

Abstract
The human tandem P domain K+ channel hTREK-1 (KCNK2) is distributed widely through the CNS. Here, whole-cell patch clamp recordings were employed to investigate the effects of hypoxia on hTREK-1 channels stably expressed in human embryonic kidney cells. Acute hypoxia caused a rapid and reversible inhibition of whole-cell K+ current amplitudes; this was PO2 dependent with a maximal inhibition achieved at 60 mmHg and below. In accordance with previous studies, hTREK-1 current amplitudes were enhanced by arachidonic acid. This effect was concentration dependent, with maximal enhancement observed at a concentration of 10 μm. Membrane deformation by the crenator trinitrophenol (to mimic cell swelling) or the cup former chlorpromazine (to mimic cell shrinkage) caused robust activation and inhibition of currents, respectively. However, current augmentation by either arachidonic acid or trinitrophenol was completely prevented during hypoxia; conversely, hypoxia blunted the inhibitory action of chlorpromazine. The abilities of arachidonic acid to augment currents and of hypoxia to completely abrogate this effect were also observed in cell-attached patches. Our data indicate that hypoxia interacts with hTREK-1, and occludes its modulation by arachidonic acid and membrane deformation. These findings also suggest that the potential neuroprotective role of TREK channels, which has recently been proposed, requires reconsideration since hTREK-1 activation is unlikely when ambient PO2 is below 60 mmHg – a situation which normally pertains in the CNS even during systemic normoxia.
The most recently discovered superfamily of K+ channels, characterised structurally by the presence of two pore-forming P domains and four transmembrane helices, is emerging as one of the most widely distributed of all known ion channel families. Termed tandem P domain K+ (K2P) channels, they form functional channels as dimers and are almost exclusively voltage insensitive (Goldstein et al. 2001; Patel & Honore, 2001b). Thus, being active at a cell's resting membrane potential, they often give rise to ‘leak’ currents, thereby strongly influencing input resistance and cellular excitability.
Recent studies have indicated that certain members of the K2P channel family can be rapidly and reversibly modulated by fluctuations in the O2 content of the local environment. For example, inhibition by hypoxia of the acid-sensitive family of K2P channels (TASK channels) appears to contribute to hypoxic depolarisation of chemosensory tissues such as the carotid body (Buckler et al. 2000) and lung neuroepithelial bodies (Hartness et al. 2001). hTASK-1 channels also display O2 sensitivity when expressed in a mammalian recombinant expression system (Lewis et al. 2001). Such responses are physiologically important, since they initiate cardiorespiratory reflexes required to optimise O2 uptake and delivery around the body (Peers & Kemp, 2001). More recently, we have demonstrated that O2-sensitive K2P channels are also present in central neurones: cerebellar granule neurones, which appear to possess at least seven K2P channel types (Talley et al. 2001), are rapidly and reversibly depolarised during hypoxia due to the specific inhibition of TASK-1 (Plant et al. 2002).
Amongst the K2P channel superfamily is one family which is uniquely regulated by multiple factors, including polyunsaturated fatty acids, such as arachidonic acid (AA), membrane distortion, heat, pH and general anaesthetics (Patel et al. 1998, 1999; Maingret et al. 1999, 2000). Such channels belong to the TREK family of K2P channels which include TREK-1, TREK-2 and TRAAK (Fink et al. 1996; Bang et al. 2000; Patel et al. 2001). In the present study, we investigate the O2 sensitivity of a recently cloned human orthologue of TREK, hTREK-1 (Meadows et al. 2000), stably expressed in human embryonic kidney (HEK 293) cells. hTREK-1 as used here is structurally similar to the rodent orthologues, but possesses an extended C-terminal tail (Meadows et al. 2000) compared with the original cloning of murine TREK-1 (Fink et al. 1996). Importantly, we report the influence of hypoxia on the responses of this channel to known modulators. Our results indicate that regulation of the activity of hTREK-1 is strongly dependent on local O2 levels, an observation which is particularly pertinent to such channels when they are expressed in the CNS, where normal PO2 values have been reported to be as low as 20 mmHg (Hoffman et al. 2000).
Methods
Cell culture
The full length hTREK-1 (KCNK2) was cloned and stably expressed in human embryonic kidney (HEK 293) cells as previously described (Meadows et al. 2000), using a vector (pcDNA3.1/V5-His-TOPO) containing a His6 and V5 epitope for immunocytochemical verification of protein expression (data not shown). Cells were maintained in Earle's minimal essential medium (containing l-glutamine) supplemented with 10 % fetal calf serum, 1 % antibiotic-antimycotic, 1 % non-essential amino acids, 0.2 % gentamicin and 0.1 % Geneticin G-418 sulphate (all purchased from Gibco BRL, Paisley, Strathclyde, UK) in a humidified incubator gassed with 5 % CO2–95 % air. Cells were passaged every 7 days in a ratio of 1:25 using Ca2+- and Mg2+-free phosphate-buffered saline (Gibco BRL).
Electrophysiology
Cells were grown for 24 h on glass coverslips before being transferred to a continuously perfused (5 ml min−1) recording chamber (volume ca 200 μl) mounted on the stage of an inverted microscope. For whole-cell patch clamp recordings, the standard perfusate was composed of (mm): 135 NaCl, 5 KCl, 1.2 MgCl2, 5 Hepes, 2.5 CaCl2, 10 d-glucose, 30 sucrose (pH 7.4). Whole-cell K+ currents were recorded at room temperature (21 ± 1 °C) using a pipette solution composed of (mm): 10 NaCl, 117 KCl, 2 MgCl2, 11 Hepes, 11 EGTA, 1 CaCl2 and 2 Na2 ATP (pH 7.2). When filled with this solution, pipettes had resistance of 5–7 MΩ. Hypoxic solutions were bubbled with N2(g) for at least 30 min prior to perfusion of cells, which produced no shift in pH. PO2 was measured (at the cell) using a polarised carbon fibre electrode (Mojet et al. 1997) and ranged between 100 and 20 mmHg, as required. Normoxic solutions were either equilibrated with room air or bubbled with medical air. Switching from either of these normoxic solutions to hypoxic solutions (± modulators) produced a shift in bath temperature of less than 1 °C. Arachidonic acid (AA; 10 nm to 100 μm), trinitrophenol (TNP; 400 μm) and chlorpromazine (CPZ; 10 μm) were applied via the perfusate. Current recordings were made using an Axopatch 200A amplifier and Digidata 1320 A/D interface (Axon Instruments, Union City, CA, USA).
To evoke whole-cell K+ currents standard ramp-step protocols were used; briefly, cells were voltage-clamped at −70 mV and were ramped from −100 mV to +60 mV over 500 ms or 1 s. Cells were then sequentially stepped from the holding potential to 0 mV and +60 mV for 200 ms each. This protocol was repeated at a frequency of 0.1 Hz.
Cell-attached recordings were also obtained using patch pipettes filled with standard extracellular solution. In these experiments, pipette potential was held at 0 mV, and 1 s voltage ramps (from +30 mV to −130 mV, which mimics the whole-cell voltage protocol in which resting membrane potential was clamped to −70 mV) were applied at a frequency of 0.1 Hz. Data were acquired as for whole-cell recordings, and exemplar traces in Fig. 4 have been inverted in accordance with convention. Data are presented as example traces together with means ±s.e.m. Time series plots were constructed from the current amplitudes measured over the last 10 ms of the +60 mV step. Statistical comparisons were made using Student's paired t test.
Figure 4. hTREK1 modulation in the cell-attached configuration.
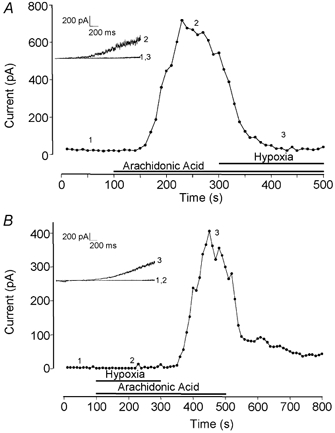
A, example time series plot of current amplitude determined using a cell-attached recording and measured at a pipette potential of −130 mV (assumed to be +60 mV if membrane potential is taken as −70 mV). Periods of exposure to 10 μm AA, and to hypoxia in the continued presence of AA, are indicated by the horizontal bars. Numbered current traces in the inset correspond to the numbered points on the time series. B, as A, except that the cell was initially exposed to both hypoxia and 10 μm AA, then to AA under normoxic conditions. Numbered current traces in the inset correspond to the numbered points on the time series.
BCECF fluorescence
Relative pHi was measured using a standard ratiometric method (see, for example, Frampton & Orchard, 1992) following incubation of cells with BCECF-AM (2 μm, Molecular Probes, Eugene, OR, USA) for 1 h. Fluorescence was measured from at least four separate cells in each of three fields from different cell preparations using a monochromator-based imaging system mounted on an inverted microscope. Excitation wavelengths were alternated between 440 and 490 nm at 0.25 Hz (200 ms exposures) and emitted light measured at 510 nm. Experiments were carried out on cells perfused as described in the electrophysiology section. Since no significant changes were observed during the experimental manoeuvres described herein, the fluorescence ratio was not calibrated, and is expressed in ratio units.
Results
Arachidonic acid (AA; 10 μm) caused a rapid and reversible enhancement of whole-cell K+ currents recorded from HEK 293 cells stably expressing hTREK-1 (Fig. 1A); this is a characteristic response of both human and rodent TREK-1 channels (Patel et al. 1998, 2001; Meadows et al. 2000). In this series of experiments, K+ current density at +60 mV was significantly enhanced by more than twofold from 92.5 ± 7.7 to 198.5 ± 27.6 pA pF−1 by AA application (n = 6 cells, P < 0.01). Another defining characteristic of murine TREK channels is that they can be modulated by membrane distortion (Patel et al. 1998). To investigate this possibility in the human orthologue, we employed the membrane deformers TNP and CPZ. TNP, a chemical crenator, was able to evoke significant hTREK-1 activation from 165.2 ± 31.2 to 211.9 ± 35.6 pA pF−1 (n = 5, P < 0.05; Fig. 1B) whilst CPZ, a chemical cup former, caused robust channel suppression from 283.7 ± 83.3 to 32.4 ± 7.4 pA pF−1 (n = 5, P < 0.05; Fig. 1C). These observations are entirely compatible with reciprocal regulation by membrane stretch and shrinkage. Figure 1D (upper traces) demonstrates for the first time in TREK channels of any species that hTREK-1 is sensitive to acute changes in PO2. Reduction of PO2 to ∼20 mmHg resulted in rapid and reversible suppression of whole-cell K+ currents by ca 40 % from 110.5 ± 13.9 to 68.0 ± 10.8 pA pF−1 (n = 10, P < 0.001). In contrast, untransfected HEK 293 cells (Fig. 1D, lower traces) exhibited very low K+ channel activity and no significant reduction in current when they were exposed to the hypoxic perfusate. During normoxia, whole-cell K+ current density was 9.37 ± 4.48 pA pF−1 compared with 9.40 ± 4.48 pA pF−1 during hypoxia (n = 6). These data represent the first direct evidence that hTREK-1 is an O2-sensitive channel. Furthermore, this suppression of channel activity by reduced O2 availability was graded and maximal at PO2 values at, or below, ∼60 mmHg (n = 25 paired experimental protocols where PO2 was reduced from 150 mmHg to the values indicated in Fig. 1E).
Figure 1. Recombinant hTREK-1 is O2 sensitive when expressed in HEK 293 cells.
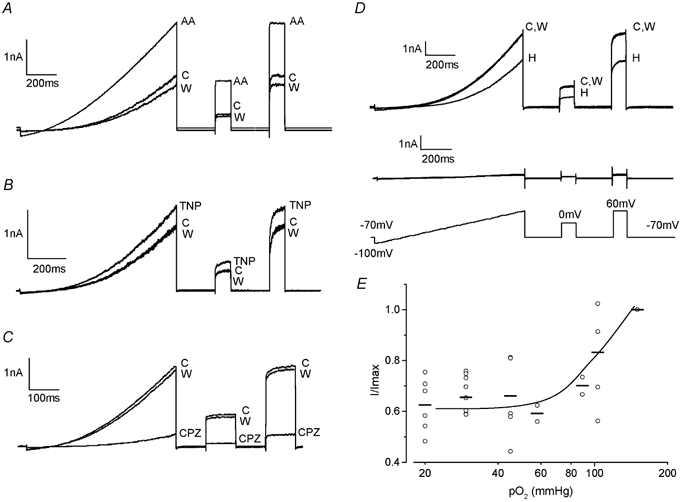
A, exemplar whole-cell currents evoked by the ramp-step protocol shown in the lower panel of D during perfusion of normoxic control solution (C), in the presence of 10 μm arachidonic acid (AA), and during washout (W) as indicated. B, as A, except that the cell was exposed to perfusate containing TNP (400 μm) rather than AA. C, as A except that the cell was exposed to perfusate containing CPZ (10 μm) rather than AA. D, exemplar whole-cell currents evoked by the ramp-step protocol shown before (C), during (H) and after (W) exposure to hypoxic solution, in hTREK-1 transfected HEK 293 cells (upper traces) and wild-type HEK 293 cells (lower traces). Shown below these traces is the ramp-step protocol used to evoke all currents illustrated. E, plot of normalised current amplitudes (I/Imax) versus chamber PO2 levels (on a log10 scale). At each PO2 level examined, the fractional inhibition (observed during a hypoxic challenge from 150 mmHg to the PO2 level shown) is plotted for individual cells (○), and the mean effect is indicated by the horizontal bars. The line was drawn by eye.
Figure 2A shows an example time-series plot obtained from measuring current amplitude at +60 mV evoked by the step depolarisations of the voltage protocol shown in Fig. 1D. Exposure of the cell to hypoxia caused a rapid and rapidly reversible inhibition of current amplitude. Following restoration of normoxia, 10 μm AA evoked a dramatic enhancement of K+ current amplitude, as previously reported in Fig. 1A. However, during a second exposure to hypoxia, co-application of AA failed to cause activation, and current amplitude returned to that seen during hypoxia alone. Indeed, AA was only able to activate K+ currents when normoxia was restored. Mean, normalised data showing the effects of hypoxia, AA and both modulators together are plotted in Fig. 2B. In the presence of 10 μm AA, exposure to hypoxia reduced K+ current density from 83.6 ± 5.9 to 56.7 ± 9.3 pA pF−1 (n = 7, P < 0.01), a value not significantly different from that seen under hypoxic conditions in the absence of AA (Fig. 1D). When AA was pre-bubbled with N2(g) for more than 30 min before being re-equilibrated with air for at least 10 min, it was still able to elicit activation, showing that AA potency as a hTREK-1 modulator was not affected by the bubbling protocol (data not shown and compatible with our previous report on a different cell line (Hartness et al. 2001)). The activation of whole-cell K+ currents by AA was concentration dependent, with the effects being maximal at 10 μm (Fig. 2C). Mean EC50 value was 0.99 ± 0.73 μm with a Hill coefficient of 1.02 ± 0.91 (d.f. = 26). Most importantly, the activation by AA was completely abolished by hypoxia at all concentrations (Fig. 2C). In agreement with previous studies (Meadows et al. 2000), activation of hTREK-1 by AA was voltage dependent, declining with increasing depolarisation whilst co-application of hypoxia removed this voltage dependence (data not shown).
Figure 2. Hypoxia occludes arachidonic acid activation of hTREK-1.
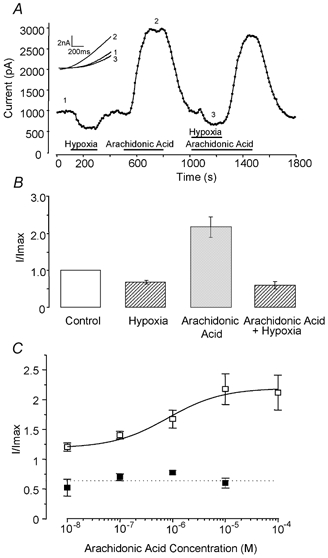
A, typical time-series plot of current amplitude measured at +60 mV showing the effects of hypoxia, and AA (10 μm) in the presence and absence of hypoxia (∼ 20 mmHg). Periods of exposure to AA and hypoxic perfusate are indicated by the horizontal bars. Numbered current traces in the inset correspond to the numbered points on the time series. B, mean, normalised current amplitudes recorded at +60 mV in control (normoxic) solutions and in the conditions indicated beneath each bar. C, concentration-response curves for AA-evoked, normalised whole-cell currents under normoxic (□) and hypoxic (▪) conditions. Each point plots mean, normalised current enhancement (with vertical s.e.m. bars, taken from 5–7 cells) due to exposure to varying AA concentrations. The dotted line indicates the mean normalised current amplitude evoked by hypoxia alone (taken from B).
Activation of hTREK-1 by application of TNP was, like the actions of AA, also abolished during perfusion with hypoxic solution. Figure 3A plots K+ current amplitudes from an example cell and illustrates this rapid enhancement of current amplitude during application of TNP. Subsequent exposure to hypoxia in the continued presence of TNP fully reversed this augmentation and brought current levels down to below those seen in normoxia (from 211.9 ± 35.6 to 140.2 ± 25.6 pA pF−1; n = 5, P < 0.05). The effect of a supramaximal concentration of TNP (4 mm) evoked current enhancement which was no greater than that observed using 400 μm TNP and hypoxia fully reversed this augmentation (n = 4; data not shown). Mean, normalised data illustrating the effects of 400 μm TNP, hypoxia and the combined effect of these two modulators are given in Fig. 3B. Conversely, application of the cup-former CPZ elicited marked and rapid current suppression, an effect which was partially and significantly blunted by co-application with hypoxic perfusate (from 32.4 ± 7.4 to 67.4 ± 10.7 pA pF−1; n = 5, P < 0.01; Fig. 3C and D).
Figure 3. Hypoxia occludes modulation of hTREK-1 by chlorpromazine and trinitrophenol.
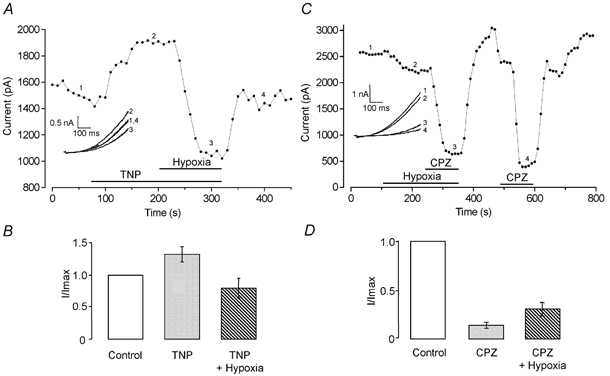
A, typical time series plot of current amplitude measured at +60 mV showing the effects of TNP in normoxia, and the complete reversal of its effects by hypoxia. Periods of exposure to TNP and hypoxic perfusate indicated by the horizontal bars. Numbered current traces in the inset correspond to the numbered points on the time series. B, mean, normalised current amplitudes recorded at +60 mV in control (normoxic) solutions and in the conditions indicated beneath each bar. C, typical time series plot of current amplitude measured at +60 mV showing the effects of hypoxia and of CPZ in the presence and absence of hypoxia. Periods of exposure to CPZ and hypoxic perfusate indicated by the horizontal bars. Numbered current traces in the inset correspond to the numbered points on the time series. D, mean, normalised current amplitudes recorded at +60 mV in control (normoxic) solutions and in the conditions indicated beneath each bar.
The marked augmentation of whole-cell K+ currents by AA was also observed in cell-attached patches (Fig. 4). During these recordings, currents observed under normoxic conditions were negligible in amplitude (34.7 ± 13.1 pA at a pipette potential of +60 mV; n = 4 patches), but application of AA (10 μm) to the perfusate caused a rapid and dramatic augmentation of currents to 277.9 ± 109.9 pA (n = 4, Fig. 4A). As was observed during whole-cell recordings, exposure to hypoxia in the continued presence of AA caused a complete reversal of this augmentation (mean amplitude 30.6 ± 8.5 pA). Importantly, when cells were exposed to AA in the presence of hypoxia (at a PO2 value similar to that reported for human CNS (Hoffman et al. 2000)), no significant augmentation in current was seen (16.7 ± 7.9 vs. 14.0 ± 9.1 pA; n = 4; Fig. 4B). However, when normoxia was restored in the continued presence of AA, current activation was again apparent (to 190.7 ± 54.5 pA; Fig. 4B). In parallel studies under identical recording conditions, we also monitored intracellular pH with the fluoroprobe BCECF. During exposure to hypoxia, BCECF fluorescence increased slightly from 0.716 ± 0.034 to 0.736 ± 0.034 ratio units (n = 3), a rate of change which was indistinguishable from the difference between pre- and post-normoxic periods (data not shown), showing that pHi is not acutely altered during hypoxic challenge.
Collectively, these data indicate that recombinant hTREK-1 is O2 sensitive and, importantly, that hypoxia occludes the effects of potential physiological modulators.
Discussion
The present study demonstrates that the human homologue of TREK-1 (hTREK-1), when stably expressed in HEK 293 cells, is an O2-sensitive K+ channel; this is the first observation of O2 sensitivity of any TREK, or TREK-related K2P channel (such as TRAAK and TREK-2). Thus, hTREK-1 can be considered the latest addition to the increasing list of ion channels which display acute O2 sensitivity (Peers, 1997; Patel & Honore, 2001a; Lopez-Barneo et al. 2001). Like rodent TREK channels, the activity of the human orthologue is modulated by AA and chemical distortion of the membrane (Figs 1–3). Chemical distortion was achieved using the crenator TNP to mimic cell swelling and the cup-former CPZ to mimic cell shrinkage. We found application of these agents to be a reliable and repeatable method by which to mimic mechanical stretch. Use of these agents also allowed direct and favourable comparisions with whole-cell data obtained from the murine orthologue (Patel et al. 1998).
As demonstrated for other K+ channels, hypoxic inhibition of hTREK-1 was rapid and freely reversible (e.g. Fig. 2A), with the rate of hypoxic inhibition probably being limited by the time course of O2 removal from the recording chamber when perfusate was switched to one equilibrated with N2; the degree of bubbling necessary to obtain the level of hypoxia employed in this study never produced more than 1 °C change in bath temperature, regardless of whether the normoxic solution was air equilibrated or bubbled with medical air. Since the hTREK-1 channel employed here was originally found to be located primarily within the CNS (Meadows et al. 2000), with some limited peripheral distribution in cardiac (Tan et al. 2002) and arterial smooth muscle (Koh et al. 2001), our results have important implications for neuronal excitability, where normal human brain PO2 is ∼20 mmHg (Hoffman et al. 2000).
At presently, the molecular mechanisms underlying O2 sensing by any ion channel remain to be fully determined, and various mechanisms have been suggested (Lopez-Barneo et al. 2001; Peers & Kemp, 2001; Patel & Honore, 2001a). Similarly, the structural requirements for O2 sensing by specific channels are largely (although not entirely (Fearon et al. 2000)) unknown. The present study demonstrates that activation of hTREK-1 by AA and by TNP is completely inhibited during hypoxia and this inhibition completely removes any voltage dependence of activation (data not shown). Interestingly, hypoxia is not as efficient at preventing channel inhibition by CPZ, although the inhibitory effect is significantly blunted by low PO2 (Fig. 3). This suggests that the mode of channel inhibition by hypoxia may not be as straightforward as regulation by other modulators and implies that there may be multiple interactions at different sites in the channel. Indeed, we could hypothesise that the extra 41 amino acids in the C-terminal of our human homologue may be intimately involved in hypoxic interactions. However, this suggestion is confounded by the fact that, although the original mTREK-1 sequence does indeed lack these additional residues (Fink et al. 1996), a later study has demonstrated that mTREK-1 is of an identical length to hTREK-1 (Meadows et al. 2000) but which of these sequences was employed in later mTREK-1 characterisation papers is not clear (e.g. Honore et al. 2002). Another possibility might be that hypoxia interferes with the crucial role of residue E306 in responding to channel activators (Honore et al. 2002). However, since we found that hypoxia was able to reverse the augmentation of channel activity caused by AA in cell-attached recordings (Fig. 3A and B), and also detected no change of pHi caused by hypoxia under these conditions (measured using BCECF), it seems unlikely that altered channel protonation can account for the inhibitory actions of hypoxia on hTREK-1.
It is noteworthy that channel activity under normoxic conditions in the absence of AA was minimal in cell-attached recordings (see insets to Fig. 4), yet whole-cell recordings revealed robust basal currents. This suggests that disruption of the intracellular milieu (presumably by disruption of the cytoskeleton as the whole-cell configuration is acheived) may cause the degree of channel activation required before inhibition by hypoxia can be observed. This suggestion is further supported by the fact that, under whole-cell conditions, AA caused only an approximate 2.5-fold augmentation of currents (Fig. 2), whilst in cell-attached recordings, current augmentation was much greater (Fig. 4). This is the first report of the activity of the human orthologue of TREK-1 in cell-attached patches, which more closely mimic the physiological condition, and our data suggest that rather than merely acting as a background channel, hTREK-1 may be more intimately involved in regulated control of human neuronal excitability.
Our findings that hypoxia prevents channel activation by AA or membrane distortion have important implications for channel function and impact strongly on the proposed role of this channel in central neurones. It has been suggested that TREK-1 may have a pathophysiological role during ischaemic episodes (Honore et al. 2002) based on three lines of evidence: (a) the neuroprotective agent riluzole opens TREK-1 (Duprat et al. 2000); (b) ischaemic episodes evoke changes in a number of central neuronal microenvironmental parameters including an increase in AA concentration; and (c) AA is neuroprotective (Lauritzen et al. 2000). In this proposal, activation of TREK channels during ischaemia would lead to neuronal hyperpolarisation which would decrease glutamate release thereby reducing the likelihood of neuronal damage due to excitotoxicity (Honore et al. 2002). Although this is an attractive explanation for the neuroprotective mechanism of agents such as riluzole and AA, our findings cast doubt on such a proposed scheme for the following reasons: (a) in the CNS, PO2 is normally as low as 20 mmHg (Hoffman et al. 2000); and (b) at this PO2 level, and even at levels as high as 60 mmHg, activation by AA (and TNP) is not possible. This new information suggests strongly that the proposed neuroprotective role provided by TREK activation (Honore et al. 2002) requires reconsideration.
Acknowledgments
This work was supported by The Wellcome Trust and The British Heart Foundation. We also thank Ian Smith and John Boyle for their help with monitoring intracellular pH.
References
- Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem. 2000;275:17412–17419. doi: 10.1074/jbc.M000445200. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Patel AJ, Fink M, Romey G, Lazdunski M. The neuroprotective agent riluzole activates the two P domain K+ channels TREK-1 and TRAAK. Mol Pharmacol. 2000;57:906–912. [PubMed] [Google Scholar]
- Fearon IM, Varadi G, Koch S, Isaacsohn I, Ball SG, Peers C. Splice variants reveal the region involved in oxygen sensing by recombinant human L-type Ca2+ channels. Circ Res. 2000;87:537–539. doi: 10.1161/01.res.87.7.537. [DOI] [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 1996;15:6854–6862. [PMC free article] [PubMed] [Google Scholar]
- Frampton JE, Orchard CH. The effect of a calmodulin inhibitor on intracellular [Ca2+] and contraction in isolated rat ventricular myocytes. J Physiol. 1992;453:385–400. doi: 10.1113/jphysiol.1992.sp019234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- Hartness ME, Lewis A, Searle GJ, O'Kelly I, Peers C, Kemp PJ. Combined antisense and pharmacological approaches implicate hTASK as an airway O2 sensing K+ channel. J Biol Chem. 2001;276:26499–26508. doi: 10.1074/jbc.M010357200. [DOI] [PubMed] [Google Scholar]
- Hoffman WE, Wheeler P, Edelman G, Charbel FT, Torres NJ, Ausman JI. Hypoxic brain tissue following subarachnoid hemorrhage. Anesthesiology. 2000;92:442–446. doi: 10.1097/00000542-200002000-00026. [DOI] [PubMed] [Google Scholar]
- Honore E, Maingret F, Lazdunski M, Patel AJ. An intracellular proton sensor commands lipid- and mechano-gating of the K+ channel TREK-1. EMBO J. 2002;21:2968–2976. doi: 10.1093/emboj/cdf288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SD, Monaghan K, Sergeant GP, Ro S, Walker RL, Sanders KM, Horowitz B. TREK-1 regulation by nitric oxide and cGMP-dependent protein kinase. An essential role in smooth muscle inhibitory neurotransmission. J Biol Chem. 2001;276:44338–44346. doi: 10.1074/jbc.M108125200. [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Blondeau N, Heurteaux C, Widmann C, Romey G, Lazdunski M. Polyunsaturated fatty acids are potent neuroprotectors. EMBO J. 2000;19:1784–1793. doi: 10.1093/emboj/19.8.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A, Hartness ME, Chapman CG, Fearon IM, Meadows HJ, Peers C, Kemp PJ. Recombinant hTASK1 is an O2-sensitive K+ channel. Biochem Biophys Res Comm. 2001;285:1290–1294. doi: 10.1006/bbrc.2001.5310. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honore E. TREK-1 is a heat-activated background K(+) channel. EMBO J. 2000;19:2483–2491. doi: 10.1093/emboj/19.11.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honore E. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem. 1999;274:26691–26696. doi: 10.1074/jbc.274.38.26691. [DOI] [PubMed] [Google Scholar]
- Meadows HJ, Benham CD, Cairns W, Gloger I, Jennings C, Medhurst AD, Murdock P, Chapman CG. Cloning, localisation and functional expression of the human orthologue of the TREK-1 potassium channel. Pflugers Arch. 2000;439:714–722. doi: 10.1007/s004249900235. [DOI] [PubMed] [Google Scholar]
- Mojet MH, Mills E, Duchen MR. Hypoxia-induced catecholamine secretion in isolated newborn rat adrenal chromaffin cells is mimicked by inhibition of mitochondrial respiration. J Physiol. 1997;504:175–189. doi: 10.1111/j.1469-7793.1997.175bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Honore E. Molecular physiology of oxygen-sensitive potassium channels. Eur Respir J. 2001a;18:221–227. doi: 10.1183/09031936.01.00204001. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001b;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci. 1999;524:422–426. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998;17:4283–4290. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honore E. Lipid and mechano-gated 2P domain K+ channels. Curr Opin Cell Biol. 2001;13:422–428. doi: 10.1016/s0955-0674(00)00231-3. [DOI] [PubMed] [Google Scholar]
- Peers C. Oxygen-sensitive ion channels. Trends Pharmacol Sci. 1997;18:405–408. doi: 10.1016/s0165-6147(97)01120-6. [DOI] [PubMed] [Google Scholar]
- Peers C, Kemp PJ. Acute oxygen sensing: diverse but convergent mechanisms in airway and arterial chemoreceptors. Respir Res. 2001;2:145–149. doi: 10.1186/rr51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Kemp PJ, Peers C, Henderson Z, Pearson HA. Hypoxic depolarization of central neurones by specific inhibition of TASK-1. Stroke. 2002;33:2324–2328. doi: 10.1161/01.str.0000027440.68031.b0. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci. 2001;21:7491–7505. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JH, Liu W, Saint DA. TREK-like potassium channels in rat cardiac ventricular myocytes are activated by intracellular ATP. J Memb Biol. 2002;185:201–207. doi: 10.1007/s00232-001-0123-0. [DOI] [PubMed] [Google Scholar]