

Abstract
Peripheral pain thresholds are regulated by the actions of inflammatory mediators. Some act through G-protein-coupled receptors on voltage-gated sodium channels. We have found that a low-threshold, persistent tetrodotoxin-resistant Na+ current, attributed to NaV1.9, is upregulated by GTP and its non-hydrolysable analogue GTP-γ-S, but not by GDP. Inclusion of GTP-γ-S (500 μm) in the internal solution led to an increase in maximal current amplitude of > 300 % within 5 min. In current clamp, upregulation of persistent current was associated with a more negative threshold for action potential induction (by 15–16 mV) assessed from a holding potential of −90 mV. This was not seen in neurones without the low-threshold current or with internal GDP (P < 0.001). In addition, persistent current upregulation depolarized neurones. At −60 mV, internal GTP-γ-S led to the generation of spontaneous activity in initially silent neurones only when persistent current was upregulated. These findings suggest that regulation of the persistent current has important consequences for nociceptor excitability.
Small (≤ 25 μm in apparent diameter) sensory neurones in dorsal root ganglia (DRG) include the cell bodies of nociceptive C-fibres (e.g. Baker & Wood, 2001). Many inflammatory mediators (for example PGE2, ATP and 5-HT) are known to increase neuronal excitability through G-protein-coupled receptors (England et al. 1996; Gold et al. 1996; Irnich et al. 2001), and can induce sensitization to non-noxious stimuli (e.g. Taiwo & Levine, 1992). The G-protein-mediated effects on the current generated by the tetrodotoxin-resistant (TTX-r) Na+ channel NaV1.8 involve a negative shift in activation voltage dependence through channel phosphorylation, promoting repetitive firing (England et al. 1996). Physiological experiments (Brock et al. 1998; Michaelis et al. 1998; Strassman & Raymond, 1999) and the sensory deficits of NaV1.8 null mutant mice (Akopian et al. 1999b) have demonstrated the importance of this TTX-r Na+ channel in nociception and pain pathways. Another TTX-r Na+ current present in small diameter sensory neurones, and attributed to NaV1.9, exhibits ultra-slow kinetics and an activation threshold around −65 mV (Cummins et al. 1999), which is substantially more negative than that of transient Na+ currents in the same neurones. This current can be recorded in the presence of TTX in NaV1.8 null mutant neurones, although it is also present in wild-type, and because of its slow and partial inactivation we refer to it as persistent current. While its kinetics have been thought to be too slow to contribute to action potential generation (Herzog et al. 2001), the persistent current has been suggested to contribute to setting a resting membrane potential that is much more positive than the K+ ion equilibrium potential (Herzog et al. 2001).
In vitro studies have indicated that many small diameter sensory neurones have a resting potential near −60 mV (e.g. Wang et al. 1994). Transmission in small diameter axons may lead to activity-related reductions in conduction velocity, which can be explained by hyperpolarization, brought about by electrogenic Na+ pumping (Serra et al. 1999). In larger demyelinated axons, Na+ pumping can generate sufficient hyperpolarization to cause impulse conduction failure (Bostock & Grafe, 1985). The membrane potential in small diameter axons may therefore become more negative in an activity-dependent manner, increasing the contribution of persistent current to excitability.
Because other ion channels, including Na+ channels, are regulated by G-protein signalling pathways, we tested the hypothesis that activation of G-proteins by intracellular GTP or GTP-γ-S might also regulate NaV1.9. We found that including GTP or its non-hydrolysable analogue GTP-γ-S in the intracellular media led to substantial upregulation of the current over a few minutes, an effect not obtained with GDP. Upregulation of the current resulted in an increase in membrane excitability, and spontaneous action potential generation. Some of these observations have been reported previously in abstract form (Baker et al. 2002).
METHODS
Tissue culture
DRG cultures were prepared from adult wild-type and NaV1.8 null mutant mice, and from 3-week-old Sprague-Dawley rats. The animals were killed by cervical dislocation, in accordance with the UK Animals (Scientific Procedures) Act 1986. Primary sensory neurones were isolated using enzymatic dissociation of pooled DRGs (dispase/collagenase; Sigma, Poole, Dorset, UK) and maintained in culture for 1–2 days, as described elsewhere (Baker & Bostock, 1997). Results obtained from wild-type neurones isolated from rat were indistinguishable from those from neurones isolated from mouse. The NaV1.8 null mutant was used in order to isolate the low-threshold Na+ current in voltage-clamp recordings in the presence of external TTX.
Electrophysiology and solutions
Conventional whole-cell patch-clamp recordings in both voltage-clamp and current-clamp modes were made from neurones of up to 25 μm apparent diameter, using an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA) driven from a PC generating pulse protocols (pCLAMP 6, Axon Instruments). Data were stored on a PC for off-line analysis. In voltage clamp, TTX-r Na+ currents were recorded in relative isolation by including pharmacological blockers of both K+ and Ca2+ currents in the media, and by including 250 nm TTX in the extracellular solution. For voltage-clamp recordings, the external solution contained (mm): NaCl 43.3, tetraethylammonium chloride 96.7, Hepes 10, CaCl2 2.1, MgCl2 2.12, 4-aminopyridine 0.5, CsCl 10, KCl 7.5, CdCl2 0.02, TTX 0.25 × 10−3. The internal solution contained (mm): CsCl 145, EGTA(Na) 3, tetraethylammonium chloride 10, Hepes 10, CaCl2 1.21, ATP(Mg) 3. In addition, the internal solution contained 500 μm GTP (Li), GTP-γ-S (Li) or GDP (Na). Control solutions contained 500 μm GDP (Na) plus 2.5 mm LiCl, or 2.5 mm LiCl only. The external and internal solutions were both buffered to pH 7.2–7.3 with the addition of CsOH. A single micromolar range IC50 was expected for Cd2+ block of high-threshold, voltage-activated (HVA) Ca2+ currents (Shafer, 1998) and a concentration of 20 μm was expected to give a substantial block (e.g. 90 %). Residual HVA Ca2+ currents were sometimes observed, although they ran down on a similar time scale to the upregulation of the low-threshold persistent current. Low-threshold Ca2+ currents were not seen even before Na+ current upregulation, and have been reported to be either absent from small diameter DRG neurones, or small (Scroggs & Fox, 1992; Cardenas et al. 1999). In current-clamp recordings (input resistance ≥ 60 MΩ, associated with a passive membrane time constant of 1 ms or longer), quasi-physiological solutions were used without blockers. The solutions contained (mm): external: NaCl 140, Hepes 10, CaCl2 2.1, MgCl2 2.12, KCl 2.5; and internal: KCl 143, EGTA (Na) 3, Hepes 10, CaCl2 1.21, MgCl2 1.21, ATP(Mg) 3. The internal solution also contained GTP, GTP-γ-S or GDP as above. The pH of the external and internal solutions was adjusted to 7.2–7.3 with the addition of NaOH. All chemicals were Analar grade (>> 99 % purity) (BDH, Lutterworth, Leicestershire, UK) or obtained from Sigma. TTX was obtained from Alomone Labs (TCS Biologicals, Botolph Claydon, Buckinghamshire, UK) and Sigma.
Electrodes were made from thin-walled glass (Harvard Apparatus, Edenbridge, Kent, UK), and had a resistance of between 2 and 3 MΩ when initially filled with recording solution. In both voltage-clamp and current-clamp experiments, series-resistance compensation was set between 70 and 75 %, with a nominal feedback lag of 12 μs. In voltage-clamp recordings, the holding potential was −110 mV, and incrementing clamp steps were preceded by a 20 ms duration pre-pulse to −130 mV. Current records were the average of the responses to three consecutive families of clamp steps. Leak subtraction was achieved on-line, using a P/N protocol, where five reverse polarity clamp steps were averaged to generate the leakage record. For measurement of input conductance, the change in holding current was measured when the membrane potential was stepped to −120 mV from −110 mV, i.e. a potential range over which the low-threshold persistent current is not expected to operate. In current clamp, steady polarizing currents were applied to set holding potentials, and were adjusted periodically. For continuous recording of membrane potential, the value was repeatedly sampled over 5 ms every 2 s, while the holding current was set to zero. Records were filtered at 5 kHz using a 4-pole Bessel filter.
Data analysis
The Na+ currents in Fig. 1 and Fig. 3 were converted to conductance values using the equation:
where G is whole-cell peak conductance, I is the whole-cell peak current, Vm is the membrane potential and ENa is the equilibrium potential for sodium, taken as +45 mV. The Boltzmann equation used to describe the voltage dependence of activation was of the form:
where Gmax is the maximal conductance, V1/2 is the potential at half-maximal activation, and ag is the slope factor in millivolts.
Figure 1. GTP increases the amplitude of persistent Na+ current in NaV1.8 null mutant neurones.
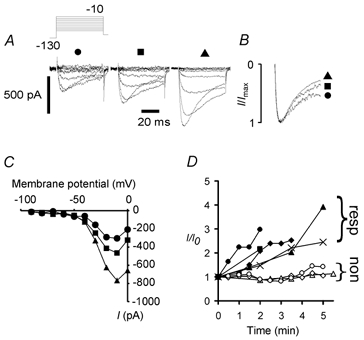
A, with 500 μm GTP in the internal solution the amplitude of low-threshold current increased dramatically over 7 min by more than 100 % (from left: 0, 2, 7 min). The voltage-clamp protocol is shown in the inset (mV). B, normalized peak currents at 0, 2 and 7 min (•, ▪, ▴, respectively) indicating no major changes in current activation kinetics and a slight increase in the amount of inactivation with time. C, the voltage dependence of current activation was essentially unchanged despite the large increase in current amplitude from 0 to 7 min. D, 5 of 8 neurones exhibited substantial upregulation over the first 5 min of voltage clamp. The 5 neurones responding (resp) are represented by •, ▪, ♦, ▴ and ×; the 3 non-responding (non) neurones are represented by open symbols.
Figure 3. Upregulation of persistent current in a NaV1.8 null mutant neurone following exposure to 500 μm GTP-γ-S.
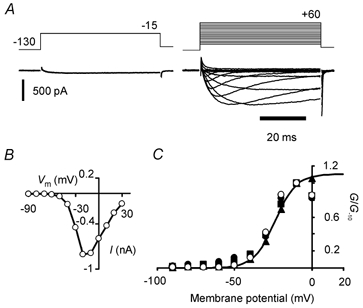
A: left, maximal inward current recorded at −15 mV as soon as possible following membrane rupture. Right, persistent current family evoked by incrementing voltage-clamp steps 12 min later. B, peak current (I) versus membrane potential (Vm) relation for the GTP-γ-S-upregulated current in A. Current activated at potentials more negative than −50 mV. C, normalized peak conductance versus membrane potential plot for the same neurone as in A and B (○). Also shown are normalized peak conductance versus membrane potential plots for the neurone in Fig. 1A (symbols for 0, 2 and 7 min: •, ▪, and ▴, respectively). The data are normalized to conductance values at −10 mV because the increasing rate of inactivation and residual K+ currents at positive potentials begin to limit inward current amplitudes at early times. The smooth curve fitted to data at 7 min (▴) is a Boltzmann function drawn with best-fit parameters (V1/2 = −23.8 mV, ag = 6.85 mV). All conductances were calculated assuming ENa = +45 mV.
Wherever possible data are expressed as means ±s.e.m. Experiments were performed at room temperature (22–25 °C).
Measurement of threshold
In current-clamp experiments, excitability was assessed by repeat estimations of voltage threshold in neurones with a long membrane time constant (> 1 ms). The threshold was determined from the responses to a family of incrementing applied currents at the start and at the end of a continuous sequence of recording, commencing as rapidly as possible after membrane rupture and attaining the whole-cell configuration. Recordings were made every 30 s for up to 15 min. Single ascending exponentials were superimposed on subthreshold voltage responses to 10 ms duration constant currents, using a least-squares method (Fig. 4A). Where the response was a subthreshold depolarization, the electrotonic waveform produced could be well described by a single exponential. With greater depolarization the response deviated, and incorporated extra depolarization caused by the recruitment of Na+ channels. The potential at which the electrotonic waveform became both non-passive and led to action potential generation was defined as the threshold potential. This method of measuring threshold overcame the effects of a variable input resistance and produced consistent values.
Figure 4. Measurement of voltage threshold and GTP-γ-S-related fall in voltage threshold for action potential induction.

A, depolarizing currents of 200 and 300 pA applied to an example wild-type neurone gave rise to subthreshold electrotonic responses. Thick curves are exponentials drawn according to best-fit parameters derived from the initial portion of the response. An action potential was elicited by 400 pA. B, internal GTP-γ-S in a NaV1.8 null mutant neurone gave rise to a reduction in voltage threshold (tested from a holding potential of −90 mV), and prolonged applied depolarizations. The reduction in threshold took place over 13 min with intracellular GTP-γ-S. Enhanced low threshold current gave rise to profound prolongation of previously subthreshold depolarizations (arrow), consistent with a substantial increase in low-threshold current amplitude and slow current kinetics (before and after threshold reduction, left and right panels, respectively). C, both NaV1.8 null mutant and wild-type (WT) dorsal root ganglion neurones exhibited falls in threshold when persistent inward current was upregulated by internal GTP-γ-S (□, n = 3 for both NaV1.8 null and wild-type; means ±s.e.m.), but where no persistent current could be recorded, the threshold increased slightly (▪, n = 16 and 11 for Nav1.8 null and wild-type, respectively; means ±s.e.m., P < 0.0001, Student's two-tailed unpaired t test, pooled data), strongly implying that the presence of the current gave rise to the threshold fall. No such fall in voltage threshold was found in 15 neurones with internal GDP.
RESULTS
Effect of GTP and analogues on TTX-r currents
We examined the effects of GTP and analogues on TTX-r currents in voltage-clamp experiments on NaV1.8 null mutant mouse sensory neurones. TTX-sensitive currents were eliminated from the recordings by including 250 nm TTX in the bathing solution. With the K+ currents blocked, it proved possible to determine from the outset whether a neurone exhibited persistent current or not, even though the current was small when recording began. The low-threshold persistent TTX-r current could be detected easily in these cells because of the absence of the major TTX-r channel isoform NaV1.8, although wild-type mouse and rat neurones also generated the same current (cf. Fig. 5). We defined the persistent current as a TTX-r current, present at −50 mV, that activated with very slow kinetics. Because the activation threshold was some 25 mV more negative than that for NaV1.8, this current could also be examined in wild-type neurones. Inclusion of GTP in the pipette solution caused a substantial upregulation of persistent current in five out of eight neurones in which current was generated when first voltage clamped, without obvious change in the current activation voltage dependence or kinetics (Fig. 1). Not all currents underwent rapid upregulation, possibly because GTP was hydrolysed before upregulation occurred. In neurones in which persistent current was generated at the start of recording, inclusion of 500 μm GTP-γ-S (a non-hydrolysable analogue of GTP) in the intracellular solution produced a dramatic increase in the amplitude of the current of up to one order of magnitude (in recordings lasting up to 20 min) in all neurones tested (n = 7). In contrast, with GDP, current amplitude was small and stable (n = 10; Fig. 2A and B). As GTP and GTP-γ-S were added to the internal solution as Li+ salts, control voltage-clamp recordings were made over 3–10 min in 13 NaV1.8 null mutant neurones with 2.5 mm Li+ in the internal solution, and without guanosine phosphates. A persistent current was generated by four neurones, none of which exhibited the rapid upregulation found with GTP-γ-S (Fig. 2C). In addition, recordings were made from eight neurones with internal solution containing 2.5 mm Li+ plus 500 μm GDP. In three of these neurones persistent current was found, but it did not exhibit upregulation (Fig. 2D). At 3 min, six of seven neurones with internal GTP-γ-S already showed an increase in current amplitude of more than 80 %. With Li+ controls, none of the neurones (0 of 7) showed an increase at 3–3.5 min (P = 0.005, Fisher exact test), suggesting that Li+ was not responsible for the upregulation. The proportion of these control neurones generating low-threshold persistent current was 7/21 (33.3 %).
Figure 5. Persistent current upregulation alters the voltage threshold and leads to accommodation breakdown.
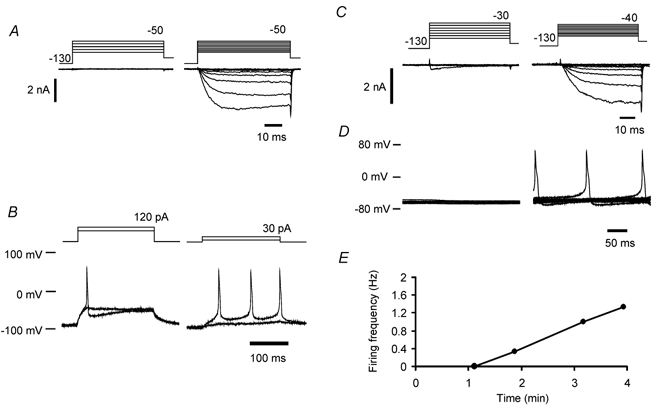
A, voltage-clamp recordings using quasi-physiological solutions from a wild-type neurone before and after persistent current upregulation (left and right panels, respectively). Persistent current began to activate over a potential range that was more negative than the threshold for K+ current recruitment. B, responses to 200 ms duration ‘just‘ subthreshold and supra-threshold depolarizations from a holding potential of −90 mV, before (left) and after (right) current upregulation. Upregulation of persistent current lowered voltage and current threshold and gave rise to repetitive firing (same neurone as in A). Repetitive firing occurred in this neurone at a frequency of 13 Hz, where the most negative potential during the interspike interval was −77 mV. C, voltage-clamp recordings using quasi-physiological solutions in an example wild-type neurone. At the start of recording a transient Na+ current began to activate at −30 mV (left). After upregulation of NaV1.9, a persistent current appeared (right). D, current-clamp recordings showing 10 sequentially recorded traces at 1 min (left) and 3 min (right) after the whole-cell configuration had been achieved (same neurone as in C). The holding current was not greater than −20 pA, and was adjusted to maintain membrane potential at close to −60 mV. E, example of the time dependence of discharge frequency. Same neurone as in C and D.
Figure 2. Internal GTP-γ-S but not GDP leads to substantial persistent current upregulation in Nav1.8 null mutant neurones.

A, internal GTP-γ-S (500 μm) consistently increased maximal current amplitude on average by > 300 % over 5 min (•, n = 7, means ±s.e.m.). In neurones from NaV1.8 null mutants, current amplitude did not increase with intracellular GDP (500 μm) for recordings over 2–40 min (found in a total of 10 neurones); data for 4 neurones are shown (○, means ±s.e.m.). B, with H7 (100 μm), ATP (3 mm) and GTP-γ-S in the internal solution, the mean current increased in amplitude (♦, n = 7, means +s.e.m.), but with H7 (200 μm) and GTP-γ-S only in the pipette solution the upregulation was significantly reduced (⋄, n = 5, means - s.e.m.; ANOVA, P < 0.001). Some s.e.m. values were smaller than the symbol size. C, internal solution containing 2.5 mm Li+ did not cause upregulation of the persistent current in 4 neurones (•, ▪, ♦, ▴; mean data plotted as ○). D, Li+ (2.5 mm) with GDP (500 μm) also did not induce upregulation of the current (n = 3; ▪, ♦, individual neurones; mean data ±s.e.m. plotted as ○).
Persistent current undergoing upregulation after exposure to GTP-γ-S is presented in Fig. 3A; the current-membrane potential relation for peak current and the peak normalized conductance for the same neurone after upregulation are plotted in Fig. 3B and C, respectively. The normalized conductances for the neurone in Fig. 1A–C, at 0, 2 and 7 min after exposure to GTP, were not different from one another or from data obtained from this neurone exposed to GTP-γ-S (Fig. 3C). These plots show that the activation threshold for the current was more negative than −50 mV, some 20 mV or more negative than the threshold expected for NaV1.8.
Persistent current upregulation and the effects of a non-selective protein kinase inhibitor
As channel phosphorylation might have played a role in inducing upregulation, we began to investigate the effects of removal of ATP and introduction of a protein kinase inhibitor to the internal solution. Large upregulation of the persistent current by GTP-γ-S was recorded without ATP in the pipette solution in three wild-type neurones (7.8 ± 0.61-fold increase within 5 min), an increase that could be observed at −30 mV and also at more negative potentials. While these data showed that substantial upregulation can take place in wild-type neurones, as sufficient endogenous ATP might remain after intracellular dialysis, the involvement of a protein kinase was not ruled out. In order to address this question, the non-selective protein kinase inhibitor 1-(5-isoquinolinesulfonyl)-2-methylpiperazine (H7) was also added to the pipette solution. Upregulation was not abolished by 100 μm H7, in the presence of 3 mm ATP and 500 μm GTP-γ-S (n = 7, NaV1.8 null mutant; Fig. 2B), although the mean maximal increase was reduced. The average increase over 5 min was smaller (approximately 150 % rather than 300 %), and this reduction was accompanied by an increase in the variability of upregulation. In two neurones, the persistent current underwent an increase larger than 150 % over 5 min, indicating that even in the nominal presence of the inhibitor, upregulation could be substantial. However, when the concentration of inhibitor was increased to 200 μm and ATP was removed from the pipette solution (n = 5, NaV1.8 null mutant), the average upregulation was significantly diminished in comparison to the data for GTP-γ-S with ATP (two-way ANOVA with Tukey test, between groups P < 0.001, at 0.5 and 1 min P < 0.05, Student's two-tailed and one-tailed, unpaired t tests, respectively), suggesting the involvement of a protein kinase.
Persistent current upregulation changes excitability
To examine how the upregulation of persistent current could change neuronal excitability, current-clamp experiments were carried out using quasi-physiological solutions and in the absence of TTX. When current-clamp solutions were used upregulation could occur from a vanishingly small persistent current; this may be because K+ currents contribute to the total current recorded, even at negative potentials. In this case, upregulation had to take place before the current was observed. In some current-clamp experiments, neurones were held at −90 mV, eliminating variations in membrane potential. By repeatedly estimating the voltage threshold for action potential induction (Fig. 4A), we found that a time-dependent increase in excitability occurred after exposure to intracellular GTP-γ-S only when the neurones were subsequently found to generate the low-threshold persistent Na+ current in voltage clamp. Because the current began to activate at potentials that were very similar to the threshold for K+ current activation, they were not overwhelmed by the outward current and could be detected as inward currents with slow activation kinetics that had amplitudes from −400 pA to over −2 nA at −50 mV. We also observed that subthreshold depolarizations could be dramatically prolonged by the upregulated current (Fig. 4B). The more negative voltage threshold is consistent with persistent current enhancement, and was seen in both wild-type and NaV1.8 null mutant neurones (Fig. 4C). The voltage threshold for action potential induction was estimated to change by −15.64 mV in wild-type (n = 3) and by −15.83 mV (n = 3) in the Nav1.8 null mutant (total n = 6; mean change =−15.74 ± 2.24 mV), for neurones with internal GTP-γ-S and a persistent Na+ current. The proportion of wild-type and Nav1.8 null mutant neurones generating the current was 21 % (3/14) and 16 % (3/19), respectively. More negative thresholds were not observed in neurones lacking the upregulated current (threshold increased by 3.72 ± 1.29 mV, n = 27; P < 0.0001, Student's two-tailed unpaired t test). Internal GDP also failed to alter voltage threshold (threshold fell by 1.05 ± 0.46 mV, 6 NaV1.8 null mutant neurones and 9 wild-type neurones, total n = 15, P < 0.001 compared with GTP-γ-S-exposed neurones generating persistent current, Student's two-tailed unpaired t test).
In addition to a decrease in threshold, upregulation of the persistent current (Fig. 5A) gave rise to a breakdown in accommodation and resulted in repetitive firing to a prolonged (200 ms) ‘just‘ supra-threshold stimulus in the same neurone (Fig. 5B), from a holding potential of −90 mV. For the neurone shown in Fig. 5A and B, the repetitive firing occurred when the most negative membrane potential value between action potentials was within the range −77 to −67 mV. In a total of 11 wild-type neurones held at −60 mV (a typical resting potential for small neurones; e.g. Wang et al. 1994), inclusion of GTP-γ-S in the pipette caused the upregulation of persistent current in four neurones (Fig. 5C), and led to the generation of spontaneous activity (Fig. 5D and E). Only in those neurones where persistent current was recorded in voltage clamp did spontaneous activity ensue at −60 mV, at firing frequencies up to > 1 Hz. No activity was recorded without persistent current activation. The spontaneous activity was driven by a slowly increasing depolarization preceding an action potential for many tens of milliseconds (Fig. 5D). The potential range over which such potential changes occurred corresponded to the most negative part of the activation range for the persistent current, and is consistent with the proposition that upregulated persistent current is able to drive the spontaneous activity.
Wild-type neurones on average exhibited a more stable voltage threshold after exposure to GTP-γ-S in the absence of the persistent current than the NaV1.8 null mutant neurones, where the voltage threshold became more positive (+0.86 ± 1.54 versus+5.68 ± 1.78 mV, n = 11 and 16 in wild-type and Nav1.8 null mutant neurones, respectively; P = 0.052, Student's two-tailed unpaired t test). NaV1.8 undergoes protein kinase A-dependent changes in activation kinetics and voltage dependence that tend to increase repetitiousness (England et al. 1996; Gold et al. 1996). We propose that the effects of GTP analogues on NaV1.8 could thus make a contribution to maintaining excitability, and this may be a reflection of this expected action of protein kinase A. However, the difference between wild-type and Nav1.8 null mutant neurones was not statistically significant, and dramatic changes in excitability can clearly occur without the involvement of NaV1.8 and correlate with NaV1.9 upregulation.
Upregulation of persistent current depolarizes neurones
As previously predicted (Herzog et al. 2001), the persistent current would be expected to contribute to setting the resting membrane potential, because the current generates an activation-inactivation gating overlap current (Cummins et al. 1999; Herzog et al. 2001; Dib-Hajj et al. 2002). The reliable upregulation of the current we describe here allowed us to test whether low-threshold persistent current upregulation alone can depolarize a neurone. In two neurones showing a substantial increase in current amplitude at −50 mV (example neurone in Fig. 6), we found that a depolarization of about 10 mV could ensue. This shift in membrane potential was not seen in the absence of persistent current upregulation. The depolarizing shift in resting potential could not be explained by a fall in input resistance, and became maximal within the range of membrane potentials at which an overlap current would be expected to flow.
Figure 6. Upregulation of persistent current by GTP-γ-S affects membrane potential.
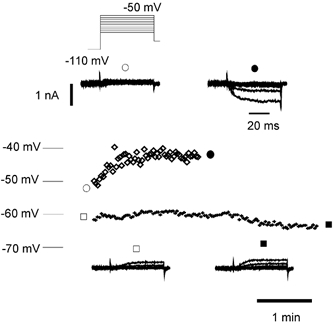
Top, upregulation of persistent current by GTP-γ-S depolarized a wild-type neurone. Membrane potential recorded in current clamp plotted over 2 min is shown (⋄), with voltage-clamp recordings (inset) from the same neurone before (○) and after (•) current upregulation. Input conductance fell from 6.96 to 5.93 nS over the same period. Bottom, in another wild-type neurone (+), there was no current upregulation over 4 min and no depolarization. Inset, voltage-clamp records from the same neurone plotted before (□) and after (▪) membrane potential recording. The calibration bars in the top inset also apply to the bottom inset.
DISCUSSION
TTX-r Na+ channels are known to play a role in nociceptive pathways (reviewed by Baker & Wood, 2001; Fang et al. 2002). Immunocytochemical evidence places NaV1.9 Na+ channels at sensory nerve endings in the cornea (Black & Waxman, 2002), suggesting that they may contribute to electrical excitability at the receptive endings of small diameter afferents. The evidence provided by Fjell et al. (2000) and Fang et al. (2002) is consistent with a role for this channel in nociceptive pathways, because it is expressed only in fibres classified as nociceptive (including C-fibres, Aδ and a few Aα/β fibres), and not in low-threshold mechanoreceptive afferents in rat dorsal root ganglia. Our present observations suggest a special role for the low-threshold persistent Na+ current (attributed to NaV1.9) that may be important for the induction and maintenance of inflammatory pain.
We hypothesized that activating G-proteins by including GTP or GTP-γ-S in the intracellular solution might also regulate NaV1.9 and have presented evidence that the persistent Na+ current is substantially upregulated by G-protein activation. This modulation can give rise to changes in membrane excitability sufficient to cause spontaneous activity at a membrane potential near −60 mV. The data clearly indicate that if a sensory neurone is hyperpolarized to potentials more negative than the activation threshold for the persistent current, GTP-dependent upregulation of the current can substantially increase excitability. The current amplifies and prolongs an imposed depolarization and is likely to operate in this way at potentials more negative than that at which it is fully activated. Because of the slow activation kinetics of the current, it would be most effective at enhancing an imposed depolarization sustained over many milliseconds. Furthermore, at potentials within the most negative portion of the potential range of activation, the upregulated current appears to be sufficiently large to drive spontaneous activity, at least in neuronal somata. The upregulated current does not exhibit substantially changed kinetics. One possibility is that non-conducting (covert) Na+ channels become operable in the presence of GTP. The time scale of current increase suggests that trafficking into the membrane or some other post-translational modification is likely to underlie this effect. The persistent current is reported to undergo a voltage-dependent ultra-slow inactivation (Cummins et al. 1999), although it is unknown whether exit from this state could be controlled by a GTP-dependent mechanism.
Immunocytochemical evidence suggests that only half of the small diameter dorsal root ganglion neurones may express NaV1.9 (consistent with the localization of SNS2; Amaya et al. 2000). The expression of NaV1.9 revealed by in situ hybridization in small diameter DRG neurones in vivo (Dib-Hajj et al. 1998; reviewed by Dib-Hajj et al. 2002) suggests that expression is widespread. Fang et al. (2002) reported that over 60 % of nociceptive neurones express NaV1.9-like immunoreactivity. In the present in vitro experiments using quasi-physiological solutions, the proportion of small diameter, Nav1.8 null mutant neurones generating low-threshold persistent current after exposure to internal GTP-γ-S was about 1 in 6 (≈16 %). For wild-type, the proportion was 1 in 4. The prevalence of the current in control voltage-clamp recordings from NaV1.8 null mutant neurones was 1 in 3 (≈33 %). The proportion of Nav1.8 null mutant neurones generating the persistent current in voltage-clamp and physiological solutions was not significantly different (P = 0.281, Fisher exact test), but we cannot entirely rule out the possibility that differences in the recording solutions may play some part in determining the proportion of neurones generating persistent current, or that small inward currents are simply easier to discern with voltage-clamp solutions that block K+ currents. Any apparent discrepancy between functional data on the one hand and immunocytochemical and molecular data on the other may be due to the channel being expressed at regions of the cell other than the soma, and thus not contributing to somatic currents, or because the channel can exist in both functional and non-functional states that cannot be distinguished by immunocytochemical techniques.
We have estimated that the Cs+ block of residual K+ currents in similar neurones has a time constant close to 1 min (data not shown), and this indicates that an important rate-limiting process for current upregulation is likely to be the diffusion of GTP-γ-S from the electrode. While our data are consistent with the involvement of a protein kinase in the regulatory pathway, one possible reason why a high concentration of kinase inhibitor was required in comparison to that used in other studies (e.g. 50 μm, Chernov et al. 2001; 10 μm, Alroy et al. 1999) could be that no time was allowed for the inhibitor to equilibrate with the cell interior before the introduction of GTP-γ-S. The GTP-binding proteins involved in the current regulatory pathway are unknown. Various combinations of βγ-subunits have been found to enhance the amplitude of the minor, persistent portion of TTX-sensitive currents generated by NaV1.2 in a heterologous system (Ma et al. 1997).
The recent identification of a TTX-r sodium current in the hippocampus that is involved in neurotrophin-evoked depolarization has highlighted another role for NaV1.9 (Blum et al. 2002). Brain-derived neurotrophic factor (BDNF) applied to hippocampal neurones evokes currents of up to 100 pA that are TTX resistant but blocked by 10 nm saxitoxin (Blum et al. 2002). These currents are dependent on the presence of the brain-derived neurotrophic factor receptor (TrkB), and may play an important role in the establishment of long-term potentiation (LTP) (Kovalchuk et al. 2002; Messaoudi et al. 2002). By contrast, the voltage-gated persistent sodium currents present in sensory neurones are larger (up to several nanoamps), are found in TrkB-negative neurones and are not blocked by saxitoxin at concentrations of up to 1 μm (data not shown). Persistent and BDNF-gated sodium currents have both been observed in transfected HEK293 cells using cDNA clones encoding NaV1.9 with or without TrkB (Blum et al. 2002; Dib-Hajj et al. 2002). However, the distinct properties of the two currents suggest that different molecular entities must underlie BDNF-gated and persistent sodium currents. Sodium channels are known to alter their properties in response to a variety of post-transcriptional regulatory steps, ranging from neurotrophin-regulated trans-splicing (Akopian et al. 1999a) to phosphorylation (Fitzgerald et al. 1999). In addition, accessory co-factors are known to alter the properties and distribution of sodium channels (Isom, 2001), and some factors are essential for particular isoform expression, for example P11 and NaV1.8 (Okuse et al. 2002). Thus tissue-specific modifications of NaV1.9 such as trans-splicing, or interactions with accessory subunits, could explain the discrepancies between the two types of current that have been ascribed to NaV1.9. Whatever the molecular mechanism, the regulation of expression and gating of NaV1.9-encoded currents is likely to play an important role both in pain pathways, and in hippocampal neuronal signalling.
In the experiments described here, the upregulated persistent current acts to increase the excitability and reduce accommodation in potential nociceptive neurones with high membrane potentials. We ascribe the reduction in accommodation to the fall in voltage threshold that allows action potential induction from potentials that minimally activate K+ currents. The loss of accommodation allows spontaneous activity at a holding potential near −60 mV. The current upregulation is related to G-protein activation, and may represent an important mechanism in the induction of inflammatory pain. One possible consequence of increased excitability at nerve endings brought about by the upregulation of the persistent current could be the recruitment of silent nociceptors to an active state (e.g. Schmidt et al. 1995). These studies suggest that blocking the current may have important effects on hyperalgesia and pain sensation.
Acknowledgments
We thank the MRC and the Wellcome Trust for support.
REFERENCES
- Akopian AN, Okuse K, Souslova V, England S, Ogata N, Wood JN. Trans-splicing of a voltage-gated sodium channel is regulated by nerve growth factor. FEBS Lett. 1999a;445:177–182. doi: 10.1016/s0014-5793(99)00126-x. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999b;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Alroy G, Su H, Yaari Y. Protein kinase C mediates muscarinic block of intrinsic bursting in rat hippocampal neurons. J Physiol. 1999;518:71–79. doi: 10.1111/j.1469-7793.1999.0071r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- Baker MD, Bostock H. Low-threshold, persistent sodium current in rat large dorsal root ganglion neurons in culture. J Neurophys. 1997;77:1503–1513. doi: 10.1152/jn.1997.77.3.1503. [DOI] [PubMed] [Google Scholar]
- Baker MD, Chandra SY, Wood JN. Up-regulation of Na+ current attributed to NaV1. 9 (NaN) changes firing properties of small diameter sensory neurones. J Physiol. 2002;544:74. P. [Google Scholar]
- Baker MD, Wood JN. Involvement of Na+ channels involved in pain pathways. Trends Pharmacol Sci. 2001;22:27–31. doi: 10.1016/s0165-6147(00)01585-6. [DOI] [PubMed] [Google Scholar]
- Black JA, Waxman SG. Molecular identities of two tetrodotoxin-resistant sodium channels in corneal axons. Exp Eye Res. 2002;75:193–199. doi: 10.1006/exer.2002.2014. [DOI] [PubMed] [Google Scholar]
- Blum R, Kafitz KW, Konnerth A. Neurotrophin-evoked depolarization requires the sodium channel Na(V)1. 9. Nature. 2002;419:687–693. doi: 10.1038/nature01085. [DOI] [PubMed] [Google Scholar]
- Bostock H, Grafe P. Activity-dependent excitability changes in normal and demyelinated rat spinal root axons. J Physiol. 1985;365:239–257. doi: 10.1113/jphysiol.1985.sp015769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock JA, McLachlan EM, Belmonte C. Tetrodotoxin-resistant impulses in single nociceptor nerve terminals in guinea-pig cornea. J Physiol. 1998;512:211–217. doi: 10.1111/j.1469-7793.1998.211bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas CG, Mar LP, Vysokanov AV, Arnold PB, Cardenas LM, Surmeier DJ, Scroggs RS. Serotonergic modulation of hyperpolarization-activated current in acutely isolated rat dorsal root ganglion neurons. J Physiol. 1999;518:507–523. doi: 10.1111/j.1469-7793.1999.0507p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernov MV, Bean LJ, Lerner N, Stark GR. Regulation of ubiquitination and degradation of p53 in unstressed cells through C-terminal phosphorylation. J Biol Chem. 2001;276:31819–31824. doi: 10.1074/jbc.M103170200. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19:RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj S, Black JA, Cummins TR, Waxman SG. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 2002;25:253–259. doi: 10.1016/s0166-2236(02)02150-1. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci U S A. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Djourhi L, Black JA, Dib-Hajj SD, Waxman SG, Lawson SN. The presence and role of the tetrodotoxin-resistant sodium channel NaV1. 9 (NaN) in nociceptive primary afferent neurons. J Neurosci. 2002;22:7425–7433. doi: 10.1523/JNEUROSCI.22-17-07425.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ. cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J Physiol. 1999;516:433–446. doi: 10.1111/j.1469-7793.1999.0433v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell J, Hjelmstrom P, Hormuzdiar W, Milenkovic M, Aglieco F, Tyrell L, Dib-Hajj S, Waxman SG, Black J. Localization of the tetrodotoxin-resistant sodium channel NaN in nociceptors. Neuro Report. 2000;11:199–202. doi: 10.1097/00001756-200001170-00039. [DOI] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Waxman SG. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J Neurophysiol. 2001;86:1351–1364. doi: 10.1152/jn.2001.86.3.1351. [DOI] [PubMed] [Google Scholar]
- Irnich D, Burgstahler R, Bostock H, Grafe P. ATP affects both axons and Schwann cells of unmyelinated C fibres. Pain. 2001;92:343–350. doi: 10.1016/S0304-3959(01)00277-9. [DOI] [PubMed] [Google Scholar]
- Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic induction of BDNF-mediated long-term potentiation. Science. 2002;295:1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein βγ subunits. Neuron. 1997;19:443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR. Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci. 2002;22:7453–7461. doi: 10.1523/JNEUROSCI.22-17-07453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis M, Vogel C, Blenk KH, Arnarson A, Janig W. Inflammatory mediators sensitize acutely axotomized nerve fibers to mechanical stimulation in the rat. J Neurosci. 1998;18:7581–7587. doi: 10.1523/JNEUROSCI.18-18-07581.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuse K, Malik-Hall M, Baker MD, Poon W-YL, Kong H, Chao MV, Wood JN. Annexin II light chain regulates sensory neuron specific sodium channel expression. Nature. 2002;417:653–656. doi: 10.1038/nature00781. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Schmelz M, Forster C, Ringkamp M, Torebjork E, Handwerker H. Novel classes of responsive and unresponsive C nociceptors in human skin. J Neurosci. 1995;15:333–341. doi: 10.1523/JNEUROSCI.15-01-00333.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS, Fox AP. Multiple Ca2+ currents elicited by action potential waveforms in acutely isolated adult rat dorsal root ganglion neurons. J Neurosci. 1992;12:1789–1801. doi: 10.1523/JNEUROSCI.12-05-01789.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra J, Campero M, Ochoa J, Bostock H. Activity-dependent slowing of conduction differentiates functional subtypes of C fibres innervating human skin. J Physiol. 1999;515:799–811. doi: 10.1111/j.1469-7793.1999.799ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer TJ. Effects of Cd2+, Pb2+ and CH3Hg+ on high voltage-activated calcium currents in pheochromocytoma (PC12) cells: potency, reversibility, interactions with extracellular Ca2+ and mechanisms of block. Toxicol Lett. 1998;99:207–221. doi: 10.1016/s0378-4274(98)00225-2. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA. Electrophysiological evidence for tetrodotoxin-resistant sodium channels in slowly conducting dural sensory fibers. J Neurophysiol. 1999;81:413–424. doi: 10.1152/jn.1999.81.2.413. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Serotonin is a directly-acting hyperalgesic agent in the rat. Neuroscience. 1992;48:485–490. doi: 10.1016/0306-4522(92)90508-y. [DOI] [PubMed] [Google Scholar]
- Wang Z, Van Den Berg RJ, Ypey DL. Resting membrane potentials and excitability at different regions of rat dorsal root ganglion neurons in culture. Neuroscience. 1994;60:245–254. doi: 10.1016/0306-4522(94)90218-6. [DOI] [PubMed] [Google Scholar]