

Abstract
5-Hydroxydecanoate (5-HD) inhibits ischaemic and pharmacological preconditioning of the heart. Since 5-HD is thought to inhibit specifically the putative mitochondrial ATP-sensitive K+ (KATP) channel, this channel has been inferred to be a mediator of preconditioning. However, it has recently been shown that 5-HD is a substrate for acyl-CoA synthetase, the mitochondrial enzyme which ‘activates’ fatty acids. Here, we tested whether activated 5-HD, 5-hydroxydecanoyl-CoA (5-HD-CoA), is a substrate for medium-chain acyl-CoA dehydrogenase (MCAD), the committed step of the mitochondrial β-oxidation pathway. Using a molecular model, we predicted that the hydroxyl group on the acyl tail of 5-HD-CoA would not sterically hinder the active site of MCAD. Indeed, we found that 5-HD-CoA was a substrate for purified human liver MCAD with a Km of 12.8 ± 0.6 μm and a kcat of 14.1 s−1. For comparison, with decanoyl-CoA (Km∼3 μm) as substrate, kcat was 6.4 s−1. 5-HD-CoA was also a substrate for purified pig kidney MCAD. We next tested whether the reaction product, 5-hydroxydecenoyl-CoA (5-HD-enoyl-CoA), was a substrate for enoyl-CoA hydratase, the second enzyme of the β-oxidation pathway. Similar to decenoyl-CoA, purified 5-HD-enoyl-CoA was also a substrate for the hydratase reaction. In conclusion, we have shown that 5-HD is metabolised at least as far as the third enzyme of the β-oxidation pathway. Our results open the possibility that β-oxidation of 5-HD or metabolic intermediates of 5-HD may be responsible for the inhibitory effects of 5-HD on preconditioning of the heart.
Preconditioning of the heart by means of brief (∼5 min) periods of ischaemia or various pharmacological agents, such as diazoxide and volatile anaesthetics, reduces the cellular damage which follows a prolonged ischaemic insult (Cope et al. 1997; Schulz et al. 2001). Mitochondrial KATP channels are thought to play a central role in mediating these phenomena (Gross & Fryer, 1999; Gross, 2000; Sato & Marbán, 2000). An important piece of evidence for implicating mitochondrial KATP channels as mediators of preconditioning is the consistent inhibitory effect of 5-hydroxydecanoate (5-HD). This drug, originally investigated for its antiarrhythmic actions (Notsu et al. 1992), has been shown to inhibit both ischaemic and pharmacological preconditioning (Gross & Fryer, 1999; Gross, 2000; Sato & Marbán, 2000). Hence, elucidation of the molecular action(s) of 5-HD may provide a clue to the elusive mechanism of preconditioning. Garlid et al. (1997) reported that 5-HD inhibited K+ flux via diazoxide-activated mitochondrial KATP channels. In further work, Jabùrek et al. (1998) found that 5-HD inhibited K+ flux via mitochondrial KATP channels in heart and liver mitochondria with a half-maximal inhibitory concentration of 45–75 μm. These studies have fostered the notion that 5-HD is a specific inhibitor of the mitochondrial KATP channel.
Recently, Hanley et al. (2002a) have shown that 5-HD is a substrate for acyl-CoA synthetase, the mitochondrial enzyme which thioester-links fatty acids with coenzyme A (CoA), a reaction referred to as fatty acid ‘activation’. This observation opened the possibility that the activated form of 5-HD, 5-HD-CoA (5-hydroxydecanoyl-CoA), might be responsible for inhibiting preconditioning. By analogy with other acyl-CoA esters, the possible targets of 5-HD-CoA include KATP channels, protein kinase C and the ADP/ATP translocase (Paucek et al. 1996; Knudsen et al. 1999; Liu et al. 2001). Another possibility is that 5-HD-CoA may serve as a substrate for medium-chain acyl-CoA dehydrogenase (MCAD), the enzyme which catalyses the initial oxidative step of mitochondrial β-oxidation. The structure and catalytic mechanism of MCAD have been extensively investigated. X-ray crystallographic structures of both pig liver and human liver MCAD have been determined at 2.4 and 2.75 Å (0.24 and 0.275 nm) resolution, respectively (Kim et al. 1993; Lee et al. 1996). These structural data revealed that the substrate-binding pocket, which is lined with hydrophobic amino acid residues, extends almost to the centre of the protein. Substrate binds to the oxidised enzyme, forming an enzyme-substrate complex, and two reducing equivalents are transferred to the bound FAD moiety (dehydrogenation step), yielding the enoyl-CoA product (for review, see Thorpe & Kim, 1995). The product dissociates after two molecules of electron transfer protein (ETF), the natural electron acceptor for MCAD (Roberts et al. 1996), have re-oxidised the enzyme. ETF transfers electrons to ubiquinone, thereby linking MCAD with the respiratory chain.
In the present study, we show that MCAD catalyses the conversion of 5-hydroxydecanoyl-CoA to 5-HD-enoyl-CoA. We further show that 5-HD-enoyl-CoA is a substrate for the second enzyme of the β-oxidation pathway, enoyl-CoA hydratase. Since the reaction rates were comparable to those of decanoate derivatives, our results raise the possibility that 5-HD may be completely oxidised in mitochondria. These findings have some important implications for our understanding of the mechanisms underlying ischaemic or pharmacological preconditioning.
METHODS
Preparation of 5-hydroxydecanoyl-CoA
5-Hydroxydecanoyl-CoA (5-HD-CoA) was synthesised using commercially available acyl-CoA synthetase (EC 6.2.1.3; Sigma). 5-Hydroxydecanoate (RBI, Sigma), coenzyme A and Na2ATP were added to Tris buffer solution containing (mm): 100 Tris, 9.1 EDTA and 1.8 EDTA (pH adjusted to 7.5 with HCl). Coenzyme A concentration before and after the reaction was determined using DTNB (5,5′-dithiobis-(2-nitrobenzoic acid)), which reacts with free thiol groups and generates 2-nitro-5-thiobenzoate (peak absorbance, 412 nm). 5-HD-CoA was separated using a Waters HPLC system which incorporated an Alltech C18-column (0.1 cm × 25 cm, 10 μm, Econosil). Methanol and 20 mm potassium phosphate solution (pH 7.0) were used as solvents. Electrospray ionisation mass spectrometry was used to confirm that the isolated peak was 5-HD-CoA (Hanley et al. 2002a).
Medium-chain acyl-CoA dehydrogenase
Recombinant human liver MCAD (medium-chain acyl-CoA dehydrogenase; EC 1.3.99.3) was expressed and purified as previously described (Peterson et al. 1995), whereas pig kidney MCAD was extracted and purified from tissue (Johnson et al. 1992). All experiments were performed at 25 °C in buffer solution containing (mm): 50 potassium phosphate, 0.3 EDTA and 10 % glycerol (pH 7.6). Activity of MCAD was routinely assayed by monitoring reduction of ferricenium hexafluorophosphate (FcPF6) at 300 nm (ε= 4.3 mm−1 cm−1) using a Perkin-Elmer Lambda 3B spectrophotometer. The cuvette had a path length of 1 cm and contained a magnetic stirrer. Decanoyl-CoA (ε= 4.3 mm−1 cm−1 at 259 nm) or 5-HD-CoA (ε assumed to be the same as for decanoyl-CoA) was used as substrate.
Molecular model of 5-hydroxydecanoyl-CoA bound to MCAD
A molecular model was used to depict both (R)- and (S)-5-HD-CoA bound to pig liver medium-chain acyl-CoA dehydrogenase. The X-ray crystallographic structure of this enzyme in the absence (pdb3mdd.pdb) and presence (pdb3mde.pdb) of octenoyl-CoA was downloaded from the Brookhaven Protein Data Bank. MCAD-bound octenoyl-CoA (C8-CoA) was modified to decanoyl-CoA (C10-CoA) and energy minimisation of the enzyme-C10-CoA complex was performed as previously described (Peterson et al. 2001). A hydroxyl group was added in either the (R)- or (S)-configuration at position C-5 to generate a model of a MCAD-5-HD-CoA complex and to predict whether the hydroxyl group could sterically hinder catalysis. Amino acid residues located within 4 Å of the ligand were identified. Data were displayed on a Silicon Graphics Indigo XZ workstation.
Enoyl-CoA hydratase
The enoyl-CoA esters decenoyl-CoA and 5-hydroxydecenoyl-CoA (5-HD-enoyl-CoA) were prepared enzymatically using MCAD and FcPF6 as previously described (Kumar & Srivastava, 1994) and purified via preparative HPLC. Purified 5-HD-enoyl-CoA was not metabolised by MCAD, indicating that it was not contaminated with 5-hydroxydecanoyl-CoA. Commercially available bovine liver enoyl-CoA hydratase (Fluka) was added to standard buffer solution and enzyme activity, using either decenoyl-CoA (Km 300 μm; Waterson & Hill, 1972) or 5-HD-enoyl-CoA as substrate, was monitored by measuring absorbance at 263 nm (Waterson & Hill, 1972).
RESULTS
Molecular model of (S)-5-hydroxydecanoyl-CoA bound to MCAD
We have studied the question whether 5-HD-CoA can be metabolised by β-oxidation in the same way as decanoyl-CoA, a natural substrate of MCAD. In order to predict whether the hydroxyl group of 5-HD-CoA, which can have either an (R)- or (S)-configuration, could hinder catalysis, we incorporated 5-HD-CoA in a molecular model of pig liver MCAD. Figure 1A shows (S)-5-hydroxydecanoyl-CoA ((S)-5-HD-CoA) bound to a subunit of the homotetrameric enzyme MCAD. The atoms of (S)-5-HD-CoA are colour-coded whereas the protein is shown as a yellow ribbon. We identified the amino acid residues located within 4 Å (solid yellow structures) of the ligand (Fig. 1B). Glu-376 (labelled in Fig. 1B) is the active site base which abstracts the α-proton of the acyl-CoA substrate. After the α-proton is abstracted, the β-hydrogen is transferred to the N-5 position of the isoalloxazine ring of FAD (flavin adenine dinucleotide). The model suggests that the hydroxyl group of 5-HD-CoA, whether in the (R)- (not illustrated) or (S)-configuration (Fig. 1B), can be easily accommodated by the ligand-binding pocket of MCAD. Furthermore, the model indicates that 5-HD-CoA would not sterically hinder the active site.
Figure 1. Molecular model of MCAD-(S)-5-hydroxydecanoyl-CoA complex.
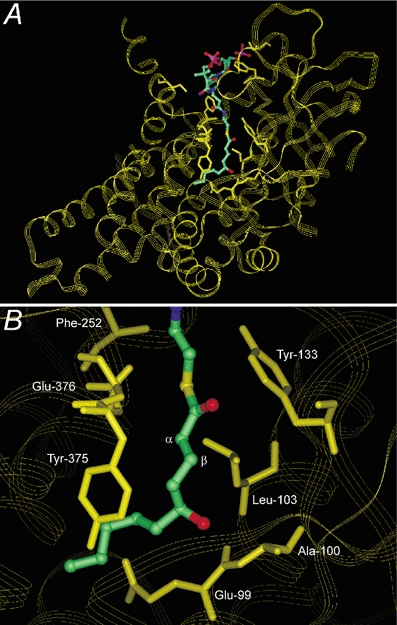
A, structure of pig liver MCAD monomer in complex with modelled ligand (S)-5-HD-CoA. Amino acid residues within 4 Å of the ligand are shown (solid yellow structures). B, enlarged view of the MCAD-(S)-5-HD-CoA complex showing relation of the (S)-5-hydroxyl group to labelled residues in the fatty acyl binding pocket. Glu-376 is the active site base which abstracts the α-proton from the acyl-CoA substrate whereas the β-hydrogen is transferred to FAD (not depicted).
5-Hydroxydecanoyl-CoA is a substrate for MCAD
Based on our above model and the known broad substrate specificity of MCAD (Peterson et al. 1995), we predicted that 5-HD-CoA would be a potential substrate for MCAD. In the experiments shown in Fig. 2, 5-HD-CoA was freshly synthesised using acyl-CoA synthetase, which was subsequently removed by ultrafiltration. The amount of 5-HD-CoA formed was determined by measuring the change in coenzyme A (CoA) concentration. Figure 2 shows that 5-HD-CoA was indeed a substrate for MCAD. In fact, maximal turnover rates of both liver (Fig. 2A and B) and kidney (Fig. 2C and D) MCAD were higher when 5-HD-CoA was used as substrate compared to decanoyl-CoA. In the case of liver MCAD, the apparent kcat was 10.3 s−1 with 5-HD-CoA as substrate (Fig. 2A) and 6.4 s−1 with decanoyl-CoA as substrate (Fig. 2B). The latter value is in line with the value of 7.4 s−1 reported by Peterson et al. (1995). Similar results were obtained using kidney MCAD: the apparent kcat was 10.7 s−1 with 5-HD-CoA (Fig. 2C) and kcat 7.8 s−1 with decanoyl-CoA as substrate (Fig. 2D). In accordance with these results, Peterson et al. (1995) reported a kcat value of 8.6 s−1 with decanoyl-CoA as substrate for MCAD.
Figure 2. 5-Hydroxydecanoyl-CoA is a substrate for human liver and pig kidney MCAD.
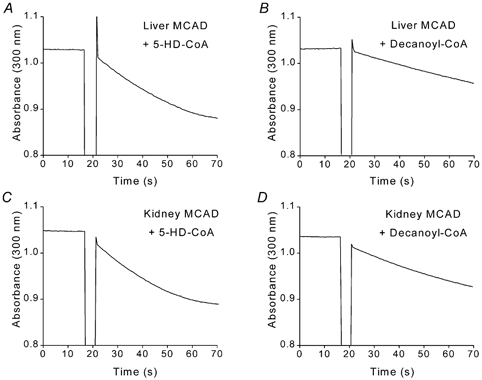
A and B, purified human liver MCAD (50 nm) was added (indicated by interruption in trace) to solution containing electron acceptor (FcPF6) and saturating concentrations (43 μm) of either 5-HD-CoA (A) or decanoyl-CoA (B). A decrease in absorbance at 300 nm indicates enzyme activity. C and D, purified pig kidney MCAD (50 nm) was added to solution containing either 5-HD-CoA (C) or decanoyl-CoA (D), each at a concentration of 43 μm.
Kinetics of 5-HD-CoA interaction with MCAD
The kinetics of human liver MCAD interacting with the acyl-CoA substrates octanoyl-CoA and decanoyl-CoA have been characterized (Peterson et al. 1995). In order to determine the kinetics of human liver MCAD interacting with 5-HD-CoA we purified 5-HD-CoA by preparative HPLC. Figure 3A shows the rate of MCAD-catalysed reaction as a function of [5-HD-CoA]. 5-HD-CoA was added from a stock solution of 2.6 mm in potassium phosphate (20 mm) solution (pH 7.0). The Km was 12.8 ± 0.6 μm and Vmax (maximal rate) was 23.6 μm s−1 (kcat, 14.1 s−1). Data were fitted using the Michaelis-Menten equation, V=Vmax[S]/([S]+Km), where V is rate and [S] is substrate concentration. For comparison, with decanoyl-CoA as substrate, the Km was found to be 4-fold lower (∼3 μm; Peterson et al. 1995), whereas the kcat was about half (6.4 s−1; Fig. 2B).
Figure 3. Kinetics of MCAD-catalysed dehydrogenation of 5-HD-CoA and metabolism of the product 5-hydroxydecenoyl-CoA.
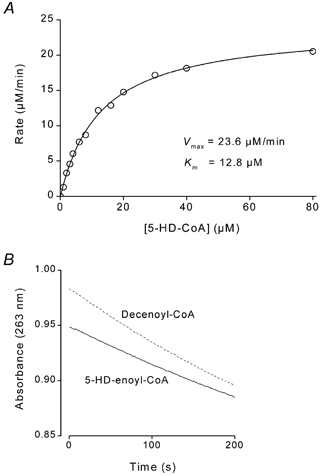
A, steady-state activity of human liver MCAD was determined at various concentrations of purified 5-HD-CoA. Km was 12.8 ± 0.6 μm and Vmax 23.6 μm min−1. Data were fitted using the Michaelis-Menten equation (see Results). B, bovine liver enoyl-CoA hydratase was activated by addition of either decenoyl-CoA (dashed line) or purified 5-hydroxydecenoyl-CoA (continuous line). Enzyme activity was monitored by measuring absorbance at 263 nm.
The enoyl-CoA product 5-hydroxydecenoyl-CoA is a substrate for enoyl-CoA hydratase
Dehydrogenation of 5-HD-CoA via MCAD yields the enoyl-CoA product 5-hydroxydecenoyl-CoA (5-HD-enoyl-CoA). We next tested whether purified 5-HD-enoyl-CoA was a substrate for enoyl-CoA hydratase, the second enzyme of the mitochondrial β-oxidation pathway. Enzyme activity was monitored by measuring absorbance at 263 nm (Waterson & Hill, 1972). When 85 μm decenoyl-CoA was added to 12 μm bovine liver hydratase, the enzyme activity was 0.021 min−1 (Fig. 3B). Similarly, with 5-HD-enoyl-CoA as substrate (added from a 2.2 mm stock solution), enoyl-CoA hydratase activity was 0.018 min−1 (Fig. 3B). In this case, the product of the hydratase reaction is l-3,5-dihydroxydecanoyl-CoA (l-3,5-DHD-CoA; see Fig. 4A).
Figure 4. Schematic diagrams of the metabolism of 5-HD and its uptake into the mitochondrial matrix.
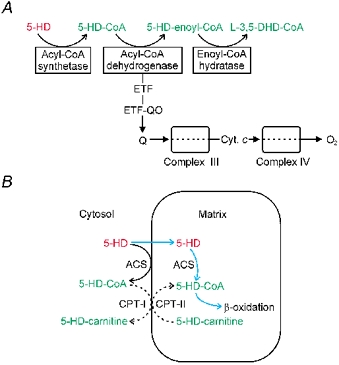
A, schematic summary showing activation and β-oxidation of 5-hydroxydecanoate. Medium-chain acyl-CoA dehydrogenase transfers electrons to the electron transport chain via electron transfer flavoprotein (ETF) and ETF oxidoreductase (ETF-QO). B, schematic diagram showing that, typical for medium-chain fatty acids, 5-HD can be activated via ACS (acyl-CoA synthetase) either on the mitochondrial outer membrane or in the matrix. Whether extramitochondrial 5-HD-CoA serves as a substrate for CPT-I (carnitine palmitoyltransferase-I) is not known.
DISCUSSION
Metabolism of 5-HD-CoA
5-Hydroxydecanoate, which blocks preconditioning, is generally presumed to be a selective mitochondrial KATP channel inhibitor. However, the recent report that 5-HD is thioester linked to CoA via acyl-CoA synthetase (Hanley et al. 2002a), producing 5-HD-CoA, undermines this assumption. In the present study, we have now shown that 5-HD-CoA is dehydrogenated by purified MCAD, the first enzyme of the β-oxidation pathway. This result could be predicted from our molecular model of a MCAD-5-HD-CoA complex, which showed that the hydroxyl group on the acyl tail would be unable to sterically hinder the active site. Moreover, we also found that the enoyl-CoA product of this reaction, 5-HD-enoyl-CoA, was a substrate for the next enzyme of the β-oxidation pathway (enoyl-CoA hydratase), yielding l-3,5-dihydroxydecanoyl-CoA. The above results are schematically summarised in Fig. 4A.
The Km of human liver MCAD was about 4-fold higher with 5-HD-CoA as substrate than with decanoyl-CoA as substrate, indicating that 5-HD-CoA binds more weakly to the enzyme. Hence, the hydroxyl group on the acyl tail of 5-HD-CoA evidently decreases the efficiency with which this ligand, compared to decanoyl-CoA, binds to the catalytic pocket of MCAD. However, the Vmax of MCAD was about 2-fold higher with 5-HD-CoA instead of decanoate as substrate. The most likely explanation for the higher Vmax is that the weaker binding of 5-HD-CoA facilitates the rate-limiting step of the enzyme, the dissociation of the reaction product (5-HD-enoyl-CoA) from the active site (Peterson et al. 1998). Taken together, the 4-fold higher Km and 2-fold higher Vmax of MCAD with 5-HD-CoA as substrate, compared to decanoyl-CoA, suggests that the rate of enzyme turnover would be similar at lower concentrations of either substrate whereas, at higher substrate concentrations, turnover rate would be about 2-fold greater with 5-HD-CoA.
We also showed that purified 5-HD-enoyl-CoA was a substrate for enoyl-CoA hydratase, which suggests that 5-HD may be completely metabolised. Assuming this is the case, and taking into account that 5-HD is a racemic mixture, the last step of β-oxidation, catalysed by β-ketothiolase (EC 2.3.1.16), would yield acetyl-CoA and a racemic mixture of 3-hydroxyoctanoyl-CoA. l-3-Hydroxyoctanoyl-CoA (the l-isomer) is not a metabolic end-product since it is a normal substrate for l-3-hydroxyacyl-CoA dehydrogenase (EC 1.1.1.35), the penultimate enzyme of the β-oxidation pathway. This enzyme is enantioselective and therefore d-3-hydroxyoctanoyl-CoA (the d-isomer) would first need to be converted to l-3-hydroxyoctanoyl-CoA via 3-hydroxyacyl-CoA epimerase before it could be metabolised. However, 3-hydroxyacyl-CoA epimerase activity has been shown to be weak in heart mitochondria (Chu et al. 1984). If d-3-hydroxyoctanoyl-CoA is not degraded by β-oxidation, the acyl group would probably exit the mitochondria as the free acid (after thioesterase-catalysed hydrolysis) or as the carnitine derivative, after transfer to carnitine via carnitine acyltransferase.
Implications for preconditioning
Despite extensive study, the mechanisms underlying the protection of the heart against ischaemic damage have remained unclear. There are three main mechanisms that have been proposed to explain the cardioprotection conferred by drugs (pharmacological preconditioning) or by brief episodes of ischaemia (ischaemic preconditioning) (Gross & Fryer, 1999; Grover & Garlid, 2000; Toyoda et al. 2000; Sanada et al. 2001; Suzuki et al. 2002; Hanley et al. 2002a, b; Standen, 2002): (i) activation of sarcolemmal KATP channels, (ii) activation of mitochondrial KATP channels, and (iii) inhibition of components of the respiratory chain. Since 5-HD has been shown to inhibit both ischaemic and pharmacological preconditioning (Gross & Fryer, 1999; Gross, 2000; Sato & Marbán, 2000) elucidation of its molecular action(s) may provide a clue to the mechanism of preconditioning.
The data reported here, together with the results of Hanley et al. (2002a), extend the possible mechanisms by which 5-HD could block preconditioning: (i) the putative mitochondrial KATP channel may be inhibited by cytosolic 5-HD or 5-HD-CoA; (ii) intermediates of 5-HD metabolism may inhibit the β-oxidation pathway or exert some other cellular action; (iii) 5-HD may be completely metabolised via the β-oxidation pathway, which is linked to the respiratory chain. This link may provide a bypass to respiratory chain inhibition at complex I or complex II (Hanley et al. 2002a). β-Oxidation of 5-HD would also explain the observation that 5-HD reverses the opening of sarcolemmal KATP channels induced by blocking glucose metabolism in ventricular myocytes (Notsu et al. 1992). (iv) Application of 5-HD may lead to a switch of metabolism from glucose to fatty acid oxidation, which may impair recovery of cardiac function after ischaemia (Lopaschuk, 1997).
Like other short- or medium-chain fatty acids, 5-HD can traverse the mitochondrial inner membrane. Subsequently, 5-HD can be activated by medium-chain acyl-CoA synthetase present in the matrix, as illustrated in Fig. 4B (blue arrows), and metabolised by β-oxidation (Fig. 4A). 5-HD-CoA produced by acyl-CoA synthetase at the mitochondrial outer membrane, however, is membrane impermeant and needs to access the matrix via carnitine palmitoyltransferase-I (CPT-I) before it can be metabolised. Whether extramitochondrially activated 5-HD serves as a substrate for CPT-I is not known (Fig. 4B, interrupted lines), although this is likely the case.
In conclusion, our results suggest that metabolism of 5-HD may be responsible for its inhibitory effect on preconditioning. Thus, 5-HD should no longer be considered a suitable pharmacological tool for the identification of mitochondrial ATP-sensitive potassium channels.
Acknowledgments
We thank Professor Horst Schulz (Department of Chemistry, City University of New York) for helpful discussion.
REFERENCES
- Chu CH, Kushner L, Cuebas D, Schulz H. The activity of 3-hydroxyacyl-CoA epimerase is insufficient to account for the rate of linoleate oxidation in rat heart mitochondria. Evidence for a modified pathway of linoleate degradation. Biochem Biophys Res Commun. 1984;118:162–167. doi: 10.1016/0006-291x(84)91081-7. [DOI] [PubMed] [Google Scholar]
- Cope DK, Impastato WK, Cohen MV, Downey JM. Volatile anesthetics protect the ischemic rabbit myocardium from infarction. Anesthesiology. 1997;86:699–709. doi: 10.1097/00000542-199703000-00023. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D'Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- Gross GJ. The role of mitochondrial KATP channels in cardioprotection. Basic Res Cardiol. 2000;95:280–284. doi: 10.1007/s003950050004. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res. 1999;84:973–979. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- Grover GJ, Garlid KD. ATP-sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Mickel M, Löffler M, Brandt U, Daut J. KATP channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002a;542:735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002b;544:687–693. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabùrek M, Yarov-Yarovoy V, Paucek P, Garlid KD. State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J Biol Chem. 1998;273:13578–13582. [PubMed] [Google Scholar]
- Johnson JK, Wang ZX, Srivastava DK. Mechanistic investigation of medium-chain fatty acyl-CoA dehydrogenase utilizing 3-indolepropionyl/acryloyl-CoA as chromophoric substrate analogues. Biochemistry. 1992;31:10564–10575. doi: 10.1021/bi00158a020. [DOI] [PubMed] [Google Scholar]
- Kim J-JP, Wang M, Paschke R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc Natl Acad Sci U S A. 1993;90:7523–7527. doi: 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen J, Jensen MV, Hansen JK, Fæbergeman NJ, Neergaard TBF, Gaigg B. Role of acylCoA binding protein in acylCoA transport, metabolism and cell signaling. Mol Cell Biochem. 1999;192:95–103. doi: 10.1007/978-1-4615-4929-1_11. [DOI] [PubMed] [Google Scholar]
- Kumar NR, Srivastava DK. Reductive half-reaction of medium-chain fatty acyl-CoA dehydrogenase utilizing octanoyl-CoA/octenoyl-CoA as a physiological substrate/product pair: similarity in the microscopic pathways of octanoyl-CoA oxidation and octenoyl-CoA binding. Biochemistry. 1994;33:8833–8841. doi: 10.1021/bi00195a027. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Wang M, Paschke R, Nandy A, Ghisla S, Kim JJ. Crystal structures of the wild type and the Glu376Gly/Thr255Glu mutant of human medium-chain acyl-CoA dehydrogenase: influence of the location of the catalytic base on substrate specificity. Biochemistry. 1996;35:12412–12420. doi: 10.1021/bi9607867. [DOI] [PubMed] [Google Scholar]
- Liu GX, Hanley PJ, Ray J, Daut J. Long-chain acyl-coenzyme A esters and fatty acids directly link metabolism to KATP channels in the heart. Circ Res. 2001;88:918–924. doi: 10.1161/hh0901.089881. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD. Alterations in fatty acid oxidation during reperfusion of the heart after myocardial ischemia. Am J Cardiol. 1997;80(3A):11A–16A. doi: 10.1016/s0002-9149(97)00453-0. [DOI] [PubMed] [Google Scholar]
- Notsu T, Tanaka I, Takano M, Noma A. Blockade of the ATP-sensitive K+ channel by 5-hydroxydecanoate in guinea pig ventricular myocytes. J Pharmacol Exp Ther. 1992;260:702–708. [PubMed] [Google Scholar]
- Paucek P, Yarov-Yarovoy V, Sun X, Garlid KD. Inhibition of the mitochondrial KATP channel by long-chain acyl-CoA esters and activation by guanine nucleotides. J Biol Chem. 1996;271:32084–32088. doi: 10.1074/jbc.271.50.32084. [DOI] [PubMed] [Google Scholar]
- Peterson KL, Galitz DS, Srivastava DK. Influence of excision of a methylene group from Glu-376 (Glu376 Asp mutation) in the medium chain acyl-CoA dehydrogenase-catalyzed reaction. Biochemistry. 1998;37:1697–1705. doi: 10.1021/bi972590s. [DOI] [PubMed] [Google Scholar]
- Peterson KL, Sergienko EE, Wu Y, Kumar NR, Strauss AW, Oleson AE, Muhonen WW, Shabb JB, Srivastava DK. Recombinant human liver medium-chain acyl-CoA dehydrogenase: purification, characterization, and the mechanism of interactions with functionally diverse C8-CoA molecules. Biochemistry. 1995;34:14942–14953. doi: 10.1021/bi00045a039. [DOI] [PubMed] [Google Scholar]
- Peterson KM, Gopalan KV, Nandy A, Srivastava DK. Influence of Glu-376 Gln mutation on enthalpy and heat capacity changes for the binding of slightly altered ligands to medium chain acyl-CoA dehydrogenase. Protein Sci. 2001;10:1822–1834. doi: 10.1110/ps.51401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DL, Frerman FE, Kim J-JP. Three-dimensional structure of human electron transfer flavoprotein to 2. 1-Å resolution. Proc Natl Acad Sci U S A. 1996;93:14355–14360. doi: 10.1073/pnas.93.25.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada S, Kitakaze M, Asanuma H, Harada K, Ogita H, Node K, Takashima S, Sakata Y, Asakura M, Shinozaki Y, Mori H, Kuzuya T, Hori M. Role of mitochondrial and sarcolemmal KATP channels in ischemic preconditioning of the canine heart. Am J Physiol Heart Circ Physiol. 2001;280:H256–263. doi: 10.1152/ajpheart.2001.280.1.H256. [DOI] [PubMed] [Google Scholar]
- Sato T, Marbán E. The role of mitochondrial KATP channels in cardioprotection. Basic Res Cardiol. 2000;95:285–289. doi: 10.1007/s003950070047. [DOI] [PubMed] [Google Scholar]
- Schulz R, Cohen MV, Behrends M, Downey JM, Heusch G. Signal transduction of ischemic preconditioning. Cardiovasc Res. 2001;52:181–198. doi: 10.1016/s0008-6363(01)00384-4. [DOI] [PubMed] [Google Scholar]
- Standen NB. Cardioprotection by preconditioning: KATP channels, metabolism, or both? J Physiol. 2002;542:666. doi: 10.1113/jphysiol.2002.025601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe C, Kim J-JP. Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 1995;9:718–725. doi: 10.1096/fasebj.9.9.7601336. [DOI] [PubMed] [Google Scholar]
- Toyoda Y, Friehs I, Parker RA, Levitsky S, McCully JD. Differential role of sarcolemmal and mitochondrial KATP channels in adenosine-enhanced ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2000;279:H2694–2703. doi: 10.1152/ajpheart.2000.279.6.H2694. [DOI] [PubMed] [Google Scholar]
- Waterson RM, Hill RL. Enoyl coenzyme A hydratase (crotonase). Catalytic properties of crotonase and its possible regulatory role in fatty acid oxidation. J Biol Chem. 1972;247:5258–5265. [PubMed] [Google Scholar]