

Abstract
Mutation of the highly conserved leucine residue (Leu-247) converts 5-hydroxytryptamine (5HT) from an antagonist into an agonist of neuronal homomeric α7 nicotinic acetylcholine receptor expressed in Xenopus oocytes. We show here that acetylcholine (AcCho) activates two classes of single channels with conductances of 44 pS and 58 pS, similar to those activated by 5HT. However, the mean open time of AcCho-gated ion channels (11 ms) is briefer than that of 5HT-gated ion channels (18 ms). Furthermore, whereas the open time of AcCho channels lengthens with hyperpolarization, that of 5HT channels is decreased. In voltage-clamped oocytes, the apparent affinity of the α7 mutant receptor for 5HT is not modified by the presence of dihydro-β-erythroidine, which acts on the AcCho binding site in a competitive manner. This indicates a noncompetitive action of 5HT on nicotinic acetylcholine receptors. Considered together, our findings show that AcCho gates α7 mutant channels with similar conductance but with different kinetic profile than the channels gated by 5HT, suggesting that the two agonists act on different docking sites. These results will help to understand the crosstalk between cholinergic and serotonergic systems in the central nervous system.
Neuronal nicotinic acetylcholine receptors (nAcChoRs) are a family of ligand-gated channels, which are located mainly at presynaptic and postsynaptic sites in several areas of the nervous system (1–3). Because of their localization at central synapses, nAcChoRs are believed to play a prominent role during normal synaptic transmission and in processes involved in short-memory formation (1, 4) and in cognitive disorders associated with impairment of learning and memory (5, 6).
Of the several neuronal nAcChoRs, a particularly interesting one is the homomeric α7, due to its special functional and pharmacological profile. The α7 nAcChoR-channel is α-bungarotoxin-sensitive and highly permeable to Ca2+ (7–9). It exhibits fast desensitization, a nonlinear current-voltage (I–V) relation, and low affinity for acetylcholine (AcCho). Finally, it is potently inhibited by 5-hydroxhytryptamine (serotonin, 5HT) (10), suggesting a coupling between cholinergic and serotonergic neurotransmission (11–13). Threonine-for-leucine 247 substitution (L247T a7) in the channel domain renders the I–V relation linear, increases the affinity for AcCho, gives rise to an additional channel conductance, and appears to be crucial in receptor desensitization, slowing considerably the rate of AcCho-activated current decay (14). Strikingly, mutation of this highly conserved residue changes 5HT from antagonist to agonist (10). Because 5HT might act on α7 mutant receptors at a site different from that which binds AcCho (10), it was particularly interesting to compare the functional profile of channels activated by the natural transmitter with those activated by 5HT, using single-channel recording and expression in Xenopus oocytes. This also could give some clues on the functional link between the highly conserved leucine ring in the M2 channel domain (14) and the extracellular domain interacting with agonists (5). Therefore, we have recorded single-channel activity in Xenopus oocytes injected with L247T α7 subunit cDNA.
MATERIALS AND METHODS
Oocyte Injection.
Full-length cDNAs encoding the chicken L247T α7 neuronal nAcChoR subunits were kindly provided by M. Ballivet (University of Geneva, Switzerland) and expressed as previously described (14, 15). Stage VI oocytes were injected intranuclearly with cDNA clones using a pressure microinjector (Eppendorf) and a Singer Instruments micromanipulator (Somerset, U.K.). Preparation of oocytes and nuclear injection procedures were as detailed elsewhere (15, 16).
Electrophysiology.
Two to four days after injection whole-cell membrane currents were recorded in voltage-clamped oocytes using two microelectrodes filled with 3 M KCl (17). The oocytes were placed in a recording chamber (volume, 0.1 ml) and perfused continuously with oocyte Ringer (82.5 mM NaCl/2.5 mM KCl/2.5 mM CaCl2/1 mM MgCl2/5 mM Hepes, adjusted to pH 7.4 with NaOH) at controlled room temperature (20–21°C), in the presence of atropine (0.5 μM). To obtain dose-response dependencies the oocytes were held at −50 mV, and the drugs were applied to the oocyte at 3-min intervals. Each dose-response relationship was fitted with the Hill equation:
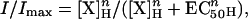 |
1 |
where [X] is the dose of the agonist, EC50 is the dissociation constant, nH is the Hill coefficient, and Imax is the maximum current response, as previously detailed (10). Drugs were dissolved in oocyte’s Ringer solution and bath applied at a flow rate of 11–12 ml/min.
Single-channel currents were recorded using the patch-clamp technique in the cell-attached mode, as reported (18–20), from the animal pole of the oocytes. 5HT-HCl was dissolved just before an experiment. Borosilicate-glass patch pipettes (1–3 MΩ tip resistance) were filled with extracellular solution containing AcCho or 5HT. Unless otherwise indicated, the agonist concentrations routinely used were those yielding a current about half the maximum (EC50), i.e., 200 nM for AcCho and 20 μM for 5HT in voltage-clamped oocytes (10). With these concentrations, the open-channel frequency was quite variable in range (0.1–20 Hz), and overlapping events were only occasionally observed. With concentrations 1–2 orders of magnitude higher, the opening frequency was substantially similar, whereas with 1–2 order of magnitude lower concentrations it was markedly reduced. Cell-attached recordings of unitary channel activity were performed using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). If no events were detected within 60 s after seal formation at 0 to 40 mV pipette potential, as well as when opening frequency was below 0.1 Hz, the patch was discarded. Typically a successful patch was stable for 5–25 min and had >200 opening transitions. Opening of channels was not observed in noninjected oocytes, nor in injected oocytes examined with a patch pipette filled with an AcCho-free solution, with the notably exception of a single patch that had low-frequency spontaneous openings of 45 pS conductance. That was probably a cholinergic channel because its conductance was different from that reported for the stretch channels (19), and voltage-clamped L247T α7 cDNA-injected oocytes exhibit a steady state spontaneous current blocked by cholinergic antagonists (E.P., F.E., T. Castrignano, and F. Grassi, unpublished work). Current recordings were filtered at 2 kHz, sampled at 10 kHz, and analyzed by pClamp 6.0.2 routines (Axon Instruments) using a threshold-crossing criterion. Events briefer than 0.2 ms were incompletely resolved and were excluded from the open-time histograms, that thus represented channel apparent mean open times. Histograms of amplitudes, apparent open times and log (shut times) were fitted with a single Gaussian function as appropriate, and with the sum of exponentials, respectively. Each histogram included from 250 to 2,000 events. Cumulative histograms of slope conductance values were fitted by the sum of two Gaussian functions using nonlinear fitting routines (included in sigma plot, Jandel, San Rafael, CA). Burst duration was studied by grouping openings separated by a specific critical time, which was calculated for each patch from the fitted parameters of the shut-time distribution (21–23). For each patch, slope conductances were obtained by least-squares linear fitting of current-voltage relations constructed by hyperpolarizing the patch potential up to 80 mV, and by depolarizing the patch membrane by up to 100 mV. The membrane potential was indirectly determined, in the cell-attached mode, by recording transmitter-activated channels at different patch potentials around the resting potential and by assuming the reversal potential (Erev) = −13 mV for AcCho, and Erev = −11 mV for 5HT (10). To avoid possible artefacts due to changes in the ionic composition of intracellular medium, values of conductances were discarded when the resting potential, extrapolated from the slope conductance, differed from the measured resting potential at the beginning and/or at the end of each experiment (−14 ± 7 mV; mean ± SD, n = 10) by more than the SD of the mean value.
Drugs and chemicals were purchased from Sigma, except for dihydro-β-erythroidine (DHβE), which was purchased from Research Biochemicals.
RESULTS
AcCho-Activated Single-channel Openings.
Single-channel recordings from L247T α7 cDNA-injected oocytes revealed a single population of channel openings, with a mean open time (τop) of 11.0 ± 1.6 ms (mean ± SEM.; 10 patches, 9 oocytes, 5 donors), made up of a briefer (τ1 = 2.6 ± 0.1 ms; 60%) and a longer (τ2 = 35 ± 6 ms; 40%) exponential components at an extrapolated membrane potential (EMP) of −52.1 ± 1.4 mV (Fig. 1A and D). Flickering activity was absent under our recording conditions, a behavior indicating no open channel block by the transmitter itself like that seen in muscle nAcChoRs (22). Opening bursts had a mean burst duration (τb) only slightly longer (13.7 ± 4.3 ms) than the open time. Channel activity and amplitude were rather stable over time at a given patch pipette potential, with only rare overlapping events. Channel clusters, with the exception of four patches, were not observed in agreement with the very low rate of desensitization of α7 mutant receptors (14). As the patch-membrane was hyperpolarized, the amount of voltage required to change the opening frequency e-fold was 45 mV, while it was 80 mV to change τop (six patches; see Fig. 1E).
Figure 1.

Properties of L247T α7 nAcChoR-channels activated by the natural transmitter AcCho. All panels refer to cell-attached patches of oocytes injected with chicken L247T α7 subunit cDNA. (A) Examples of single-channel currents at −70 mV EMP. Inward currents are represented by upward deflections. (B) Distribution of single-channel amplitudes at − 53 mV EMP, with 200 nM ACh in the patch pipette, in the same patch. Histogram is best fitted by a single Gaussian curve, with a mean of 2.42 ± 0.02 pA (n = 379). (C) Mean channel current amplitudes at different potentials plotted vs. pipette potential, and fitted by linear regression (solid line), yielding the slope conductance indicated. (D) Histogram of open durations fitted by the sum of two exponential functions, with τ1 = 2.69 ± 0.01 (67%), τ2 = 34.84 ± 0.01 (32%), and τop as indicated. Same patch as A–C. (E) Voltage-dependence of mean open time obtained from six patches (six cells, two donors). Averaged values of τop plotted vs. membrane potential. Error bars indicate SEM. The solid lines represent the least-squares fits of τop with the equation τop = τ0 exp(mV), where m indicates the slope of the linear fit. Note the increase of τop with membrane hyperpolarization.
Analyses of unitary events revealed two classes of high and low current levels, which differed in amplitude by ≈1.3-fold. The cumulative histogram of slope conductances disclosed two classes of channel conductance: a low class (γL), with a peak at 44.0 (± 1.2) pS (mean ± SD) and a high class (γH) at 58 (± 11) pS (Fig. 2; e.g., Fig. 1 B and C). In contrast to results reported elsewhere (14), both γH and γL appeared to be unrelated to the concentration of the transmitter in the patch pipette. For instance, the largest (71 pS) and smallest (38 pS) conductance values both were observed with an AcCho concentration of 300 nM; and in an oocyte with 20 μM AcCho in the patch pipette the channel conductance was 60 pS. Furthermore, (i) both γL and γH could be not related to the openings of sublevels or to channel oscillations between different modes (23); (ii) there were no transitions from higher to lower amplitude channels, and (iii) different channel populations were not detected in a given patch or in different patches from the same oocyte. In other words, each oocyte exhibited a homogeneous nAcChoR-channel population. All these observations indicate that γL and γH are associated to nAcChoR channel populations with different conducting states.
Figure 2.

Cumulative histograms of single-channel slope conductances. AcCho- and 5HT-gated channel conductances obtained in the cell-attached mode from 18 and 19 oocytes (10 donors), respectively. The histograms are best fitted by the sum of two Gaussian curves, with indicated mean conductances. Only determinations based on a minimum of five holding potentials were included. Ordinate represents number of patches in which each conductance value was observed.
5HT-Activated Currents in Voltage-Clamped Oocytes.
We previously have shown that 5HT behaves as an agonist on α7 mutant receptors and reported that its action is probably noncompetitive in nature, because the apparent affinity of AcCho for α7 mutant receptors is not modified by 5HT (10). To further address this issue, 5HT dose-current response relationships were constructed in the presence of DHβE, a drug acting competitively on the AcCho binding site of α7 wild-type receptors (24, 25). If the 5HT binding site is different from that of both AcCho and DHβE, then DHβE would be incapable of shifting the 5HT dose-response curve toward higher EC50 values. In six oocytes exposed to DHβE at a concentration of 0.1 μM, near the EC50 for α7 mutant receptor (0.21 μM; nH = 1.7; 3 oocytes; cf. ref. 25), the 5HT dose-response curve did not shift to the right (Fig. 3), as expected of a competitive agonism on the receptor. In contrast, the 20-s treatment with DHβE (0.1 μM) shifted the AcCho dose-current relation with EC50 = 0.26 μM and nH = 1.9 (5 oocytes) to EC50 value of 0.46 μM. Thus, these findings are consistent with the previously reported noncompetitive action of 5HT on α7 mutant receptors (10).
Figure 3.

Membrane currents from voltage-clamped oocytes injected with L247T α7 subunit cDNA. (A) Records of 5HT currents (I5HT) in a control (Left) and in an oocyte (Right) exposed to 0.1 μM DHβE before applying 5HT at the indicated doses (in μM). Note the current elicited by DHβE, before the 5HT current. Inward currents are represented by downward deflections. The timings of drug applications are indicated by bars above the current traces. (B) 5HT dose-I5HT relationship fitted to Eq. 1 (see Materials and Methods), in the presence of 0.1 μM DHβE (•) and in standard solution (○). Oocytes were held at −50 mV and exposed to DHβE for 20 s before eliciting I5HT in the presence of the drug. The current elicited by DHβE peaked to 90 (± 20) nA. The peak I5HT was normalized to that evoked by 0.5 mM 5HT (I5HT peak amplitude in standard solution: −1,490 ± 770 μA; mean ± SD). Each point represents the mean ± SD (n = 6; two donors for •; n = 8, three donors for ○). Control: EC50 = 20 μM; nH = 1.8. In the presence of DHβE: EC50 = 15 μM; nH = 1.8. Same values were obtained in oocytes voltage-clamped at −110 mV.
5HT-Gated Channels.
To compare the properties of the channels gated by 5HT with those gated by AcCho, cell-attached patch-clamp recordings were performed with 5HT in the patch pipette solution. The distribution of slope conductance values again revealed two classes of channel conductance, γL = 43.0 (± 2.4) pS and γH = 53.3 (± 4.6) pS, matching those found with AcCho (Fig. 2; e.g., Fig. 4 B and C). As with AcCho, the current amplitude of 5HT-gated channels routinely remained stable over all the time of the records, and neither direct transitions from different amplitude levels nor openings with a different conductance were seen from patch to patch in a single oocyte. In six oocytes where we tested AcCho and 5HT separately, we found similar conductance values. Furthermore, 5HT-gated channels gave a τop value of 18 ± 2.1 at −52.6 ± 1.5 mV EMP (mean ± SEM; 7 patches, 7 oocytes, 7 donors; Student’s t test P < 0.01 of τop data sets for AcCho-gated vs. 5HT-gated channels), with τ1 = 4.3 ± 0.7 ms (53%) and τ2 = 45 ± 6 ms (47%), both of which were considerably longer than those of AcCho-gated channels (e.g., Fig. 4D). The burst analysis of channel kinetics gave a τb value of 22.3 ± 5 ms, not very different from τop. Flickering of the channels was not observed at either positive EMP as high as +50 mV or as low as −100 mV, which was again inconsistent with an open channel block by the free 5HT molecules. Furthermore, the absence of 5HT-activated channel clusters indicates a low degree of nAcChoR desensitization by 5HT, as previously described under voltage-clamp conditions (10). Interestingly, in contrast to the lengthening of AcCho-gated channel lifetime by membrane hyperpolarization, τop of 5HT-activated channels was decreased considerably when the membrane was hyperpolarized (Fig. 4E vs. Fig. 1E). The amount of voltage required to decrease the open time by an e-fold was 85 mV. Furthermore, the 5HT-activated openings required a hyperpolarization of 50 mV to effect an e-fold increase in the opening frequency.
Figure 4.

Properties of L247T α7 nAcChoR-channels activated by 5HT. (A) Examples of single-channel currents at −59 mV EMP. (B) Distribution of single-channel amplitudes at same membrane potential, with 20 μM 5HT in the patch pipette, in the same patch. Histogram best fitted as in Fig. 1B, with mean of 2.72 ± 0.02 pA (n = 573). (C) Mean channel current amplitudes vs. pipette potential, plotted and fitted as in Fig. 1C, yielding the indicated slope conductance. Same patch as A and B. (D) Histogram of open durations fitted as Fig. 1D, with τ1 = 5.04 ± 0.02 (36%), τ2 = 27.4 ± 0.01 (64%), and τop as indicated. Same patch as A–C. (E) Voltage dependence of mean open time obtained from five patches (five cells, three donors). Values of τop, errors, and plot, as in Fig. 1E. Note the decrease of τop with hyperpolarization.
DISCUSSION
Mutation of a conserved leucine in the M2 channel domain of neuronal homomeric serotonin type 3 receptors and of heteromeric muscle nAcChoRs or neuronal γ-aminobutyric acid type A receptors, induces an increase in agonist sensitivity (26–30). The same mutation in the neuronal β2 subunit converts the antagonist cytisine into an agonist of neuronal α3β2 nAcChoR (31). Finally, this mutation in the homomeric neuronal nicotinic α7 receptor, which is considered to be an ancestor of heteromeric nAcChoRs (32), not only produces an increase in transmitter sensitivity but also converts the noncompetitive antagonist 5HT into a noncompetitive agonist (10).
We compared the properties of L247T α7 mutant-channels gated by the natural neurotransmitter AcCho with those gated by the heterologous transmitter 5HT, and found two classes of conductance, γL (44 pS) and γH (58 pS), for the channels gated by AcCho. Two classes of AcCho-activated channel conductance have been described previously, though their values differ from those reported here, particularly for γH (14). In addition to possible differences in oocyte and/or donor conditions, a further cause for such variety of conductances may result from different recording conditions (cell-attached in here vs. outside-out patches, cf., ref. 11). Differences in channel functional profile depending on recording configuration already have been reported (e.g., ref. 33).
Similar classes of conductance, γL (43 pS) and γH (53 pS), were obtained for channels activated by 5HT. However, their open time was considerably longer than that of AcCho-gated channels. Such slower kinetics cannot be attributed to increased receptor affinity for the transmitter (30), the EC50 for AcCho being lower than for 5HT (10), nor to the number of occupied binding sites (34). Our recordings were performed at sufficiently high neurotransmitter concentrations to make multiliganded unitary events the majority of the openings (34). Thus, the origin of the lengthening of the open time with 5HT as compared with AcCho is presumably due to different conformational changes in response to binding of the noncompetitive agonists. An attempt to identify the extracellular residues involved in the binding sites of nicotinic noncompetitive agonists has been described elsewhere (5).
We also report here that the membrane voltage-dependence of the lifetime of channels gated by AcCho and by 5HT is different: the open time of 5HT-gated channels becomes briefer whereas that of AcCho-gated channels lengthens with hyperpolarization. Under the assumption that the AcCho binding site as well as the docking site for the noncompetitive agonist 5HT are outside the field (34), the protein conformational change triggered in the extracellular domain could exhibit a different voltage-dependence during channel gating. Alternatively, with binding sites within the field, the receptor activation processes including the movement of transmitter molecules may be influenced differently by the electrical field. It is unlikely that docking sites are in the neck of the channel because an obvious flickering activity for both transmitters never was observed.
A relevant question arising from this study concerns the reasons why two classes of conductance for an homomeric receptor are observed. It generally is believed that a major determinant of the function of ligand-gated channels is receptor stoichiometry along with subunit disposition around the central pore and posttranslational modifications. Whereas in oocytes injected, for instance, with two neuronal cDNA subunits, eight arrangements of the resulting heteromeric nAcChoRs are possible (35); in the case of homomeric α7 nAcChoR, the constraint presented by assuming the pentameric subunit assembly predicts a unique arrangement for α7 nAcChoRs. However, it is not known definitely whether the α7 receptors are made up of five or four subunits. We can speculate that functional α7 nAcChoR can be formed by five or four subunits, leading to receptors gating different conductances. Whatever the explanation may be, the different conductances we observed depend on the oocyte itself because we regularly found a unique population of channels in each oocyte tested. Thus, the different conductances could derive from the oocyte expression system, which in some unknown way might influence channel gating (36). That would not be peculiar to the oocytes because in a human heterologous expression cell system two channel populations of α7 homomeric nAcChoRs already have been reported (37).
In conclusion, our findings suggest that the mutated leucine ring in the α7 nAcChoR renders active a docking site for 5HT, which is different from the AcCho binding site, and which triggers a change of protein conformation that leads to the activation of channels with longer open time than those gated by AcCho. Because cholinergic and serotonergic systems coexist in several regions of the central nervous system (1), these results could provide useful insights to the functional relevance of crosstalk between the two systems under physiological and pathological conditions. In particular, the agonism-antagonism switch of 5HT caused by threonine for leucine substitution might reveal intriguing interactions between cholinergic and serotonergic systems in the brain.
Acknowledgments
We thank Drs. Piotor Bregestovski, Sergio Fucile, and Enzo Wanke for critical reading of the manuscript. This work was supported by Ministero Universita’ Ricerca Scientifica Tecnologica (to F.E.), by Telethon (to E.P.), and by the National Institute of Neurological Disorders and Stroke (to R.M.).
ABBREVIATIONS
- 5HT
5-hydroxytryptamine
- I5HT
5HT-activated current
- AcCho
acetylcholine
- nAcChoR
nicotinic AcCho receptor
- L247Tα7
threonine-for-leucine 247 α7 subunit mutant
- DHβE
dihydro-β-erythroidine
- EMP
extrapolated membrane potential
- τi
- τop
mean open time
- τb
mean burst duration
- γH
high conductance channels
- γL
low conductance channels
- nH
Hill coefficient
References
- 1.McGehee D S, Role L. Curr Opin Neurobiol. 1996;6:342–349. doi: 10.1016/s0959-4388(96)80118-8. [DOI] [PubMed] [Google Scholar]
- 2.Wonnacot S. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Z, Coggan J S, Berg D K. Neuron. 1996;17:1231–1240. doi: 10.1016/s0896-6273(00)80253-6. [DOI] [PubMed] [Google Scholar]
- 4.Clarke P, Quik M, Adikofer F, Thurau K, editors. Advances in Pharmacological Sciences. Vol. 2. Basel: Birkhauser; 1995. [Google Scholar]
- 5.Maelicke A, Schrattenholz A, Schroder H. Semin Neurosci. 1995;7:103–114. [Google Scholar]
- 6.Perry E K, Morris C M, Court J A, Cheng A, Fairbairn A F, McKeith I G, Irving D, Brown A, Perry H. Neuroscience. 1995;64:385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- 7.Couturier S, Bertrand D, Matter J M, Hernandez M C, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 8.Séguela P, Wadiche J, Dineley-Miller K, Dani J A, Patrick J W. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delbono O, Gopalakrishnan M, Renganathan M, Monteggia L M, Sullivan J P. J Pharmacol Exp Ther. 1997;280:428–438. [PubMed] [Google Scholar]
- 10.Palma E, Mileo A M, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1996;93:11231–11235. doi: 10.1073/pnas.93.20.11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Colunga J J, Miledi R. Proc Natl Acad Sci USA. 1995;92:2919–2923. doi: 10.1073/pnas.92.7.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Colunga J, Miledi R. Proc Natl Acad Sci USA. 1996;93:3990–3994. doi: 10.1073/pnas.93.9.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Colunga J, Awad J N, Miledi R. Proc Natl Acad Sci USA. 1997;94:2041–2044. doi: 10.1073/pnas.94.5.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Revah F, Bertrand D, Galzi J L, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux J P. Nature (London) 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 15.Bertrand D, Cooper E, Valera S, Rungger D, Ballivet M. Methods Neurosci. 1991;4:174–193. [Google Scholar]
- 16.Palma E, Bertrand S, Binzoni T, Bertrand D. J Physiol (London) 1996;491:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miledi R. Proc R Soc London B. 1982;215:491–497. doi: 10.1098/rspb.1982.0056. [DOI] [PubMed] [Google Scholar]
- 18.Mileo A M, Palma E, Polenzani L, Limatola C, Grassi F, Eusebi F. J Neurosci Res. 1995;41:443–451. doi: 10.1002/jnr.490410403. [DOI] [PubMed] [Google Scholar]
- 19.Methfessel C, Witzemann V, Takahashi T, Mishina M, Numa S, Sakmann B. Pflügers Arch Eur J Physiol. 1986;407:577–588. doi: 10.1007/BF00582635. [DOI] [PubMed] [Google Scholar]
- 20.Miledi R, Parker I, Sumikawa K. Proc R Soc London B. 1983;218:481–484. doi: 10.1098/rspb.1983.0053. [DOI] [PubMed] [Google Scholar]
- 21.Colquhoun D, Sakmann B. J Physiol (London) 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colquhoun D, Ogden D C. J Physiol (London) 1988;395:131–159. doi: 10.1113/jphysiol.1988.sp016912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss D S, Magelby K L. J Neurosci. 1989;9:1316–1324. [Google Scholar]
- 24.Bertrand D, Bertrand S, Ballivet M. Neurosci Lett. 1992;146:87–90. doi: 10.1016/0304-3940(92)90179-b. [DOI] [PubMed] [Google Scholar]
- 25.Bertrand D, Dellivers-Thiery A, Revah F, Galzi J L, Hussy N, Mulle C, Bertrand S, Ballivet M, Changeux J P. Proc Natl Acad Sci USA. 1992;89:1261–1265. doi: 10.1073/pnas.89.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yakel J L, Lagrutta A, Adelman J P, North R A. Proc Natl Acad Sci USA. 1993;90:5030–5033. doi: 10.1073/pnas.90.11.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filatov G N, White M M. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- 28.Labarca C, Nowak M W, Zhang H, Tang L, Deshpande P, Lester H A. Nature (London) 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- 29.Chang Y, Wang R, Barot S, Weiss D S. J Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearney P C, Zhang H, Zhong W, Dougherty D A, Lester H A. Neuron. 1996;17:1221–1229. doi: 10.1016/s0896-6273(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 31.Colquhoun L M, Patrick J W. Soc Neurosci Abstr. 1996;22:1260. [Google Scholar]
- 32.Ortells M O, Lunt G G. Trends Neurosci. 1995;18:121–127. doi: 10.1016/0166-2236(95)93887-4. [DOI] [PubMed] [Google Scholar]
- 33.Clark B A, Farrant M, Cull-Candy S G. J Neurosci. 1997;17:107–116. doi: 10.1523/JNEUROSCI.17-01-00107.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auerbach A, Sigurdson W, Chen J, Akk G. J Physiol (London) 1996;494:155–170. doi: 10.1113/jphysiol.1996.sp021482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palma E, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1997;94:1539–1543. doi: 10.1073/pnas.94.4.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sivilotti L G, McNeil D K, Lewis T M, Nassar M A, Schoepfer R, Colquhoun D. J Physiol (London) 1997;500:123–138. doi: 10.1113/jphysiol.1997.sp022004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fucile S, Ragozzino D A, Giovannelli A, Grassi F, Mileo A M, Ballivet M, Alemà S, Eusebi F. Eur J Neurosci. 1997;9:480–488. doi: 10.1111/j.1460-9568.1997.tb01625.x. [DOI] [PubMed] [Google Scholar]