Suspensions of responder cells were prepared from lymph nodes (of naive mice, of OVA-primed C57BL/6 mice, and of B6.lpr/lpr mice). Splenocytes from AC-injected mice 7 days prior were used as the regulator cells. PEC were stimulated with OVA (100 µg/ml) overnight, and then harvested as stimulator cells. In this study, 5 × 106 responder cells were mixed with 5 × 106 regulator cells and 1 × 106 stimulator cells, and this inoculum (10 µl) was injected into the ear pinnae of naive syngeneic mice.
Statistical analysis
Data were analysed by using anova and Scheffe’s test. Means were considered to be significantly different when P < 0·05.
Results
ACAID-inducing capacity of TGF-β-treated PEC that are functionally deficient in CD95 or CD95L expression
The following experiments were designed to determine whether TGF-β-treated PEC taken from B6.lpr/lpr mice (CD95-deficient) and B6.gld/gld mice (CD95L-deficient) could generate an ACAID-signal in vitro. Peritoneal exudate cells (PEC) were obtained from B6.lpr/lpr and B6.gld/gld mice, cultured with TGF-β2 and OVA overnight, then injected into the tail vein of naive B6 mice. Seven days later the mice were immunized s.c. with OVA plus CFA. To detect DH, ear pinnae of the mice were challenged with OVA (200 µg/10 µl) 7 days after immunization. As the results of a representative experiment presented in Fig. 1 reveal, mice that received TGF-β2-treated, OVA-pulsed PEC from C57BL/6 or B6.lpr/lpr donors displayed impaired DH expression, whereas mice that received TGF-β2-treated, OVA-pulsed PEC from B6.gld/gld donors displayed intense DH expression (Fig. 1). These findings indicate that the immune deviation induced in mice by injection of TGF-β2-treated, OVA-pulsed PEC requires that the injected cells express CD95L.
Figure 1.
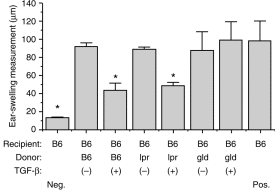
ACAID induction when PEC donors are C57BL/6, B6.lpr/lpr and B6.gld/gld mice. PEC from C57BL/6, B6.lpr/lpr and B6.gld/gld mice were incubated overnight (treatment) in the presence of TGF-β2 and OVA (5 mg/ml). The next day, the cells were washed three times with HBSS medium and injected (2 × 104) i.v. into syngeneic recipients. Seven days later these recipients were immunized with OVA (100 µg) in CFA s.c. and 1 week later their ears were challenged with OVA (400 µg). Naive mice were only ear challenged with OVA. Ear swelling responses after 24 hr are presented as mean±SE. Asterisks indicate mean values significantly lower than positive controls (P < 0·05).
ACAID-inducing capacity of TGF-β2-treated APC in mice deficient in CD95 expression
To confirm that in-vitro generated CD95L-expressing APC interact with CD95-bearing cells in the induction of immune deviation, normal C57BL/6 PEC were cultured with TGF-β2 and OVA overnight, then injected intravenously into C57BL/6 (control) or B6.lpr/lpr mice. The recipients were subsequently immunized with OVA–CFA and ear challenged as described above. The results of a representative experiment are presented in Fig. 2. As anticipated, TGF-β-treated C57BL/6 PEC induced immune deviation in naive C57BL/6 mice, but these same PEC failed to prevent the acquisition of DH following OVA immunization of B6.lpr/lpr mice. These findings indicate that recipient cells must express CD95 during induction of immune deviation by in-vitro generated APC, and they confirm that the requirement for CD95L expression on the inducing APC is related to interactions with CD95+ cells.
Figure 2.
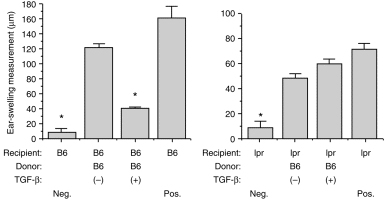
ACAID induction when recipients are C57BL/6 and B6.lpr/lpr mice. PEC from C57BL/6 mice were prepared as described in legend to Fig. 1 and injected i.v., respectively, into C57BL/6 (left graph) and B6.lpr/lpr (right graph) recipients. Seven days later, all mice were immunized and then ear challenged with OVA as described in Fig. 1. Ear swelling results are presented as mean± SE (n = 4–5). Asterisks indicate mean values significantly lower than positive controls (P < 0·05).
Capacity of TGF-β2-treated APC to induce immune deviation in CD95-deficient mice reconstituted with T cells capable of expressing CD95
Our next goal was to identify the CD95-expressing cell revealed by the previous experiments. To examine this point, we reconstituted CD95-deficient mice with T cells capable of expressing this molecule. Column purified T cells were prepared from spleen and lymph nodes of normal C57BL/6 donors and infused intravenously (4 × 107 cells/recipient) into B6.lpr/lpr mice. Simultaneously, TGF-β-treated, OVA-pulsed PEC from C57BL/6 donors were prepared, then injected intravenously into T-cell-reconstituted mice 24 hr after injection of T cells. Seven days later, the mice were immunized s.c. with OVA–CFA, then ear challenged for DH 1 week later. The results of one such experiment, displayed in Fig. 3, showed that TGF-β2-treated, OVA-pulsed PEC injected into CD95 deficient mice failed to prevent the acquisition of OVA-specific DH, whereas similar cells injected into B6.lpr/lpr mice that had been reconstituted with normal C57BL/6 T cells displayed only feeble ear swelling responses. We conclude that immune deviation can be induced in CD95-deficient mice by in-vitro generated ACAID-inducing APC only if the recipients are provided with exogenous T cells capable of expressing CD95. Thus, CD95-expressing T cells are the presumptive in vivo targets of APC treated with TGF-β2 and OVA when these cells induce immune deviation.
Figure 3.
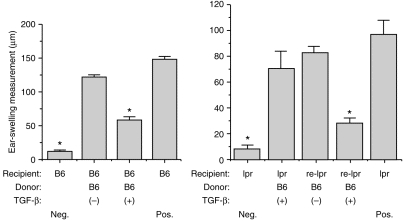
ACAID induction when recipients are C57BL/6 and T-cell reconstituted B6.lpr/lpr mice. To prepare T-cell reconstituted mice, mice received 4 × 107 naive C57BL/6 T cells intravenously 24 hr prior to use. T cells were isolated from C57BL/6 spleens and lymph nodes using T-cell enrichment columns. PEC from C57BL/6 were prepared as described in legend to Fig. 1 and injected i.v., respectively, into C57BL/6 (left graph) and T-cell reconstituted B6.lpr/lpr (right graph) recipients. Seven days later, all mice were immunized and then ear challenged with OVA as described in Fig. 1. Ear swelling results are presented as mean± SE (n = 5). Asterisks indicate mean values significantly lower than positive controls (P < 0·05).
Role of CD95–CD95L interactions in efferent suppression of soluble protein-induced ACAID
Wilbanks and Streilein have characterized the regulatory T cells that are generated in the spleens of mice with ACAID induced by heterologous protein antigens.13 As mentioned above, one population of T cells is CD4+ and prevents, in normal mice, the induction of immunity leading to the generation of DH effectors, i.e. afferent regulators. Another population of regulatory T cells is CD8+ and inhibits the expression of DH in vivo, i.e. efferent regulators. To determine whether CD95–CD95L interactions are involved in the suppression mediated by efferent regulators, we used a local adoptive transfer assay. In this assay, regulatory T cells are added to DH-generating responder T cells plus antigen-pulsed APC, and the subsequent ear swelling response measured. In the following experiments, responder T cells were obtained from OVA-primed B6.lpr/lpr mice or normal C57BL/6 mice (positive control). Splenic T cells from C57BL/6 mice in which ACAID had been induced 1 week previously by an injection of OVA into the anterior chamber were used as regulators, and OVA-pulsed C57BL/6 PEC were used as stimulators. This tripartite mixture was injected into the ear pinnae of naive C57BL/6 mice. When ear swelling responses were measured 24 and 48 hr later (see Fig. 4), the results indicated that responder cells from both normal and CD95 deficient donors were inhibited from eliciting DH in the presence of regulator T cells. Thus, CD95–CD95L interactions do not govern the ability of efferent regulator cells of ACAID to suppress the expression of DH to soluble protein antigens.
Figure 4.
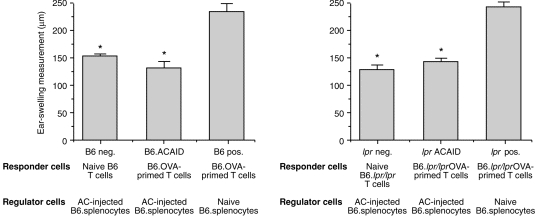
Influence of CD95 expression on capacity of regulatory T cells to suppress delayed hypersensitivity. Splenocytes from C57BL/6 mice that received an injection of OVA (50 µg) into the AC seven days earlier were mixed with OVA-pulsed C57BL/6 PEC plus responder cells from naive or OVA-primed C57BL/6 or B6.lpr/lpr mice, and then injected into the ear pinnae of naive C57BL/6 mice. Ear swelling responses were measured 24 hr later. Bars represent the mean± SE (n = 5). Asterisks indicate mean values significantly lower than positive controls (P < 0·05).
Soluble protein antigen induces ACAID in mice functionally deficient in CD95, not CD95L
Because CD95 and CD95L are required when in-vitro generated ACAID-inducing APC induce immune deviation when injected intravenously into normal mice, we predicted that mice universally deficient in either CD95 or CD95L expression should fail to acquire ACAID when antigen was injected intracamerally. To test this prediction, OVA (50 µg) was injected into the anterior chamber of one eye of wild-type, B6.gld/gld and B6.lpr/lpr mice. Seven days later, these mice were immunized s.c. with OVA plus CFA, and after 1 week their ear pinnae were challenged with injection of OVA (200 µg). Negative control mice received only ear pinnae injection of OVA. Ear swelling responses were measured 24 hr and 48 hr after challenge. The results of a representative experiment (one of three) are displayed in Fig. 5(a). As expected, wild-type C57BL/6 mice that received an AC injection of OVA prior to systemic immunization failed to acquire DH, i.e. they displayed ACAID. By contrast, CD95-deficient B6.lpr/lpr mice acquired intense OVA-specific DH even though they first received an AC injection of OVA. Unexpectedly, B6.gld/gld mice resembled wild-type mice in failing to acquire DH when pretreated with an AC injection of OVA (Fig. 5b). These paradoxical results imply that CD95, but not CD95L, is required for induction in vivo of ACAID with soluble protein antigens.
Figure 5.
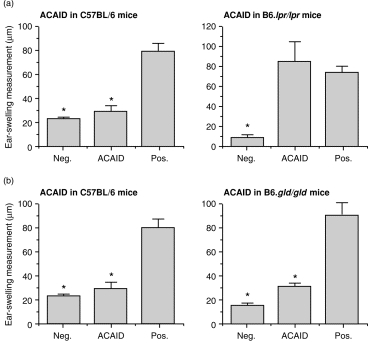
Capacity of OVA injected intracamerally to induce ACAID in mice deficient in CD95 or CD95L expression. OVA was injected into the anterior chamber of one eye of panels of normal C57BL/6 mice, and of B6.lpr/lpr (a) or B6.gld/gld (b) mice. Seven days later these mice were immunized subcutaneously with OVA plus CFA. One week later their ear pinnae, as well as pinnae of naive (negative control) mice, were challenged with an injection of OVA. Positive controls were immunized with OVA and CFA s.c. 7 days prior to intrapinnae challenge with OVA. Ear swelling responses were measured 24 hr after challenge. Bars represent the mean± SE (n = 5). Asterisks indicate mean values significantly lower than positive controls (P < 0·05).
Discussion
Griffith et al.7 first called attention to the importance of CD95/CD95L interactions in immune privilege of the eye and in the elicitation of systemic tolerance (ACAID) following injection of antigenic material into the anterior chamber. Since then, Kawashima et al.8 have reported that CD95/CD95L interactions are essential for ACAID induced by intracameral injection of allogeneic spleen cells. The results we report here indicate that CD95/CD95L interactions are also involved in immune deviation induced in mice by injection of soluble antigen-pulsed, TGF-β2-treated APC. Although all three lines of evidence implicate CD95/CD95L in the development of immune deviation following introduction of antigen into the eye, the experimental data presented by each laboratory are not consistent with a single, mechanistic explanation.
Our studies used a model system developed in our laboratory that is based on the in-vitro generation of an ACAID-inducing signal, i.e. conventional APC pretreated with TGF-β2 and pulsed with antigen in vitro. Intravenous injection of these cells into naive mice induces immune deviation similar to that elicited by injection of antigen directly into the anterior chamber of the eye.14 In the present experiments, we found that immune deviation occurred only if the ACAID-inducing APC expressed CD95L, and if the recipient expressed functional CD95. Further, CD95 expression by recipient T cells proved to be required. The simplest explanation for our results is that CD95L-bearing, OVA-pulsed, TGF-β2-treated APC encounter OVA-specific, CD95-bearing T cells in the recipient, and induce apoptosis among these responding cells. In support of this view is the recent report of Zhang et al.15 who demonstrated that CD95L+ antigen presenting cells induced antigen-specificT-cell tolerance by deleting antigen-specific T cells. These investigators suggested that CD95L+ APC at immune privileged sites might immigrate to secondary lymphoid organs where they could induce deletion among antigen-specific CD95-bearing T cells. Our results support this view and may thus explain why OVA-specific T cells that mediate delayed hypersensitivity are functionally deleted in mice with ACAID.
On the one hand, this construction is consistent with the report of Kawashima et al.8 These investigators reported that when allogeneic spleen cells induce ACAID following their introduction into the anterior chamber of the eye, the injected spleen cells must express CD95L, and the recipient must express CD95. One explanation for this outcome is that CD95L-bearing APC are present in the allogeneic spleen cell inoculum, and when they migrate to the spleen they encounter CD95+ allo-specific T cells, which are then deleted via apoptosis. On the other hand, Ferguson and his collaborators have reported that induction of ACAID by intracameral injection of HSV or syngeneic TNP-spleen cells requires that indigenous ocular cells express CD95L.7 Moreover, experimental evidence suggests that apoptosis of T cells is triggered via CD95/CD95L interactions within the eye itself, and that ACAID is the eventual outcome.
Perhaps more than one antigen-presentation mechanism gives rise to ACAID. If so, then different mechanisms may utilize CD95/CD95L interactions in different ways. The current literature contains two different views of the basis for ACAID induction. Ferguson et al.16 first reported that when TNP-spleen cells are injected into the eyes of mice, a soluble factor appears in serum within 2–3 days that can confer ACAID on naive mice into which it is injected. By contrast, Wilbanks et al.4 reported that when OVA is injected intracamerally, F4/80+ monocytic/dendritic cells appear in the peripheral blood at 2–3 days that confer immune deviation on mice into which these blood-borne cells are injected intravenously. The in-vitro generated ACAID-inducing signal used in the current experiments resembles the blood-borne leucocyte-associated signal of ACAID. Ferguson has argued that when ACAID is induced by soluble antigens, a cell-associated ACAID-inducing signal is generated, whereas when ACAID is induced by particulate antigens (TNP-spleen cells, HSV-1), a soluble ACAID-inducing signal is generated.17 In our laboratory,18 and in the laboratory of Cone and co-workers,19 the physicochemical nature of the antigen has not been found to be important in determining whether the ACAID-inducing signal is soluble or cell associated, but rather that the immune status of the recipient is pivotal. Thus, injection of antigen into the anterior chamber of eyes of mice presensitized to the antigen in question correlates with the appearance of a soluble factor in the blood. Cone and co-workers19 have provided evidence that soluble factors of this type originate from T cells. Injection of antigen, soluble or particulate, into eyes of unprimed mice uniformly leads to the appearance of a leucocyte-associated ACAID-inducing signal in the blood. Our current results are consistent with the view that the ACAID-inducing leucocyte that appears in the blood after AC injection of OVA expresses CD95L.
While this view is appealing, it fails to account for the results reported by Ferguson and his colleagues, and it does not fully explain our own experimental evidence. Particularly, our finding that OVA injected into the anterior chamber induced ACAID in CD95L-deficient mice, but not in CD95-deficient mice is paradoxical. We are at a loss to explain a mechanism in which only one partner of the CD95/CD95L pair is involved. Perhaps the explanation resides in unsuspected effects of CD95 and/or CD95L that are independent of each other. Moreover, the genetic lesion in the gld mutation has not been fully described, and there is the possibility that the lesion extends beyond the CD95L locus itself, involving other gene products.
Although our explanation for why CD95L expressed on in vitro generated ACAID-inducing APC can account for the elimination of DH-mediated CD95+ T cells, it does not address the fact that other antigen-specific T cells are still generated. For example, the spleens of mice with ACAID contain two populations of regulatory T cells, a CD4+ cell that suppresses the induction of DH, and a CD8+ cell that suppresses the expression of DH in vivo. We have recently reported that CD4+ T cells from spleens of mice that received AC injections of OVA respond to OVA stimulation in vitro by producing TGF-β, but not other anticipated cytokines (interleukin (IL)-2, IL-4, IL-10, interferon-γ (IFN-γ)). These cells are candidates for the afferent CD4+ regulatory cell described in ACAID, but at present we have no knowledge about their expression of either CD95 or its ligand. In the current experiments, we have tested the possibility that the efferent CD8+ regulatory T cell of ACAID suppresses DH expression via CD95/CD95L. Our results refute this possibility. Regulatory T cells removed from spleens of mice with ACAID were fully able to suppress DH expression in a local adoptive transfer assay in which the T cell responsible for triggering DH lacked CD95 expression (from B6.lpr/lpr donors). We infer that the T cells of ACAID that suppress DH expression accomplish this end independent of CD95 and its ligand.
Acknowledgments
We wish to thank Dr Kazuhisa Miyamoto for helpful suggestions, and we appreciate the managerial assistance of Dr Jacqueline Doherty. This work was supported by USPHS grant EY 05678.
Abbreviations
- ACAID
anterior chamber-associated immune deviation
- PEC
peritoneal exudate cells
References
- 1.Streilein JW, Niederkorn JY, Shadduck JA. Systemic immune unresponsiveness induced in adult mice by anterior chamber presentation of minor histocompatibility antigens. J Exp Med. 1980;152:1121. doi: 10.1084/jem.152.4.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Streilein JW. Immune regulation and the eye: a dangerous compromise. FASEB J. 1987;1:199. [PubMed] [Google Scholar]
- 3.Niederkorn JY, Streilein JW. Analysis of antibody production induced by allogeneic tumor cells inoculated into the anterior chamber of the eye. Transplantation. 1982;33:573. doi: 10.1097/00007890-198206000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Wilbanks GA, Streilein JW. Studies on the induction of anterior chamber-associated immune deviation (ACAID). 1. Evidence that an antigen-specific, ACAID-inducing, cell-associated signal exists in the peripheral blood. J Immunol. 1991;146:2610. [PubMed] [Google Scholar]
- 5.Wilbanks GA, Mammolenti M, Streilein JW. Studies on the induction of anterior chamber-associated immune deviation (ACAID). II. Eye-derived cells participate in generating blood-borne signals that induce ACAID. J Immunol. 1991;146:3018. [PubMed] [Google Scholar]
- 6.Wilbanks GA, Streilein JW. Macrophages capable of inducing anterior chamber associated immune deviation demonstrate spleen-seeking migratory properties. Region Immunol. 1992;4:130. [PubMed] [Google Scholar]
- 7.Griffith TS, Yu X, Herndon JM, Green DR, Ferguson TA. CD95-induced apoptosis of lymphocytes in an immune privileged site induces immunological tolerance. Immunity. 1996;5:7. doi: 10.1016/s1074-7613(00)80305-2. [DOI] [PubMed] [Google Scholar]
- 8.Kawashima H, Yamagami S, Tsuru T, Gregerson DS. Anterior chamber inoculation of splenocytes without Fas/Fas–ligand interaction primes for a delayed-type hypersensitivity response rather than inducing anterior chamber-associated immune deviation. Eur J Immunol. 1997;27:2490. doi: 10.1002/eji.1830271005. [DOI] [PubMed] [Google Scholar]
- 9.Taguchi O, Kontani K, Ikeda H, Kezuka T, Takeuchi M, Takahashi T. Tissue-specific suppressor T cells involved in self-tolerance are activated extrathymically by self-antigens. Immunology. 1994;82:365. [PMC free article] [PubMed] [Google Scholar]
- 10.Taguchi O, Nishizuka Y. Autoimmune oophoritis in thymectomized mice: T cell requirement in adoptive cell transfer. Clin Exp Immunol. 1980;42:324. [PMC free article] [PubMed] [Google Scholar]
- 11.Wilbanks GA, Streilein JW. Fluids from immune privileged sites endow macrophages with the capacity to induce antigen-specific immune deviation via a mechanism involving transforming growth factor-beta. Eur J Immunol. 1992;22:1031. doi: 10.1002/eji.1830220423. [DOI] [PubMed] [Google Scholar]
- 12.Williamson JS, Streilein JW. Impaired induction of delayed hypersensitivity following anterior chamber inoculation of alloantigens. Region Immunol. 1988;1:15. [PubMed] [Google Scholar]
- 13.Wilbanks GA, Streilein JW. Characterization of suppressor cells in anterior chamber-associated immune deviation (ACAID) induced by soluble antigen. Evidence of two functionally and phenotypically distinct T-suppressor cell populations. Immunology. 1990;71:383. [PMC free article] [PubMed] [Google Scholar]
- 14.Wilbanks GA, Mammolenti M, Streilein JW. Studies on the induction of anterior chamber-associated immune deviation (ACAID). III. Induction of ACAID depends upon intraocular transforming growth factor-beta. Eur J Immunol. 1992;22:165. doi: 10.1002/eji.1830220125. [DOI] [PubMed] [Google Scholar]
- 15.Zhang HG, Su X, Liu D, et al. Induction of specific T cell tolerance by Fas ligand-expressing antigen-presenting cells. J Immunol. 1999;162:1423. [PubMed] [Google Scholar]
- 16.Ferguson TA, Hayashi JD, Kaplan HJ. The immune response and the eye. III,. Anterior chamber-associated immune deviation can be adoptively transferred by serum. J Immunol. 1989;143:821. [PubMed] [Google Scholar]
- 17.Ferguson TA, Herndon JM. The immune response and the eye: the ACAID inducing signal is dependent on the nature of the antigen. Invest Ophthal Vis Sci. 1994;35:3085. [PubMed] [Google Scholar]
- 18.Streilein JW, Okamoto S, Hara Y, Kosiewicz M, Ksander B. Blood-borne signals that induce anterior chamber-associated immune deviation after intracameral injection of antigen. Invest Ophthalmol Vis Sci. 1997;38:2245. [PubMed] [Google Scholar]
- 19.Hadjikouti CA, Wang Y, O'rourke J, Cone RE. Intracameral injection of antigen potentiates the production of antigen-specific T cell proteins in serum after the induction of delayed-type hypersensitivity. Invest Ophthalmol Vis Sci. 1995;36:1470. [PubMed] [Google Scholar]