

Abstract
Chronic overexpression of the acetylcholine-hydrolysing enzyme acetylcholinesterase (AChE) is a notable consequence of exposure to anticholinesterase drugs or poisons. However, the physiological consequences for the resultant neuromuscular disfunction have not yet been carefully analysed. Here we report detailed dissection of the different components of neuromuscular function in transgenic mice previously shown to display motor fatigue and altered muscle morphology as a consequence of neuronal overexpression of AChE-S, the synaptic AChE variant. Transgenic diaphragm muscle presented exaggerated fatigue as a combined consequence of neurotransmission fading and muscle mechanical malfunctioning. In a tetanic stimulation protocol, transgenic muscles rapidly fatigued to a larger extent than wild-type muscles, when stimulated either directly or via the phrenic nerve. AChE overexpression involved moderate but significant aberrations of synaptic transmission with higher quantal content (measured at 0.2 mm Ca2+, 2.3 mm Mg2+). Furthermore, treatment with the anti-cholinesterase physostigmine revealed a higher amplitude and half-decay time of the transgenic quantal postsynaptic response. Our observations imply that elevated levels of neuronal AChE-S are expected to cause muscle exhaustion due to a combination of modest, multilevelled aberrations in synaptic transmission, muscle function and morphology.
Acetycholinesterase (AChE) is a key enzyme in cholinergic transmission, and altered activity levels of this protein are associated with a high probability of disrupted homeostasis in the synaptic cleft. Indeed, cholinergic imbalances in neuromuscular junctions (NMJs) are involved in several pathological situations, for example in congenital myasthenias (reviewed in Engel et al. 1999; and Ohno et al. 2001), spinal muscular dystrophy (Crawford et al. 1996) and amyotrophic lateral sclerosis (Brown et al. 1995). More common is exposure to agricultural AChE inhibitors such as the organophosphate paraoxon. Rare but devastating are the effects of chemical warfare agents such as sarin, tabun or soman, which result, among other things, in muscle fibre necrosis and muscle paralysis (reviewed in Ray, 1998; Schwarz et al. 1995). The recent increase in the use of anti-cholinesterases such as Alzheimer's disease drugs (Darreh-Shori et al. 2002) further expands such exposure, albeit to lower, chronic doses. Previous reports demonstrated that exposure to AChE inhibitors results in the accumulation of AChE in the brain (Kaufer et al. 1998) and muscle (Lev-Lehman et al. 2000). However, the physiological consequences of chronic AChE irregularities are as yet unknown. We have therefore employed transgenic mice that chronically overexpress the synaptic variant of AChE (AChE-S) in both the central nervous system and motor neurons (Andres et al. 1997 and Fig. 1 in Supplementary Material for an exampled TG NMJ). The elevated AChE activities in these mice induced multi-site dysfunctions, including cognitive deficiencies (Beeri et al. 1997; Cohen et al. 2002), intensified neuropathology markers for neurodeterioration in the brain (Sternfeld et al. 2000), and neuromotor impairments expressed as progressively exaggerated motor fatigue and altered electromyography patterns (Andres et al. 1997). A detailed dissection of this motor dysfunction may contribute to the understanding of the related clinical pathologies mentioned above.
Figure 1. Transgenic diaphragm muscle displays exaggerated contraction fatigue in tetanic nerve stimulation.
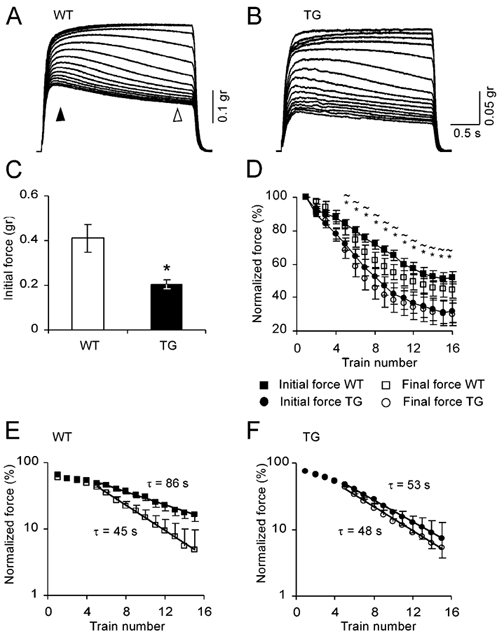
A, nerve induced contractions in WT diaphragm muscle. Contraction sessions were composed of 15 consecutive stimulation trains of 200 stimuli at 66 Hz, with 10 s intertrain intervals. B, similar to A in TG diaphragm muscle. C, averaged initial force of 6 measurements in each of 4 WT and 5 TG diaphragms (measured at the time marked by a filled arrowhead in A, means ± s.e.m.). *P < 0.05, compared with WT group, two-tailed t test. D, time course of initial and final forces normalized to the first contraction (final force was measured at the time marked by an open arrowhead in A). *P < 0.05, initial forces compared with WT values; ≈P < 0.05, final forces compared with WT values. E, semi-logarithmic plot for WT initial and final forces (steady state values subtracted). Time constants derived from the slope of the line fitted by regression to the linear part were found to be significantly different (F test, P < 0.05). F, same as E for TG muscles.
Muscle fatigue may result from impaired excitation- contraction mechanisms, neurotransmission failure or a combination of both. Neurotransmission depression was reported to make an important contribution to muscle fatigue (Aldrich et al. 1986; Kuei et al. 1990; Van Lunteren & Moyer, 1996), and may result from vesicle depletion or malfunctioning of the upstream release machinery, leading to depression of release. Postsynaptically, cumulative receptor desensitization may reduce postsynaptic potentials (PSPs). Blocking AChE activity was found to enlarge the relative fraction of neurotransmission in muscle fatigue (Panenic et al. 1999).
In transgenic mice overexpressing AChE-S, muscle morphology is irregular, including enlarged NMJ and abnormal ultrastructure of the synaptic clefts with either much deeper or very shallow folds (Andres et al. 1997). Presynaptic features were atypical as well, most prominently in higher numbers of vesicles at the transgenic presynaptic site (Andres et al. 1998). These changes were accompanied by altered transcription patterns in spinal cord motor neurons, for example premature overexpression of choline acetyltransferase mRNA, or suppression of neurexin Iβ mRNA (Andres et al. 1997). Additionally, tongue muscles from AChE-S transgenic mice display exaggerated neurite branching and disorganized fibres (Lev-Lehman et al. 2000).
The overall objective of the present study has been to elucidate which levels, from neuronal activation to muscle contraction, are involved in the induction of motor fatigue under an excess of AChE. First, we examined whether muscle properties and/or neuronal transmission are involved in the phenotype of muscle fatigue. Second, we questioned whether pre- and/or postsynaptic properties are affected by an excess of synaptic AChE.
Methods
Experiments were approved by The Hebrew University's committee for animal studies and all efforts were made to minimize both the number of animals used and their suffering. Locally grown AChE-S transgenic mice and age- and sex-matched FVB/N control mice were used throughout this study. Diaphragm muscles were obtained from 3- to 5-month-old mice (28-32 g). Animals were placed in a desiccator with an ether-saturated atmosphere and were decapitated when they stopped responding to a noxious stimulus. Hemi-diaphragms were isolated with the phrenic nerve and were placed in an experimental bath containing physiological salt solution with the following composition (mm): 124 NaCl, 5 KCl, 1.3 MgSO4, 1.2 NaH2PO4, 26 NaHCO3, 10 d-glucose and 2.4 CaCl2. Muscles were perfused continuously with a salt solution, aerated with 95 % O2-5 % CO2 at room temperature (22- 24 °C) and at a rate of 2 ml min−1.
Muscle fatigability measurements
The intact hemi-diaphragm was fixed at the rib side onto a Sylgard-coated bath. The central tendon was connected to an isometric transducer and was aligned at 45 deg with the experimental bath. The muscle was progressively stretched until a basal tension of 0.015 g was obtained, and from this length subsequent protocols were undertaken. The output of the force transducer was digitized, stored and analysed offline. Fifteen stimulation trains separated by 10 s intertrain intervals induced nerve-evoked contractions. Each train consisted of 200 stimuli at 66 Hz, 0.1 ms duration. For direct stimulation, a group of muscle fibres was stimulated with a suction electrode placed at the far end of the rib side in order to prevent electrode movements during muscle contraction.
Cut muscle preparations
Cut muscles were prepared by cutting fibres on both sides of the main intramuscular nerve branch. Recording of PSPs commenced after contractions were halted. Intracellular recordings were performed with microelectrodes of 20-30 MΩ resistance, filled with potassium acetate (3 m) and KCl (100 mm). The nerve was stimulated with six trains of 15 stimuli at 50 Hz, 0.1 ms duration, separated by 10 s intertrain intervals. This short stimulation protocol did not create accumulating decay during subsequent trains (data not shown), and therefore averaged results are presented.
Characterization of synaptic quantal responses
Quantal size and content were measured by recordings of PSPs in low calcium, high magnesium solution (0.2 and 2.3 mm, respectively). Frequency of spontaneous release was measured within a 400 ms period before the evoked releases. Quantal content was determined by the failure method (del Castillo & Katz, 1954) in evoked responses to over 600 stimuli per cell given at 0.5 Hz. With this sample size, and considering quantal content in the range of 0.07, the standard error was calculated to be no more than 15 %. Since at this probability of release the chance for multiple quanta is ≈0.002, indicating only ≈3 % of the evoked responses are multiple, quantal sizes of spontaneous and evoked release of each cell were merged. Rise time was measured from onset to peak amplitude, whereas decay time was measured by the time to 50 % reduction of PSP amplitude. Control parameters were determined from a sample of at least five NMJs per diaphragm. Fresh physostigmine (10 μm, Sigma, St Louis, MI, USA) was continuously perfused and a sample of at least seven NMJs was taken.
Statistics
All analysed data were compared by Student's two-tailed paired t test and significant differences were determined when P < 0.05. Significant linear regression analysis was determined with an F test.
Results
Both nerve and muscle contribute to the phenotype of pronounced fatigue
The first step we employed in order to examine the contribution of nerve and muscle to the fatigue phenotype was to measure muscle contraction induced by administering tetanizing stimulation either to the nerve or directly to the muscle. This approach enabled us to distinguish between the possible fatigue developed at the level of muscle excitation-contraction mechanisms and fading of neuromuscular transmission. We used nerve stimulation sessions of 15 trains which induced a development of muscle fatigue within one session, as demonstrated in Fig. 1A and B for wild-type (WT) and transgenic (TG) muscles, respectively. In the first few contractions of the series, there was a slow build-up of force in the course of the stimulation train, a phenomenon that was more pronounced in WT muscles. Gradually, fatigue appeared during the train itself, followed by a reduction in the initial force achieved at the subsequent contraction. With repetition, the contractions slowly attained a lower steady-state level. Although the general behaviour appeared similar, several differences between WT and TG muscles were observed in this pattern. First, the initial force (at 500 ms from onset of stimulation) achieved by WT muscles was 0.41 ± 0.06 g (mean ± s.e.m.), significantly higher than the 0.2 ± 0.02 g in TG (n = 6, in each of 4 WT and 5 TG muscles, P < 0.05, Fig. 1C). Second, the time course of fatigue development was clearly different in the TG and WT diaphragms, in both parameters of intra- and intercontraction fatigue. These were determined by measuring the percentage reduction of the initial and final force from the maximal force obtained at the first stimulation of the session (measured, respectively, at 0.5 and 3 s from contraction onset). WT muscles decay significantly less than TG ones at the level of initial activation, reaching 49 ± 3 % decrement at the last contraction, whereas TG muscles declined by 68 ± 3 % (P < 0.01, Fig. 1D). In contrast to wild-type muscles, transgenic muscles not only fatigued to a greater extent, but also failed to recover during the intertrain intervals. This phenomenon is demonstrated by the smaller difference attained between the initial and the final force in the TG contractions during the train (Fig. 1D). A single exponential fit to the initial force represents a clear 1.6-fold faster fatigue time constant of TG as compared with WT muscle (52.6 vs. 85.9 s, Fig. 1E vs. F, P < 0.05, F test). Interestingly, the time constant of final fatigue seems to be similar in both phenotypes (45.3 vs. 48.1 s, respectively). These results may indicate that the main difference between the TG contraction mechanism and that of the WT phenotype is a slower intertrain recovery in TG muscles.
The observed differences could originate either in the nerve or in the muscle. To distinguish between these possibilities, we measured the isotonic force generated by direct muscle fibre stimulation (see Methods). This approach yielded essentially similar patterns of contraction to those induced indirectly via the nerve (Fig. 2A and B). However, the levels of fatigue in both inter- and intratrain stimuli were lower than those measured in indirect stimuli. In contrast to the evident recovery process in TG muscle activity induced by muscle stimulation, the recovery process in the WT muscle was very slow. Fatigue that originated from muscle excitation-contraction properties reached 25 ± 2.9 % in WT and up to 40 ± 3.0 % in TG muscle at the 10th contraction (n = 6, in each of 3 WT and 3 TG muscles, P < 0.02, Fig. 2C). Interestingly, despite this difference in magnitude, the relative contribution of muscle mechanisms to the total fatigue was similar in TG and WT (68.9 % and 70.1 %, respectively, Fig. 2D). To conclude, transgenic diaphragm muscle under chronic excess of AChE suffers from augmented fatigue of both neuronal and muscle origins.
Figure 2. Transgenic diaphragm muscle displays exaggerated contraction fatigue in tetanic direct muscle stimulation.
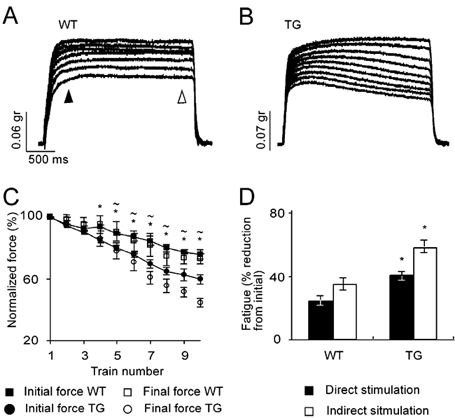
A, contractions in WT diaphragm muscles induced by delivering the stimulation directly into the muscle. The contraction session was composed of 10 consecutive stimulation trains of 200 stimuli at 66 Hz, with 10 s intertrain intervals. B, same as A for TG muscle. C, time course of initial and final forces normalized to the first contraction. Values are means ± s.e.m. of 6 measurements in each of 3 WT and 3 TG diaphragms. Forces were measured at the time marked by the filled and open arrowheads in A. *P < 0.05, initial forces compared with WT values; ≈P < 0.05, final forces compared with WT values. D, summary of the muscle- and nerve-induced fatigue, measured at the 10th contraction in direct and indirect stimulation. Note similar ratio of muscle- and nerve-induced fatigue in the TG and WT muscles. Data are given as means ± s.e.m. *P < 0.05, compared with WT group, two-tailed t test.
Neurotransmission failure is demonstrated directly at the synaptic level
To characterize the physiological behaviour of neuromuscular transmission we used cut muscle preparations. As action potentials are inactivated in the cut muscle fibres (Hubbard & Wilson, 1973), it is possible to measure synaptic transmission at physiologically high levels of release. Figure 3A presents typical trains induced in WT and TG fibres, demonstrating larger and faster decay of the PSPs in the TG fibres. No significant difference was observed in the amplitude of the first PSP in the train, suggesting no apparent effect or involvement of the elevated AChE concentrations (amplitudes were scaled in relation to the resting potentials, Fig. 3B). Figure 3C shows the subsequent PSPs in the train after normalization to the first PSP, which presented an initially small facilitation followed by a gradual decrease in the amplitude within the train, which in the case of transgenic synapses declined to a lower plateau. While no significant differences in facilitation were observed, WT fibres showed a 20 ± 2.4 % decrement, a significantly smaller magnitude than in transgenic synapses, demonstrating synaptic fade of 28 ± 2.5 % (n = 44 and 45 cells from 9 WT and 9 TG muscles, P < 0.03). Synaptic fading was also faster in the transgenic NMJ than in WT, noted by a smaller time constant of the exponential part of the amplitude decay in the trains (62.7 s in WT vs. 38.4 s in TG, Fig. 3D). Therefore, the synaptic depression during the initial stimuli of the train, as demonstrated here, could contribute only a relatively small part to the lower initial force achieved by transgenic muscle contractions (Fig. 1C).
Figure 3. TG synapses demonstrate pronounced neurotransmission depression in cut muscle preparations.
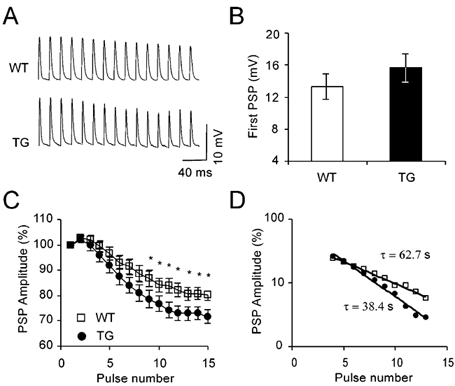
A, examples of trains of PSPs evoked in cut muscle preparations. Trains consisted of 15 stimuli at 50 Hz with 10 s intertrain intervals. For clarity, stimulation artifacts were omitted. B, average amplitude (± s.e.m.) of the first PSP in the train. In order to reduce PSP variability resulting from differences in membrane potentials, amplitudes were scaled to a driving force at a membrane potential of - 50 mV, assuming zero as the synaptic reversal potential (Linder & Quastel, 1978). n = 44 and 45 cells from 9 WT and 9 TG muscles. C, time course of PSP decay during the train. PSPs are normalized to the first PSP in the train. *P < 0.05, compared with WT group, two-tailed t test. D, semi-logarithmic plot of PSP values (extrapolated steady state levels were subtracted). Time constants derived from the slope of the regression. P < 0.05 (F test).
Characterization of neuromuscular transmission changes at the cellular level
Whether AChE over-producing NMJs demonstrate different release characteristics, and if so, whether these take place at pre- and/or post-synaptic sites, was tested by measuring quantal size and quantal content under conditions of low probability of release (0.2 mm Ca2+, 2.3 mm Mg2+). Postsynaptic quantal responses of evoked and spontaneous activity were similar in WT and TG synapses. Amplitudes were 0.98 ± 0.1 mV in WT and 1.05 ± 0.14 mV in TG (n = 75, in each of 4 WT and 5 TG muscles, P < 0.62, Fig. 4A); rise times were 0.89 ± 0.1 ms in WT and 0.97 ± 0.1 ms in TG (P < 0.79, Fig. 4B) and half-decay times were 1.68 ± 0.22 ms in WT and 1.74 ± 0.1 ms in TG (P < 0.88, Fig. 4C). The apparent morphological atrophy of TG muscle fibres could potentially lead to a biased sampling of the less injured fibres. However, at this stage of muscle deterioration, electron microscopy analysis demonstrated that the damage was limited to several myofibrils (Andres et al. 1997 and Fig. 2 in Supplementary Material) and was thus not likely to affect the electrophysiological measurement. To explore the effects of AChE's catalytic activity on these different PSP parameters, we blocked AChE with the carbamate AChE blocker, physostigmine (10 μm). At this concentration, physostigmine is minimally active as an ACh receptor blocker (Shaw et al. 1985; Dudel et al. 1999). Synaptic responses were recorded during 1.5 h of physostigmine perfusion and demonstrated quantal size increases in both amplitude and time course. Significantly larger increases in amplitude appeared in the TG animals than in WT (98 and 57 %, respectively, P < 0.05, Fig. 4A), as well as in decay time (65 and 35 %, respectively, P < 0.05, Fig. 4C). The rise time of both WT and TG mice increased significantly, yet to similar extents (41 and 32 %, respectively, Fig. 4B). The effective concentration of physostigmine at the junctional fold was probably not high enough for a complete blockade of AChE, since the decay time was not dramatically elevated, as expected by treatment with anti-cholinesterase (see, for example, Ferry & Kelly, 1988). Yet, the larger effect of physostigmine on TG miniature PSP amplitudes was consistent with the higher levels of the catalytic activity of AChE in the TG NMJs.
Figure 4. TG quantal size is similar to WT responses in control conditions, but larger following physostigmine treatment.
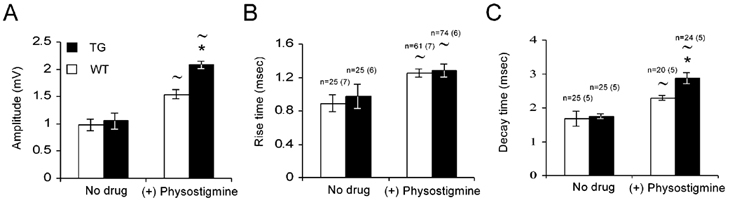
Quantal size recorded in WT and TG muscles under conditions of low probability of release (0.2 mm Ca2+, 2.3 mm Mg2+). Evoked and spontaneous responses were combined (see Methods). A sample of PSPs from several cells was taken in the presence of physostigmine (10 μm). A, PSP amplitude. B, PSP rise time (from onset to peak). C, PSPs half-decay time (from peak to half-decay). Numbers of cells analysed are indicated above the bars, number of animals in brackets (similar numbers for A and B). Data were analysed with an unpaired two-tailed t test: *P < 0.05, compared with WT results; ≈P < 0.05, compared with no drug conditions within the same group.
Transgenic NMJs exhibit higher quantal content
Presynaptic properties of the TG NMJ are particularly relevant since the genetic interference was limited in this TG line to the neuronal part (Beeri et al. 1995), and therefore excess AChE produced by the nerve terminal (Anglister, 1991) could potentially affect the levels and/or interactions in the release machinery. Quantal content was determined by the failure method of del Castillo & Katz (1954) at lower Ca2+ concentration (0.2 mm Ca2+, 2.3 mm Mg2+). Transgenic NMJs displayed higher quantal content (0.095 ± 0.01 in TG, 0.068 ± 0.006 in WT, n = 75, in each of 4 WT and 5 TG muscles, P < 0.05, Fig. 5A) and frequency of spontaneous release than WT ones (0.28 ± 0.015 Hz in TG and 0.15 ± 0.005 Hz in WT, P < 0.05, Fig. 5B). Both results are in accord with possible presynaptic changes in the TG terminal that involve either increase in the average probability of release (p) and/or increase in n (del Castillo & Katz, 1954). Treatment with physostigmine caused a pronounced decrease in both quantal content and spontaneous release in TG synapses (21 and 27 % reduction, P < 0.05), while changes in WT were not significant (Fig. 5A and B, P < 0.41). The larger effect of physostigmine on the release probabilities of TG NMJs may reasonably reflect the hypocholinergic synaptic situation. Furthermore, the sharp effect of physostigmine better supports an increase in the probability of release in the TG synapse than a change in the number of releasing sites.
Figure 5. TG synapse displays higher probability of release and different susceptibility to AChE inhibition than WT.
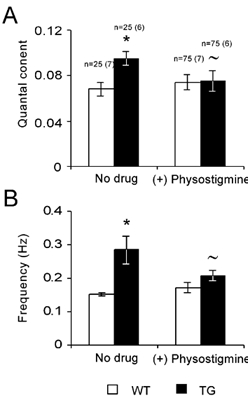
A, quantal content of WT and TG synapses, measured at conditions of low probability of release (0.2 mm Ca2+, 2.3 mm Mg2+) with and without physostigmine (10 μm). B, frequency of spontaneous release with and without physostigmine. Numbers of cells analysed are indicated above the bars, number of animals in brackets (similar numbers for A and B). Data were analysed with a two-tailed t-test: *P < 0.05, compared with WT results; ≈P < 0.05, compared with no drug conditions within the same group.
Discussion
Using transgenic mice with chronic neuronal overexpression of synaptic AChE, we found that the consequences of higher levels of AChE are widespread across multiple locations in the neuromuscular system. Interestingly, each level presented moderate but significant differences between TG and WT muscle that are likely to contribute to the fatigue phenotype collaboratively. Characterizing the transgenic muscle ex vivo, we observed an exaggerated mechanical fatigue following repetitious activation of the muscle, either directly or via the phrenic nerve. These results may explain, at least in part, the reported motor behavioural difficulties of these mice (Andres et al. 1997). Our results further demonstrate that the accentuated mechanical fatigue is due to a combination of alterations in both neuromuscular transmission and intrinsic muscle properties, such as excitation-contraction coupling processes. Synaptic fade was explicitly demonstrated in the cut muscle preparation, with more pronounced synaptic depression in the transgenic NMJs, while enhanced mechanical fatigue was demonstrated by repetitive direct stimulation of the diaphragm muscle. The emerging picture from our analysis is that synaptic homeostasis is altered with excess AChE. This subsequently affects release properties, yielding higher probability of release, larger quantal size and hypersensitivity to cholinergic, stress-like insults. The latter are consistent with neuronal hypersensitivity of hippocampal functioning under AChE excess (Kaufer et al. 1998; Meshorer et al. 2002). The multiple effects on muscle function are likely to include both direct and indirect responses to the hypo-cholinergic state.
Lower tetanic force production was evident in the transgenic muscle. Total muscle force is a build-up of recruited single fibres. In the cut muscle experiments, the PSP in the transgenic fibres decays following repetitive stimulation to a lower level than in WT. Therefore, the TG muscle probably undergoes a faster reduction in the number of recruited fibres during the train, which explains, at least in part, the lower force production of the transgenic diaphragm. Apparently, the small difference in the steady state of PSP amplitude can underlie only a relatively small part in the differences observed in the initial force, unless the steady state values are close to the threshold for action potential in the muscle fibres.
TG and WT muscles showed similar stereotypical changes in the contractions induced by repeated nerve stimulation (Hill, 1970). In the nerve stimulation protocol, which combines both neurotransmission and muscle mechanics, the main difference between TG and WT muscles could be attributed to a reduction in intertrain recovery, which is reflected in a significantly smaller difference between the initial and final forces and the lower final steady state level attained by the TG muscle. The direct stimulation revealed similar time course and fatigue processes as in nerve stimulation, demonstrating an accentuated mechanical fatigue in the TG muscle. It is important to note that intratrain fatigue is low in the nerve stimulation of TG muscle, but conspicuous under the direct stimulation. Therefore, the contribution of neurotransmission fade to the fatigue processes during the train is probably underestimated in the nerve stimulation experiment. We attribute the transgenic diaphragm aberration to an impaired recovery process, and conclude that the motor fatigue phenotype involves several sites, including both neuronal and muscle elements.
The augmented fatigue of the TG muscle mechanics may be related to the altered muscle morphology, and potentially to biochemical processes as well as to reduced muscle excitability. Cholinergic transmission-dependent modifications in excitability have been demonstrated in myasthenia gravis patients and in a rat model involving reduction in voltage-gated sodium channels (Ruff & Lennon, 1998). Since these properties are not directly associated with excess AChE catalytic activity we suggest two underlying alternatives. (1) AChE has been demonstrated to have non-catalytic, morphogenic activities, which may affect muscle function (Darboux et al. 1996; Sternfeld et al. 1998; Grifman et al. 1998; Soreq & Seidman, 2001). (2) A developmental disruption of muscle function due to chronic hypocholinergic level. A disturbance of muscle development that depends on cholinegic-altered excitation has been demonstrated in transgenic zebrafish embryo (Behra et al. 2002).
The synaptic analysis in TG muscles revealed normal quantal responses at the postsynaptic cellular level, yet with higher quantal content in the TG neuromuscular junction. AChE's content in the NMJ is contributed by both muscle and neuronal systems (Anglister, 1991). In the TG muscle, Andres et al. (1997) found that human AChE contributes 6 % of the total muscle enzyme, which reflects the neuronal-originated AChE. However, this limited contribution may be functionally more significant in elevating AChE concentrations at the NMJs. Theoretical models predict that a higher synaptic AChE concentration by itself will not significantly affect the quantal size (Anglister et al. 1994). Moreover, density calculations (Salpeter et al. 1978) indicate that AChE concentration in the mouse NMJ is already in excess of a critical lower limit. Consistent with these assumptions, we find a similar picture of quantal amplitudes and time courses in TG and WT NMJs. Nevertheless, blocking AChE activity revealed higher quantal amplitude in TG synapses than in WT, supporting excess enzyme in the NMJ and suggesting that in contrast to the theoretical prediction, this excess in the TG synapse is effective. The higher quantal amplitude may be an outcome of larger ACh content, but could also be the result of high receptor number consistent with the larger TG NMJs. Therefore, we interpret the ‘real’ amplitude as a delayed compensation response to excess AChE that retrieves the effect of single quanta to standard sizes.
Presynaptic properties are supposedly affected by raising AChE concentration, since the faster hydrolysis of released transmitter will induce less feedback inhibition on the release system (Starke et al. 1989). Thus, the higher quantal content and rate of spontaneous release observed in TG endplates could result from reduction in presynaptic autoinhibition (Parnas et al. 2000; Slutzky et al. 2001), and are likely to be a direct response to the expression of the transgene. Again, this finding is consistent with morphometric evidence pointing to higher density of presynaptic vesicles at the transgenic terminal (Andres et al. 1998), which could by itself be responsible for the higher frequency of spontaneous release (Del Castillo & Katz, 1954). If the quantal content is affected by tonic concentration of ACh in the synaptic cleft, as suggested above, then the reduction of quantal content to WT level upon physostigmine treatment indicates that ACh level in the transgenic NMJ is lower under control conditions, and is comparable to WT synapses only under conditions of AChE blockade. Support for the lower basal level of free ACh was found in the TG brain (Erb et al. 2001). It thus appears that the parameters of quantal content and frequency of spontaneous release are a direct consequence of the elevated catalytic activity of AChE in the transgenic NMJ. The in vitro synaptic situation in the TG muscle presents conditions of steady state release with higher quantal content and actual amplitude, combined with a relatively high vesicle number. Together, these probably rescue the hypo-cholinergic synaptic state under higher levels of AChE.
In conclusion, our experiments have confirmed that altered activity levels of AChE induce muscle malfunctioning beyond neuromuscular transmission. These results suggest broad physiological implications for AChE-related muscular pathologies.
Acknowledgments
This work was supported by the US Army Medical Research and Materiel Command (DAMD 17-99-1-9547), US/Israel Bi-national Science Foundation (BSF, 1999-115) and Ester Neuroscience, Ltd (to H.S.).
Supplementary material
The online version of this paper can be found at:
http://www.jphysiol.org/cgi/content/full/546/1/165
and contains supplementary material. This consists of morphological examples of (1) excess enzyme in the Tg NMJ and (2) abnormal Tg myofibrils.
References
- Aldrich TK, Shander A, Chaudhry I, Nagashima H. Fatigue of isolated rat diaphragm: role of impaired neuromuscular transmission. J App Physiol. 1986;61:1077–1083. doi: 10.1152/jappl.1986.61.3.1077. [DOI] [PubMed] [Google Scholar]
- Andres C, Beeri R, Freidman A, Lev-Lehman E, Henis S, Timberg R, Shani M, Soreq H. Acetylcholinesterase-transgenic mice display embryonic modulations in spinal cord choline acetyltransferase and neurexin Iβ gene expression followed by late-onset neuromotor deterioration. Proc Natl Acad Sci USA. 1997;94:8173–8178. doi: 10.1073/pnas.94.15.8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres C, Seidman S, Beeri R, Timberg R, Soreq H. Transgenic acetylcholinesterase induces enlargement of murine neuromuscular junctions but leaves spinal cord synapses intact. Neurochem Int. 1998;32:449–456. doi: 10.1016/s0197-0186(97)00121-6. [DOI] [PubMed] [Google Scholar]
- Anglister L. Acetylcholinesterase from the motor nerve terminal accumulates on the synaptic basal lamina of the myofiber. J Cell Biol. 1991;115:755–764. doi: 10.1083/jcb.115.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglister L, Stiles JR, Salpeter MM. Acetylcholinesterase density and turnover number at frog neuromuscular junctions, with modeling of their role in synaptic function. Neuron. 1994;12:783–794. doi: 10.1016/0896-6273(94)90331-x. [DOI] [PubMed] [Google Scholar]
- Beeri R, Andres C, Lev-Lehman E, Timberg R, Huberman T, Hani M, Soreq H. Transgenic expression of human acetylcholinesterase induces progressive cognitive deterioration in mice. Curr Biol. 1995;5:1063–1071. doi: 10.1016/s0960-9822(95)00211-9. [DOI] [PubMed] [Google Scholar]
- Beeri R, Le Novere N, Mervis R, Huberman T, Grauer E, Changoux JP, Soreq H. Enhanced hemicholinium binding and attenuated dendrite branching in cognitively impaired acetylcholinesterase-transgenic mice. J Neurochem. 1997;69:2441–2451. doi: 10.1046/j.1471-4159.1997.69062441.x. [DOI] [PubMed] [Google Scholar]
- Behra M, Cousin X, Bertrand C, Vonesch JL, Biellmann D, Chatonnet A, Strahle U. Acetylcholinesterase is required for neuronal and muscular development in the zebrafish embryo. Nat Neurosci. 2002;5:111–118. doi: 10.1038/nn788. [DOI] [PubMed] [Google Scholar]
- Brown G., Jr Amyotrophic lateral sclerosis: recent insight from genetics and transgenic mice. Cell. 1995;80:687–692. doi: 10.1016/0092-8674(95)90346-1. [DOI] [PubMed] [Google Scholar]
- Cohen O, Erb C, Ginzberg D, Pollak Y, Shohami S, Seidman S, Soreq H, Yirmiya R. Overexpression of “readthrough” acetylcholinesterase is associated with antisense suppressible behavioral impairments. Mol Psych. 2002;7:874–85. doi: 10.1038/sj.mp.4001103. [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Darboux I, Barthalay Y, Piovant M, Hipeau-Jacquotte P. The structure-function relationship in Drosophila neurotactin show that cholinesterasic domain may have adhesion properties. EMBO J. 1996;15:4835–4843. [PMC free article] [PubMed] [Google Scholar]
- Darreh-Shori T, Almkvist O, Guan ZZ, Garlind A, Strandberg B, Svensson AL, Soreq H, Hellstrom-Lindahl E, Nordberg A. Sustained cholinesterase inhibition in AD patients receiving rivastigmine for 12 months. Neurology. 2002;59:563–572. doi: 10.1212/wnl.59.4.563. [DOI] [PubMed] [Google Scholar]
- del Castillo J, Katz B. Quantal components of the endplate potential. J Physiol. 1954;124:560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudel J, Schramm M, Franke C, Ratner E, Parnas H. Block of quantal end-plate currents of mouse muscle by physostigmine and procaine. J Neurophysiol. 1999;81:2386–2397. doi: 10.1152/jn.1999.81.5.2386. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes. In: Engle AG, editor. Myasthenia Gravis and Myasthenic Syndromes. Oxford University Press; 1999. pp. 251–297. [Google Scholar]
- Erb C, Troost J, Kopf S, Schmitt U, Loffelholz K, Soreq H, Klein J. Activity-induced compensatory mechanisms facilitate hippocampal acetylcholine release in transgenic mice over-expressing human acetylcholinesterase. J Neurochem. 2001;77:638–646. doi: 10.1046/j.1471-4159.2001.00287.x. [DOI] [PubMed] [Google Scholar]
- Ferry CB, Kelly SS. The nature of the presynaptic effects of (+)-tubocurarine at the mouse neuromuscular junction. J Physiol. 1988;403:425–437. doi: 10.1113/jphysiol.1988.sp017257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifman M, Galyam N, Seidman S, Soreq H. Functional redundancy of acetylcholinesterase and neuroligin in mammalian neuritogenesis. Proc Natl Acad Sci USA. 1998;95:13935–13940. doi: 10.1073/pnas.95.23.13935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AV. First and Last Experiments in Muscle Mechanics. Cambridge University Press; 1970. The development and duration of the active state in stimulated muscle, as revealed by an applied stretch; pp. 62–75. [Google Scholar]
- Hubbard JI, Wilson DF. Neuromuscular transmission in a mammalian preparation in the absence of blocking drugs and the effect of D-tubocurarine. J Physiol. 1973;228:307–325. doi: 10.1113/jphysiol.1973.sp010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufer D, Freidman A, Seidman S, Soreq H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature. 1998;393:373–377. doi: 10.1038/30741. [DOI] [PubMed] [Google Scholar]
- Kuei JH, Shadmehr R, Sieck GC. Relative contribution of neurotransmission failure to diaphragm fatigue. J Appl Physiol. 1990;68:174–180. doi: 10.1152/jappl.1990.68.1.174. [DOI] [PubMed] [Google Scholar]
- Lev-Lehman E, Evron T, Broide RS, Meshorer E, Ariel I, Seidman S, Soreq H. Synaptogenesis and myopathy under acetylcholinesterase over expression. J Mol Neurosci. 2000;14:93–105. doi: 10.1385/JMN:14:1-2:093. [DOI] [PubMed] [Google Scholar]
- Linder TM, Quastel DMJ. A voltage clamp study of the permeability change induced by quanta of transmitter at the mouse end-plate. J Physiol. 1978;281:535–556. doi: 10.1113/jphysiol.1978.sp012438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E, Erb C, Gazit R, Pavlovsky L, Kaufer D, Glick D, Freidman A, Ben-Arie N, Soreq H. Alternative splicing and neuritic mRNA translocation under long-term neuronal hypersensitivity. Science. 2002;295:508–512. doi: 10.1126/science.1066752. [DOI] [PubMed] [Google Scholar]
- Ohno K, Tsujino A, Brengman JM, Harper CM, Bajzer Z, Udd D, Beyring R, Robb S, Kirkham FJ, Engel AG. Choline acetyletransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA. 2001;98:2017–2022. doi: 10.1073/pnas.98.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panenic R, Gisiger V, Gardiner PF. Fatigability of rat hindlimb muscles after acute irreversible acetylcholinesterase inhibition. J Appl Physiol. 1999;87:1455–1462. doi: 10.1152/jappl.1999.87.4.1455. [DOI] [PubMed] [Google Scholar]
- Parnas H, Segal L, Dudel J, Parnas I. Autoreceptors, membrane potential and regulation of transmitter release. Trends Neurosci. 2000;23:60–68. doi: 10.1016/s0166-2236(99)01498-8. [DOI] [PubMed] [Google Scholar]
- Ray D. University of Leicester, UK: IEH Reports, MRC Institute for Environment and Health; 1998. Organophosphorus esters: An evluation of chronic neurotoxic effects. [Google Scholar]
- Ruff RL, Lennon VA. End-plate voltage-gated sodium channels are lost in clinical and experimental myasthenia gravis. Ann Neurol. 1998;43:370–379. doi: 10.1002/ana.410430315. [DOI] [PubMed] [Google Scholar]
- Salpeter MM, Rogers AW, Kasprzak H, McHenry FA. Acetylcholinesterase in the fast extraocular muscle of the mouse by light and electron microscope autoradiography. J Cell Biol. 1978;78:274–285. doi: 10.1083/jcb.78.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Lowenstein-Lichtenstein Y, Glick D, Liao J, Norgaard-Pedrsen B, Soreq H. Successive orgnophosphate inhibition and oxime reactivation reveals distinct responses of recombinant human cholinesterase variants. Mol Brain Res. 1995;31:101–110. doi: 10.1016/0169-328x(95)00040-y. [DOI] [PubMed] [Google Scholar]
- Shaw KP, Aracava Y, Akaike A, Rickett DL, Albuquerque EX. The reversible cholinesterase inhibitor physostigmine has channel-blocking and agonist effects on the acetylcholine receptor-ion channel complex. Mol Pharmacol. 1985;28:527–538. [PubMed] [Google Scholar]
- Slutsky I, Silman I, Parnas I, Parnas H. Presynaptic M2 muscarinic receptors are involved in controlling the kinetics of ACh release at the frog neuromuscular junction. J Physiol. 2001;536:717–725. doi: 10.1111/j.1469-7793.2001.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq H, Seidman S. Acetylcholinesterase - new roles for an old actor. Nat Rev Neurosci. 2001;2:294–303. doi: 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- Starke K, Gathert M, Kilbinger H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol Rev. 1989;69:864–989. doi: 10.1152/physrev.1989.69.3.864. [DOI] [PubMed] [Google Scholar]
- Sternfeld M, Ming G, Song H, Sela K, Timberg R, Poo MM, Soreq H. Acetylcholinesterase enhances neurite growth and synapse development through alternative contribution of its hydrolytic capacity, core protein, and variable C-termini. J Neurosci. 1998;18:1240–1249. doi: 10.1523/JNEUROSCI.18-04-01240.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternfeld M, Shoham S, Klein O, Flores-Flores C, Evron T, Idelson GH, Kitsberg D, Patrick JW, Soreq H. Excess “read-through” acetylcholinesterase attenuates but the “synaptic” variant intensifies neurodeterioration correlates. Proc Natl Acad Sci USA. 2000;97:8647–8652. doi: 10.1073/pnas.140004597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lunteren E, Moyer M. Effects of DAP on diaphragm force and fatigue, including fatigue due to neurotransmission failure. J Appl Physiol. 1996;81:2214–2220. doi: 10.1152/jappl.1996.81.5.2214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.