

Abstract
The mechanisms that cause aging are not well understood. The oxidative stress hypothesis proposes that the changes associated with aging are a consequence of random oxidative damage to biomolecules. We hypothesized that oxidation of specific proteins is critical in controlling the rate of the aging process. Utilizing an immunochemical probe for oxidatively modified proteins, we show that mitochondrial aconitase, an enzyme in the citric acid cycle, is a specific target during aging of the housefly. The oxidative damage detected immunochemically was paralleled by a loss of catalytic activity of aconitase, an enzyme activity that is critical in energy metabolism. Experimental manipulations which decrease aconitase activity should therefore cause a decrease in life-span. This expected decrease was observed when flies were exposed to hyperoxia, which oxidizes aconitase, and when they were given fluoroacetate, an inhibitor of aconitase. The identification of a specific target of oxidative damage during aging allows for the assessment of the physiological age of a specific individual and provides a method for the evaluation of treatments designed to affect the aging process.
The aging process or senescence is characterized by a ubiquitous decline in the functional capacity of various physiological systems and is perhaps most noticeably manifested in the loss of motor ability and the stamina for sustained physical effort. One hypothesis of aging proposes that the characteristic changes of aging are a consequence of the accumulation of random oxidative damage to cellular molecules, caused by reactive oxygen or nitrogen species, produced under normal physiological conditions (1, 2). This hypothesis is supported by experimental data from many laboratories which established that cellular steady-state level of oxidatively damaged macromolecules increases with age in all species examined thus far (3–7). The increase is especially pronounced in the latter part of the life-span. Although the oxidative modifications clearly affect nuclear and mitochondrial DNA, they are also readily observed in proteins (5). Oxidatively modified proteins are generally dysfunctional, losing catalytic or structural integrity (8). Thus, oxidative damage to proteins is considered to be of key importance in the aging process. The protein carbonyl content is a general measure of oxidative damage, and substantial increases in protein carbonyls have been reported during aging in the homogenates of a variety of tissues and species (8). Such findings have contributed to the prevalent view that random modifications of protein molecules cause a generalized decrease in metabolic efficiency occurring during senescence.
However, we hypothesized that oxidative modification of a particular target or targets occurs during aging and that loss of activity of this key target initiates an amplification of oxidative damage which becomes particularly evident during the latter part of the life-span of an organism. Mitochondria are the major cellular sources of reactive oxygen species such as superoxide anion radical and hydrogen peroxide, and the rates of their generation increase during aging (9–11). In insects as well as in mammals, oxidative damage to mitochondrial proteins and DNA increases whereas oxidative phosphorylation declines during aging (12–14). Furthermore, oxidative damage to an essential mitochondrial protein could also be a critical initiator of age-related accumulation of oxidized proteins, a phenomenon that would have been difficult to detect in previous studies with whole cell homogenates (15). It was thus reasoned that if specific mitochondrial protein oxidative damage indeed plays a role in senescence and in the associated loss of motor (flight) ability in flies, such protein(s) should be detectable in mitochondria. Mitochondria from the flight muscles of adult houseflies were selected for this study because of the relatively high rate of oxygen consumption in this tissue (16) and because it is possible to identify flies of the same chronological age which differ in physiological age. The distinction between chronological and physiological age can be made because the ability to fly decreases with age and is entirely lost in the days before death. Thus, in a cohort population, flies which only walk (“crawlers”) and are unable to fly will die within a few days while those which can fly (“flyers”) will live considerably longer (17, 18).
MATERIALS AND METHODS
Materials.
Acrylamide/bis-acrylamide was purchased from Fisher Scientific. Ammonium persulfate and N,N,N′,N′-tetramethylethylenediamine were purchased from Bio-Rad. BSA, Coomassie brilliant blue R-250, DTT, 2,4-dinitrophenylhydrazine (DNPH), glycine, isocitrate, NADP+, Tris(hydroxymethyl) aminomethane, SDS, and Triton X-100 were purchased from Sigma. Vanadyl sulfate trihydrate was obtained from Aldrich. Bicinchoninic acid protein assay kit was purchased from Pierce. Mouse anti-2,4-dinitrophenyl (DNP) mAb (IgG) and rat anti-mouse IgG mAb conjugated with horseradish peroxidase were purchased from Zymed. Immobilon-P membrane was purchased from Millipore. Enhanced chemiluminescence immunochemical detection kit was obtained from Amersham. Protein standard markers were obtained from Novex. Diethylaminoethyl Sepharose Fast Flow and carboxymethyl Sepharose Fast Flow were purchased from Pharmacia Biotech.
Animals.
Male houseflies were used in all experiments. After hatching from the pupae, houseflies were separated by sex, and male flies were placed in 1-ft3 cages at 25°C and 50% relative humidity. Flies were fed on sucrose and water, which has been demonstrated in this laboratory to result in a longer life-span than a diet additionally containing lipids and/or proteins. Age-related studies were conducted in flies ranging from 5 to 15 days of age because mitochondria undergo developmental maturation during the early phase of adult life, and increased mortality begin around 15 days of age.
Isolation of Mitochondria.
Mitochondria were isolated from the thoracic flight muscles of adult flies by a modification of the procedure described by Van Den Bergh (19). Flies confined in a plastic tube were immobilized by chilling and the thoraces were separated from the head and the abdomen. Thoraces were then placed in a mortar containing homogenization buffer (5 ml/200 thoraces) consisting of 154 mM KCl, 0.16 mM KHCO3, and 1.0 mM EDTA (pH 7.0) and gently pounded. The resulting mash was centrifuged at 150 × g for 5 min and the pellet was discarded. The supernatant was further centrifuged at 3,000 × g for 10 min. The mitochondrial pellet was rinsed carefully, without disturbing the pellet, with two portions of buffer and then stored at −80°C until analysis.
Polyacrylamide Gel Electrophoresis.
Mitochondrial proteins (typically 1 mg/ml) were treated with 10 mM DNPH (in 2 M HCl) for 30 min at room temperature. The sample was then neutralized and electrophoresed under reducing conditions by SDS/PAGE according to Laemmli (20) using a Mini-PROTEAN II electrophoresis cell (Bio-Rad). Gels were stained with Coomassie brilliant blue R-250.
Immunochemical Detection.
Immunochemical detection was performed according to the methods of Keller et al. (21) and Shacter et al. (22). Following SDS/PAGE, proteins were transferred to Immobilon-P membranes with Mini Trans-Blot electrophoretic transfer cell (Bio-Rad) using transfer buffer [25 mM Tris/192 mM glycine/20% (vol/vol) methanol, pH 8.3) and 100 V (constant voltage) for 1 h. The blots were incubated with 50 ml 5% nonfat dried milk (wt/vol) for at least 1 h, and then washed three times for 10 min with Tris-buffered saline (20 mM Tris/500 mM NaCl, pH 7.5) containing 0.1% Tween-20 (TBST). Blots were incubated for 1 h at room temperature with anti-dinitrophenyl mAbs (diluted 1:1000 in TBST containing 0.2% BSA). The primary antibody was removed, and the blots were washed three times (10 min each) with TBST. The blots were then incubated in horseradish peroxidase-labeled mouse anti-rat IgG (diluted 1:10,000 in TBST containing 0.2% BSA) for 1 h at room temperature. After the blots were washed with TBST three times (10 min each), the oxidized proteins were visualized with an enhanced chemiluminescence detection kit. Proteins on the membrane were stained with Amido Black. An 84-kDa mitochondrial matrix protein exhibited a strong and an age-dependent increase in staining for protein carbonyls.
Purification and N-Terminal Microsequencing of the 84-kDa Protein.
Mitochondria, resuspended in 50 mM sodium phosphate (pH 7.4), were sonicated in Branson 2200 sonifier four times for 30 s with 1-min intervals. The sonicated sample was centrifuged at 8,250 × g for 10 min, and the supernatant further centrifuged at 80,000 × g for 40 min. The supernatant contained mitochondrial matrix, whereas the pellet contained mitochondrial membranes. The matrix fraction was 70% saturated by the addition of solid ammonium sulfate, and centrifuged at 30,000 × g for 30 min. The supernatant was dialyzed against 50 mM sodium phosphate (pH 7.4) for at least 12 h with several changes of buffer. The sample was concentrated and applied to a diethylaminoethyl Sepharose fast flow column, which was pre-equilibrated with 20 mM Tris buffer (pH 8.2). The column was washed with Tris buffer containing increasing concentration of NaCl (0–500 mM, step gradient with 20 ml of elution buffer in each step, the increment of NaCl is 100 mM, made from a 2 M stock solution). Fractions containing the 84-kDa protein were pooled together, concentrated, and applied to a carboxymethyl Sepharose fast flow column. The 84-kDa protein was eluted by 50 mM Mes buffer (pH 6.2) containing increasing concentration of NaCl (step gradient, same as above). The N-terminal amino acid sequence of this protein, blotted to Immobilon-P membrane following SDS/PAGE, was determined by automated Edman degradation on a Hewlett–Packard model G1000A protein sequencer.
Enzyme Activity Assay.
Mitochondria, resuspended in Tris⋅HCl buffer (154 mM, pH 7.4), were sonicated four times for 30 s, with 1-min interval between each sonication. Unbroken mitochondria were removed by centrifugation at 8,250 × g for 10 min at 4°C. The aconitase activity was determined in the supernatant using two different methods. Method 1 is a coupled assay in which the formation of NADPH was followed at 340 nm, using isocitrate as a substrate (23). Method 2 is based on the formation of isocitrate from cis-aconitate, measured as the decrease in absorbance at 240 nm (24). Citrate synthase was assayed according to Srere (25). Protein concentrations were determined by bicinchoninic acid assay (26) using BSA as the standard.
Densitometric Estimation of Aconitase Carbonyl Content.
BSA was used as the standard for densitometric aconitase carbonyl determination as described by Winn and Wells (27). BSA (1 mg/ml in 10 mM sodium phosphate, pH 7.4) was oxidized by a hydroxyl radical generating system consisting of 1 mM vanadyl sulfate and 1 mM H2O2 (28–31). Immediately after the addition of vanadyl sulfate, the protein was derivatized with an equal volume of 0.5 mM DNPH in 0.1 M sodium phosphate buffer (pH 6.3). The oxidized BSA carbonyl content was determined spectrophotometrically as described by Levine et al. (32). The obtained value was used for estimating aconitase carbonyls.
Treatment with Oxygen or Fluoroacetate.
Flies confined within a 1-ft3 cage were placed in a sealed Plexiglas container connected via a gas manometer to a gas cylinder containing 100% oxygen. Oxygen was bubbled through water and then passed into the Plexiglas chamber under a low, constant pressure. Exposure to ≈100% oxygen is known to increase the rates of mitochondrial superoxide and hydrogen peroxide production (33, 34), providing a model of accelerated aging, and causes death within 5 days (35, 36). For the fluoroacetate experiments, houseflies (200/cage) were given tap water with varying concentrations of fluoroacetate.
RESULTS
Identification of Mitochondrial Aconitase as a Target of Oxidative Damage During Aging.
To search for proteins which were especially sensitive to oxidative damage during aging, we used a previously developed immunochemical assay for protein carbonyl groups (21, 22). Mitochondrial matrix fraction was reacted with the carbonyl reagent, DNPH, resolved by SDS/PAGE, and oxidatively modified proteins were detected with anti-DNP antibodies. The level of protein carbonyls in the mitochondrial matrix (increased immunostain intensity) was obtained from mitochondria that were not subjected to an oxidative stress other than that imposed by age. Although a relatively large number of protein bands were visualized by Coomassie blue staining, only one major carbonyl-containing band was seen (Fig. 1A). This protein, with molecular weight of ≈84 kDa and pI of 7.3, exhibited a negative immunostain when the sample was not treated with DNPH and showed an increasing carbonyl content with increasing age (Fig. 1B). It was therefore purified and subjected to automated Edman sequencing which allowed its identification as mitochondrial aconitase (Fig. 1C).
Figure 1.
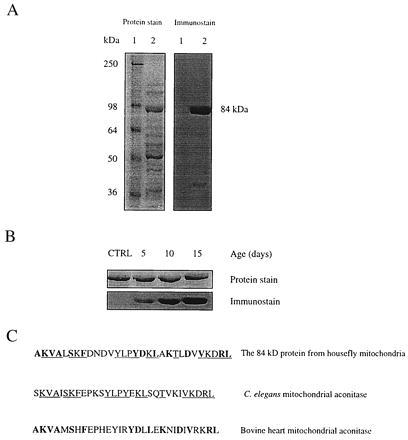
Identification of housefly mitochondrial aconitase as a target of oxidative damage. (A) Immunochemical detection of protein carbonyls in mitochondria from flight muscles of houseflies (15 days old). DNPH-treated mitochondrial matrix proteins were electrophoresed on SDS/PAGE (8.5% resolving gel) and transferred to Immobilon-P membrane. Oxidized proteins were detected immunochemically as described in the text. Lane 1 shows protein standard markers, and lane 2 contains mitochondrial matrix proteins. Protein was stained with Coomassie blue. An 84-kDa protein in the matrix exhibited a strong immunoreaction for the carbonyl groups. (B) Increase in carbonyl content of the 84-kDa protein in housefly at different ages. (Upper) The protein band stained with amido black. (Lower) The immunostain. The control (CTRL) shown is 5-day-old fly mitochondrial matrix protein without DNPH treatment. A 25-μg protein was applied onto SDS/PAGE in both A and B. (C) A computer-assisted search from protein database identified the 84-kDa protein as mitochondrial aconitase. The underlined amino acids show the N-terminal amino acid sequence homology in mitochondrial aconitase from the housefly and Caenorhabditis elegans; whereas the amino acids in bold show the homology with bovine heart mitochondrial aconitase.
Determination of Aconitase Carbonyl Content and Aconitase Activity at Different Ages.
The carbonyl content of mitochondrial aconitase was measured densitometrically (27) in 5-, 10-, and 15-day-old flies, representing young, middle-aged, and old animals (35, 36). Aconitase carbonyl content in the 15-day-old flies was 0.71 mol carbonyl/mol protein, ≈50% higher than that of 5- or 10-day-old flies (0.46 and 0.49 mol/mol, respectively). The catalytic activity of mitochondrial aconitase was measured in flies of different ages using two different methods (23, 24), which yielded the same result (Fig. 2A). Aconitase activity decreased 62% between 7 and 15 days of age, with a precipitous decline after 10 days of age. Thus, the drop in functional activity matched the increase in oxidative modification of the protein.
Figure 2.
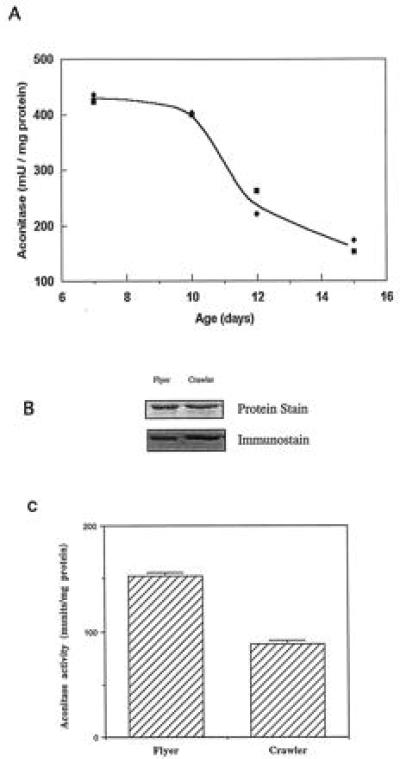
(A) Mitochondrial aconitase activity in the flight muscles of the houseflies at different ages. Two different methods were used to determine aconitase activity. Method 1 (▪) is a coupled assay in which the formation of NADPH was followed at 340 nm, using isocitrate as a substrate (23). Method 2 (⧫) is based on the formation of isocitrate from cis-aconitate, measured as the decrease in absorbance at 240 nm (24). (B) Immunochemical quantitation of aconitase carbonyls in 15-day-old flyers and crawlers, collected from the same population of flies. The procedure was the same as in Fig. 1A. (C) Aconitase enzyme activity in mitochondria from the flight muscles of 15-day-old flyers and crawlers, collected from the same population of flies. Enzyme activity was determined by method 1.
Aconitase Activity and Physiological Age.
A universal feature of senescence in flies is the loss of flying ability a few days prior to death, permitting the identification of senescent and nonsenescent individuals, which are still able to fly (17, 18). Thus, in a cohort of the same chronological age, one can separate flies of relatively younger and older physiological age. A comparison of carbonyl content and aconitase activity was made between 15-day-old flies that had lost the ability to fly (crawlers) and their cohorts which retained the ability to fly (flyers). Aconitase carbonyl content was distinctly lower in flyers compared with crawlers (Fig. 2B) whereas enzyme activity was 70% higher in the flyers (Fig. 2C). Thus, oxidative inactivation of aconitase was inversely related to the physiological age of the flies.
Effects of Hyperoxia/Fluoroacetate on Aconitase Inactivation and Aging.
The above correlations support the hypothesis that oxidative damage to aconitase is mechanistically important in the aging process. We tested this idea experimentally in two different fashions. In the first, flies were exposed to hyperoxia; in the second, flies were treated with fluoroacetate, an inhibitor of aconitase (37). In response to hyperoxic exposure, the intensity of immunostaining of aconitase carbonyls increased progressively (Fig. 3A), with 23, 72, and 70% increase, respectively, for 24-, 48-, and 72-h oxygen exposed flies as compared with control flies. Accordingly, aconitase activity was remarkably decreased, with less than 10% activity remaining after 72-h exposure (Fig. 3B). This was not simply a generalized effect, as citrate synthase activity was only slightly affected by the same treatment. The increasing effect of hyperoxia on aconitase immunostain intensity was over the constitutive levels in 6-day-old flies.
Figure 3.
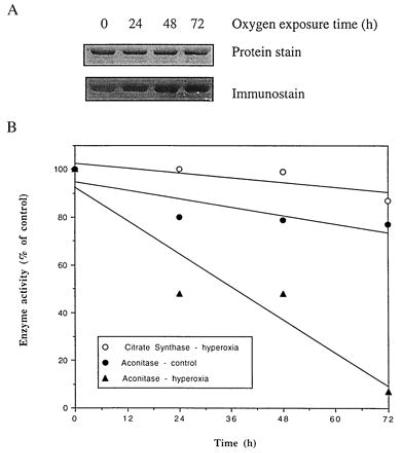
(A) Immunochemical detection of aconitase carbonyls in houseflies exposed to 100% ambient oxygen. The assay was performed as described in Fig. 1. (B) Effect of 100% ambient oxygen on the activities of mitochondrial aconitase and citrate synthase. Six-day-old flies were exposed to 100% oxygen for indicated times. Aconitase activity was determined by method 1, as described in the legend of Fig. 2. Citrate synthase was assayed according to Srere (25).
To test the relationship of aconitase activity to life-span, flies were exposed to different concentrations of fluoroacetate in their drinking water. This aconitase inhibitor caused a dose-dependent decrease in life-span (Fig. 4). Under our experimental conditions, the maximum life-span of the nontreated houseflies was 47 days, shortened to 38, 30, and 17 days by addition of 1, 50, and 100 μM fluoroacetate to the drinking water.
Figure 4.
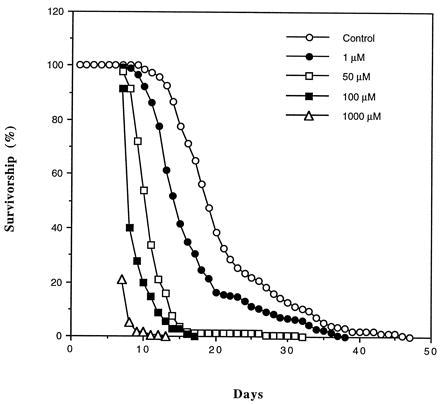
Survivorship curves of adult male houseflies administered with different concentrations of fluoroacetate in their drinking water. Flies were housed in 1-ft3 cages with 200 flies per cage. A concentration of 1 mM resulted in 80% mortality within 24 h. Data are based on the mortality of 200 flies in each population.
DISCUSSION
These studies show that oxidative damage to protein accumulates selectively during aging of the fly. In the mitochondrial matrix, aconitase is both oxidatively modified and inactivated during normal aging. Experimental inactivation of aconitase by either hyperoxia or fluoroacetate shortened life-span, indicating a causal relationship between aconitase activity and life-span.
Aconitase catalyzes the interconversion of citrate and isocitrate in the citric acid cycle, a reaction essential to normal metabolic function. In housefly, aconitase was estimated to account for ≈15% of the total mitochondrial matrix proteins (data not shown). Therefore, it might be argued that it is the high abundance of aconitase in mitochondrial matrix that leads to our identification of this protein. This, apparently, is not the case, because another highly abundant protein in the matrix, with a molecular weight of 50 kDa (Fig. 1A), did not exhibit any detectable carbonyl groups. In addition, cytosolic aconitase did not exhibit oxidative modification (data not shown). The relative abundance of aconitase raises the question whether the accumulation of inactive molecules actually has functional importance. If aconitase activity became rate-limiting, then citrate should accumulate during aging. Build-up of citrate during aging of the fly was indeed demonstrated by Zahavi and Tahori (38) some 30 years ago. It seems reasonable to suggest that the decrease in aconitase activity of the magnitude (62%) observed here would deleteriously affect oxidative phosphorylation.
The particular susceptibility of mitochondrial aconitase to oxidative damage may be related to the iron-sulfur cluster [4Fe-4S] in its active site (39). Studies in vitro established that aconitase is particularly sensitive to reaction with superoxide (40–44), which causes release of one iron atom from the cluster (45). Under normal redox conditions, the release of aconitase iron by superoxide anion and the resultant inactivation of its activity can be reversed (46). However, elevation in the rate of superoxide generation, occurring during aging, can lead to a corresponding enhancement in the production of H2O2 and hydroxyl free radical. The latter may cause carbonylation and an irreversible inactivation of aconitase through metal-dependent, site-specific oxidations, according to the mechanism proposed by Stadtman (47).
Inactivation of aconitase may block normal electron flow to oxygen, leading to an accumulation of reduced metabolites such as NADH. This condition has been termed “reductive stress,” and can cause an increased production of reactive oxygen species through autoxidation of the reduced metabolites, thus further increasing oxidative damage to macromolecules (48, 49). Therefore, oxidative inactivation of aconitase can initiate a cascade with the potential to cause a dramatic increase in the cellular burden of oxidative damage. This can account for the exponential increase in oxidized proteins during aging, which has been documented in flies, rodents, and humans (8).
The physiologic age of flies could be distinguished from their chronological age by assessment of the extent of oxidative modification of aconitase or by assay of its catalytic activity. The observation that serum citrate of rabbits increased with age is consistent with age-dependent inactivation of aconitase in mammals (38). If the same or some other similar correlations can be demonstrated in different species, such measurements may provide a general surrogate (or biomarker) measure of physiologic age. Such a surrogate would be useful in assessing interventions designed to affect the aging process. It also follows that interventions which specifically protect mitochondrial aconitase from oxidative inactivation would be predicted to prolong life-span. Any treatments that directly or indirectly accelerate the oxidation of aconitase would be predicted to decrease life-span.
The increase in the steady-state level of oxidatively modified aconitase may be due either to an increased rate of oxidation or to a decreased rate of removal of the modified protein by proteolysis (8). Regardless of the causes, the build-up of inactive forms of enzymes which occupy crucial positions in intermediary metabolism will certainly impair cellular function. Aconitase is central to carbohydrate and energy metabolism. Similarly, glutamine synthetase affects a panoply of pathways (50). Its activity declines in the brain of aging mammals, paralleling an increase in oxidative modification of proteins (51). Treatment of older animals with an antioxidant spin-trap restored glutamine synthetase activity to the level of younger animals (51). Treatment also decreased protein oxidation, increased protease activity, and, most notably, improved cognitive function of the animals. It will be of interest to determine the effect of treatment on aconitase activity. Finally, although aconitase is a selective target of oxidative damage during aging, the present results do not warrant the conclusion that aconitase is a causal factor of senescence in the housefly or other organisms. Such an inference may be drawn only if retardation of the oxidative modification of aconitase also delays senescence.
Acknowledgments
We thank B. Sohal for technical assistance in the enzyme activity assay of aconitase. This work was supported by Grants RO1AG7657 and RO1AG13563 from the National Institute on Aging, National Institutes of Health.
ABBREVIATIONS
- 2
4-DNPH, dinitrophenylhydrazine
- TBST
Tris-buffered saline containing 0.1% Tween-20
- DNP
2,4-dinitrophenyl
References
- 1.Harman D. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Harman D. Proc Natl Acad Sci USA. 1981;78:7124–7128. doi: 10.1073/pnas.78.11.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheldahl J A, Tappel A L. Exp Gerontol. 1974;9:33–41. doi: 10.1016/0531-5565(74)90005-9. [DOI] [PubMed] [Google Scholar]
- 4.Park J W, Ames B N. Proc Natl Acad Sci USA. 1988;85:7467–7470. doi: 10.1073/pnas.85.20.7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadtman E R. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 6.Orr W C, Sohal R S. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 7.Sohal R S, Weindruch R. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine R L, Stadtman E R. In: Handbook of the Biology of Aging: Protein Modifications with Aging. Schneider E L, Rowe J W, editors; Schneider E L, Rowe J W, editors. Orlando: Academic; 1996. pp. 184–197. [Google Scholar]
- 9.Nohl H, Hegner D. Eur J Biochem. 1978;82:563–567. doi: 10.1111/j.1432-1033.1978.tb12051.x. [DOI] [PubMed] [Google Scholar]
- 10.Farmer K J, Sohal R S. Free Radical Biol Med. 1989;7:23–29. doi: 10.1016/0891-5849(89)90096-8. [DOI] [PubMed] [Google Scholar]
- 11.Sohal R S, Sohal B H. Mech Ageing Dev. 1991;57:187–202. doi: 10.1016/0047-6374(91)90034-w. [DOI] [PubMed] [Google Scholar]
- 12.Sohal R S. In: Comprehensive Insect Physiology, Biochemistry and Pharmacology: Aging in Insects. Kerkut G A, Gilbert L I, editors; Kerkut G A, Gilbert L I, editors. Oxford: Pergamon; 1985. pp. 595–631. [Google Scholar]
- 13.Hansford R G. Biochim Biophys Acta. 1983;726:41–80. doi: 10.1016/0304-4173(83)90010-1. [DOI] [PubMed] [Google Scholar]
- 14.Shigenaga M K, Hagen T M, Ames B N. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cabiscol E, Levine R L. J Biol Chem. 1995;270:14742–14747. doi: 10.1074/jbc.270.24.14742. [DOI] [PubMed] [Google Scholar]
- 16.Davis R A, Fraenkel G. J Exp Biol. 1940;17:402–407. [Google Scholar]
- 17.Ragland S S, Sohal R S. Exp Gerontol. 1973;8:135–145. doi: 10.1016/0531-5565(73)90003-x. [DOI] [PubMed] [Google Scholar]
- 18.Sohal R S, Toy P L, Allen R G. Mech Ageing Dev. 1986;36:71–77. doi: 10.1016/0047-6374(86)90140-5. [DOI] [PubMed] [Google Scholar]
- 19.Van Den Bergh S G. Methods Enzymol. 1967;10:117–122. [Google Scholar]
- 20.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Keller R J, Halmes N C, Hinson J A, Pumford N R. Chem Res Toxicol. 1993;6:430–433. doi: 10.1021/tx00034a007. [DOI] [PubMed] [Google Scholar]
- 22.Shacter E, Williams J A, Lim M, Levine R L. Free Radical Biol Med. 1994;17:429–437. doi: 10.1016/0891-5849(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 23.Kennedy M C, Emptage M H, Dreyer J L, Beinert H. J Biol Chem. 1983;258:11098–11105. [PubMed] [Google Scholar]
- 24.Fasler B, Lowenstein J M. Methods Enzymol. 1969;13:26–30. [Google Scholar]
- 25.Srere P A. Methods Enzymol. 1969;13:3–11. doi: 10.1016/s0076-6879(76)44004-1. [DOI] [PubMed] [Google Scholar]
- 26.Smith P K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk D C. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 27.Winn L M, Wells P G. Free Radical Biol Med. 1997;22:607–621. doi: 10.1016/s0891-5849(96)00340-1. [DOI] [PubMed] [Google Scholar]
- 28.Keller R J, Sharma R P, Grover T A, Piette L H. Arch Biochem Biophys. 1988;265:524–533. doi: 10.1016/0003-9861(88)90157-9. [DOI] [PubMed] [Google Scholar]
- 29.Keller R J, Coulombe R J, Sharma R P, Grover T A, Piette L H. Free Radical Biol Med. 1989;6:15–22. doi: 10.1016/0891-5849(89)90154-8. [DOI] [PubMed] [Google Scholar]
- 30.Carmichael A J. FEBS Lett. 1990;261:165–170. doi: 10.1016/0014-5793(90)80662-3. [DOI] [PubMed] [Google Scholar]
- 31.Shi X L, Sun X Y, Dalal N S. FEBS Lett. 1990;271:185–188. doi: 10.1016/0014-5793(90)80402-5. [DOI] [PubMed] [Google Scholar]
- 32.Levine R L, Garland D, Oliver C N, Amici A, Climent I, Lenz A-G, Ahn B-W, Shaltiel S, Stadtman E R. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- 33.Turrens J F, Freeman B A, Levitt J G, Crapo J D. Arch Biochem Biophys. 1982;217:401–410. doi: 10.1016/0003-9861(82)90518-5. [DOI] [PubMed] [Google Scholar]
- 34.Turrens J F, Freeman B A, Crapo J D. Arch Biochem Biophys. 1982;217:411–421. doi: 10.1016/0003-9861(82)90519-7. [DOI] [PubMed] [Google Scholar]
- 35.Sohal R S, Agarwal S, Dubey A, Orr W C. Proc Natl Acad Sci USA. 1993;90:7255–7259. doi: 10.1073/pnas.90.15.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sohal R S, Dubey A. Free Radical Biol Med. 1994;16:621–626. doi: 10.1016/0891-5849(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 37.Lauble H, Kennedy M C, Emptage M H, Beinert H, Stout C D. Proc Natl Acad Sci USA. 1996;93:13699–13703. doi: 10.1073/pnas.93.24.13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zahavi M, Tahori A S. J Insect Physiol. 1965;11:811–816. doi: 10.1016/0022-1910(65)90160-5. [DOI] [PubMed] [Google Scholar]
- 39.Kent T A, Dreyer J L, Kennedy M C, Huynh B H, Emptage M H, Beinert H, Munck E. Proc Natl Acad Sci USA. 1982;79:1096–1100. doi: 10.1073/pnas.79.4.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardner P R, Nguyen D D, White C W. Proc Natl Acad Sci USA. 1994;91:12248–12252. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardner P R, Raineri I, Epstein L B, White C W. J Biol Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 42.Gardner P R, Fridovich I. J Biol Chem. 1991;266:19328–19333. [PubMed] [Google Scholar]
- 43.Castro L, Rodriguez M, Radi R. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- 44.Verniquet, F., Gaillard, J., Neuburger, M. & Douce, R. (1991) Biochem. J. 643–648. [DOI] [PMC free article] [PubMed]
- 45.Flint D H, Smyk R E, Tuminello J F, Draczynska L B, Brown O R. J Biol Chem. 1993;268:25547–25552. [PubMed] [Google Scholar]
- 46.Patel M, Day P J, Crapo J D, Fridovich I, McNamara J O. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 47.Stadtman E R. Free Radical Biol Med. 1990;9:315–325. doi: 10.1016/0891-5849(90)90006-5. [DOI] [PubMed] [Google Scholar]
- 48.Stadtman E R. Exp Gerontol. 1988;23:327–347. doi: 10.1016/0531-5565(88)90036-8. [DOI] [PubMed] [Google Scholar]
- 49.Van den Enden M K, Nyengaard J R, Ostrow E, Burgan J H, Williamson J R. Invest Ophthalmol Vis Sci. 1995;36:1675–1685. [PubMed] [Google Scholar]
- 50.Stadtman E R, Ginsburg A. In: The Enzymes: The Glutamine Synthetase of Escherichia coli Structure and Control. Boyer P D, editor; Boyer P D, editor. New York: Academic; 1974. pp. 755–807. [Google Scholar]
- 51.Carney J M, Starke R P, Oliver C N, Landum R W, Cheng M S, Wu J F, Floyd R A. Proc Natl Acad Sci USA. 1991;88:3633–3636. doi: 10.1073/pnas.88.9.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]