

Abstract
In the present study we report that a replication-defective adenovirus construct of GMF cDNA (GMF-V) induced overexpression of GMF protein in neuroblastoma (N18) cells caused cytotoxicity and loss of cell viability. A significant increase in activation of GSK-3β occurred after infection with GMF-V when compared with mock and lacZ controls. Overexpression of GMF also increased caspase-3 activity, an early marker of apoptosis. Depletion of GMF gene by introducing GMF-specific siRNA (GsiRNA) completely blocked both activation of GSK-3β and caspase-3 activation whereas a control scrambled siRNA (CsiRNA) had no effect. A cell-permeable peptide inhibitor of GSK-3β, and lithium completely prevented GMF-dependent activation of caspase-3. These results demonstrate that GSK-3 mediates activation of the death domain caspase by GMF overexpression. We also show that the phosphorylation of GSK-3-dependent site of Tau was a consequence of GMF-overexpression in N18 cells. Taken together our results imply that GMF is involved in the signaling leading to the activation of GSK-3β and caspase-3 in N18 cells and strongly suggest its involvement in neurodegeneration since, GSK-3β is known to hyperphosphorylate tau which is associated with the neurotoxicity of neurofibrillary tangles in Alzheimer's disease.
Keywords: Glia maturation factor (GMF), neuroblastoma (N18) cells, glycogen synthase kinase-3β (GSK-3β), Caspase-3, Alzheimer's disease (AD)
1. Introduction
The Glia Maturation Factor (GMF), a highly conserved brain-specific protein, was isolated, sequenced and cloned in our laboratory (Kaplan et al., 1991; Lim et al., 1989; Lim et al., 1990; Zaheer et al., 1993; Zaheer et al., 1995a; Zaheer et al., 1995b). The functional property of GMF was largely unknown. Our research efforts in recent years have demonstrated an immunomodulatory function for GMF. We also established that GMF is an intracellular regulator of stress-activated signal transduction by demonstrating that GMF activates p38 MAP kinase and transcription factor NF-κB in astrocytes (Lim and Zaheer, 1996; Lim et al., 1998; Lim et al., 2000; Zaheer and Lim, 1996; Zaheer and Lim, 1997; Zaheer and Lim, 1998; Zaheer et al., 2002; Zaheer et al., 2001; Zaheer et al., 2007b). We reported that the overexpression of GMF in astrocytes leads to the destruction of primary oligodendrocytes, the myelin-forming cells in the central nervous system, by interactions between astrocytes, microglia, and oligodendrocytes. These results suggested a novel pathway of GMF initiated cytotoxicity of brain cells, and implicated its involvement in the pathogenesis of inflammatory demyelinating diseases of the central nervous system such as multiple sclerosis/ experimental autoimmune encephalomyelitis (Menon et al., 2007; Zaheer et al., 2007a; Zaheer et al., 2007b; Zaheer et al., 2007c; Zaheer et al., 2007d). In the present report we show that the overexpression of GMF in neuroblastoma N18 cells directly induces caspase-dependent apoptosis mediated by GSK-3β activation.
2. Results
2. 1. Overexpression of GMF in neuroblastoma cells
The cultures of mouse neuroblastoma, N18 cells were infected with a replication-defective adenovirus containing a full length GMF cDNA (GMF-V). For control infections or mock infections, the adenovirus containing cytoplasmic lacZ cDNA (lacZ) or no viral vector (mock) was used following the identical procedures. Cells were incubated with 20 m.o.i. (multiplicity of infectivity) in a serum-free and antibiotic free medium for 4 h. The expression of GMF protein was assessed by Western blotting using a specific anti-GMF antibody (G2-09). The GMF-V transfected cells revealed a significant increase in GMF protein levels (Fig. 1A) and GMF mRNA expression as estimated by RT-PCR (Fig.1B). Since, cells treated with larger than 20 MOI dose exhibited no further increase in GMF protein expression (results not shown), we used 20 MOI. in all subsequent experiments. Control LacZ (20 MOI.) and mock transfections had no effect on GMF expression. The levels of β-actin remained unchanged under all of our experimental conditions.
Fig. 1.

Overexpression of GMF in mouse neuroblastoma N18 cells after transfection with GMF/adenovirus construct (GMF-V). Mouse N18 cells were plated into 24-well plates (1×105 cells per well) and grown in DMEM/F12 containing 5% fetal bovine serum (complete medium). Replication-defective human adenovirus vector containing a full length GMF cDNA (GMF-V) or cytoplasmic lacZ cDNA (LacZ) at 20 MOI (multiplicity of infectivity) were added to cells in serum-free and antibiotic free DMEM/F12 medium for 4 h. Cells were rinsed and allowed to grow in fresh complete medium for another 24 h. Mock-transfected controls were processed identically in the absence of virus. Cell lysates (20 μg protein per lane) were subjected to SDS-polyacrylamide gel electrophoresis followed by electroblotting. The blots were probed with anti-GMF (G2-09) antibody and anti-β-actin antibody. Actin served as internal marker showing equal sample loading. For siRNA mediated down-regulation of GMF expression in N18, cells were transfected with duplex siRNA targeted against GMF (GsiRNA 10 nM), or with a control scrambled sequence (CsiRNA 10 nM). Cells were transfected with siRNA 4 h prior to adenovirus transfections. Lane 1, mock infection; lane 2, lacZ; lane 3, GMF-V; lane 4, CsiRNA; and lane 5, GsiRNA. (A) Western blot. (B) RT-PCR. The data shown are representative of at least three experiments.
2. 2. siRNA mediated down regulation of GMF in N18 cells
To selectively down regulate the expression of GMF protein, we designed GMF-specific (GsiRNA) and control scrambled siRNA (CsiRNA) duplexes. The conditions for cell culture, ratio of siRNA and the transfection reagent were optimized. The N18 cells were transfected with siRNA duplexes (10 nM) 4 h prior to the addition of GMF-V. Analysis of GMF protein (Fig. 1A, lane 5), and GMF mRNA (Fig. 1B, lane 5) from cells showed efficient down regulation of GMF expression in by GsiRNA. Transfection with control scrambled CsiRNA had no effect on GMF expression (Fig. 1A and 1B, lane 4).
2. 3. Reduced cell viability by the overexpression of GMF in N18 cells
The metabolic reduction of MTT by active cells was determined by measuring the absorbance of a formazan product at 570 nm. The number of surviving cells is directly proportional, or in other words the reduction in cell viability as a result of apoptosis or necrosis is inversely proportional to the absorbance of the formazan product formed. No significant differences in cell viability at 24 h of exposure time were noted between cells infected with 2.5-20 MOI of either GMF-V or control LacZ (Fig. 2). The cell viability decreased significantly only after 48-96 h of GMF-V exposure at 10 and 20 MOI (p< 0.05). However, treatment of cells with control LacZ did not change the cell viability at any concentrations up to 96 h (Fig. 2). These results suggest that overexpression of GMF in neuroblastoma N18 cells is correlated with the increase in the loss of cell viability.
Fig. 2.
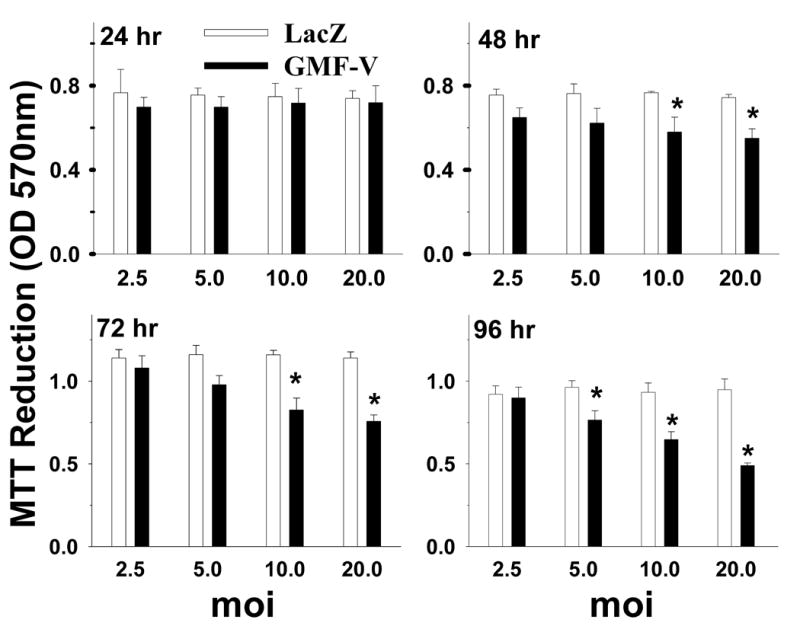
Over expression of GMF in N18 cells is correlated with the increase in the loss of cell viability. N18 cells (5× 104 cells/well) were plated into 96-well plates and transient transfection to overexpress GMF was carried by adding GMF-V or LacZ-V, at 2.5, 5, 10 and 20 MOI viral doses for 24, 48, 72, or 96 h. Cell survival was measured by MTT assay as described in Methods. The results show a time and dose-dependent effect of GMF-V as compared with control LacZ. The data shown are mean ± SD of three separate experiments. * p< 0.05 (GMF-V versus control LacZ).
2. 4. GMF-overexpressing N18 cells undergo apoptosis
To detect the possible involvement of apoptosis in the GMF-induced reduction in N18 cell survival, we observed morphological and apoptotic features using the PI staining as well as a DNA fragmentation assay using gel electrophoresis (Fig. 3). Overexpression of GMF produced a decrease in cell number (Fig. 3B) compared to LacZ (Fig. 3A). Results show apoptotic nuclei and condensed chromatin in cells treated with GMF-V (Fig. 3D) as compared to normal nuclei in control LacZ treated cells (Fig. 3C). Results show DNA laddering, a characteristic feature of apoptosis, in GMF-V transfected N18 cells (Fig. 3E, lane 3). LacZ or mock transfection had no such effect. These results demonstrate that GMF overexpression causes apoptosis in N18 cells.
Fig. 3.
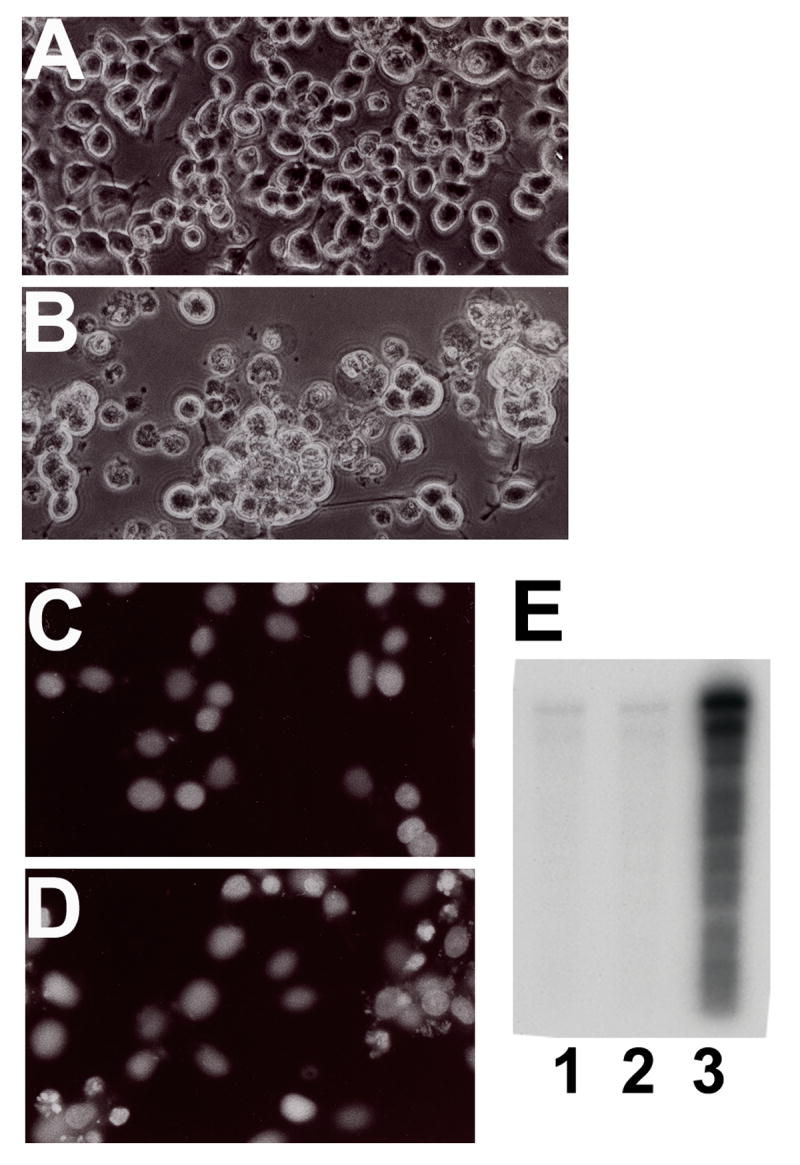
N18 cells transfected with GMF-V undergo apoptosis. Cells were transfected with GMF-V or control LacZ (20 MOI) for 72 h. Note higher number of cell death in GMF-V transfected cells (B) compared to control LacZ (A). Nuclear condensation (D) and DNA fragmentation (E) in GMF-V transfected N18 cells. The cells were stained with propidium iodide (after RNase treatment) to reveal the nuclei. Cells transfected with control LacZ (C) or with GMF-V (D). DNA laddering (E) was detected by electrophoresis as described in Methods. Lane 1, mock; lane 2, LacZ; and lane 3, GMF-V. Results show that the cell death in GMF-V transfected cells is typical of apoptosis.
2. 5. GMF overexpression activates GSK-3β and inhibits Akt in neuroblastoma cells
Recent reports suggest that glycogen synthase kinase-3β (GSK-3β) affects many cellular functions, including the cell cycle and apoptosis. Therefore, we examined whether GSK-3 is involved in GMF-induced cell death in N18 cells. A significant dose-dependent increase in activation of GSK-3β occurred after infection with GMF-V when compared with mock and lacZ controls (Fig. 4). Depletion of GMF gene by introducing GMF-specific siRNA (GsiRNA) completely blocked activation of GSK-3β whereas a control scrambled siRNA (CsiRNA) had no effect. A significant reduction in GSK-3β and Akt phosphorylation are also observed in N18 cells over expressing GMF due to GMF-V transfection when compared with mock and lacZ infections (Fig. 5). There were no changes in total Akt levels. Reduction in phospho-serine9 GSK-3β and phospho-serine473 Akt is consistent with activation of GSK-3β and Akt inactivation, respectively. More over, overexpression of GMF also increased the phosphorylation of a neuronal microtubule-associated protein Tau in N18 cells. Western blots using specific phosho-tau antibodies show a significant increase in tau phosphorylation at GSK-3β specific sites (Ser-396 and Ser-404). Mock and LacZ controls had no such effects. Total tau levels remained unchanged (Fig. 6). The results demonstrate that GMF is involved in the signaling leading to the activation of GSK-3β and tau phosphorylation in neuroblastoma cells.
Fig. 4.

Overexpression of GMF activates GSK-3β kinase in N18 cells. Cells were infected with LacZ (20 MOI), GMF-V (5, 10, and 20 MOI), and GMV-V (20 MOI) in the presence of GMF-specific siRNA (GsiRNA, 20 nM) or control scrambled siRNA (CsiRNA, 20 nM) and harvested at 48 h. GSK-3β kinase was immunoprecipitated from cell lysates and subjected to immune-complex kinase assay using glycogen synthase peptide-2 as the substrate, as described in Methods. The results show a significant dose-dependent increase in activation of GSK-3β kinase in GMF-V transfected cells and significant blocking of GMF-induced GSK-3β activation by GMF-specific siRNA. (siRNA was added 4 h before infection). The figure is representing three independent experiments. p< 0.001, *GMF-V versus control mock and LacZ); # GMF-V versus GsiRNA.
Fig. 5.
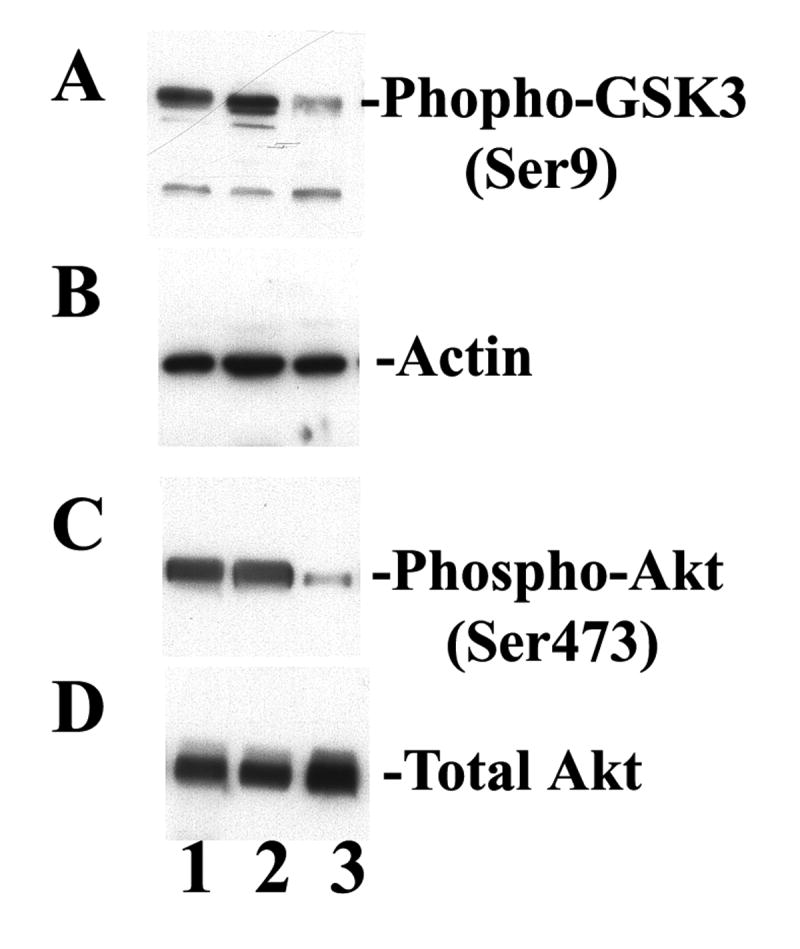
GMF overexpression alters GSK-3β and Akt phosphorylation. Immunoblots show reduced phospho-GSK-3β (A) and reduced phospho-Akt (C) levels in GMF-V (lane 3) compared to control mock (lane 1) and LacZ (lane 2) treated N18 cells. Note no changes in total Akt levels (D). Actin served as a loading control (B).
Fig. 6.
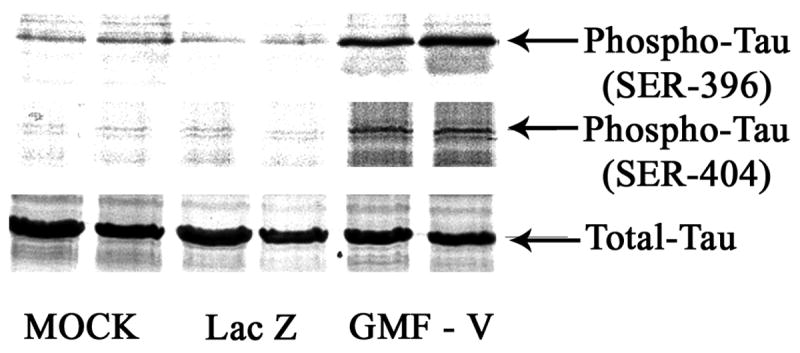
Overexpression of GMF increased Tau phosphorylation in N18 cells. Western blots using specific phosho-tau antibodies show a significant increase in tau-phoshorylation at Ser-396 and Ser-404. Mock and LacZ controls had no such effects. Total tau levels remained unchanged.
2. 6. GMF-dependent activation of caspase-3
Caspase-3 is an important downstream effector of the cell death, and its activation has been reported in many apoptotic processes. To determine whether caspases were activated in GMF-overexpressing N18 cells, we assessed the activation of caspase-3 by monitoring the cleavage of a caspase-3 substrate. A significant GMF-V dose-dependent increase in the caspase-3 activity was detected (P < 0.001) compared to mock and control LacZ treatments (Fig. 7). GMF-dependent caspase-3 activity was completely blocked by GMF-specific siRNA (GsiRNA) whereas control scrambled siRNA (CsiRNA) had no effect on caspase activity. These results demonstrate that overexpression of GMF in N18 cells increased caspase-3 activity, an early marker of apoptosis. To further understand the apoptotic pathway, we analyzed the mRNA expression levels of caspase-1, caspase-2, caspase-6, and caspase-9 by quantitative real-time RT-PCR using highly specific oligonucleotide primers as described in Experimental Procedures. We detected significant up-regulation of caspase-1, caspase-6, and caspase-9 in N18 cells over expressing GMF due to GMF-V transfection when compared with lacZ infections (Fig. 8).
Fig. 7.
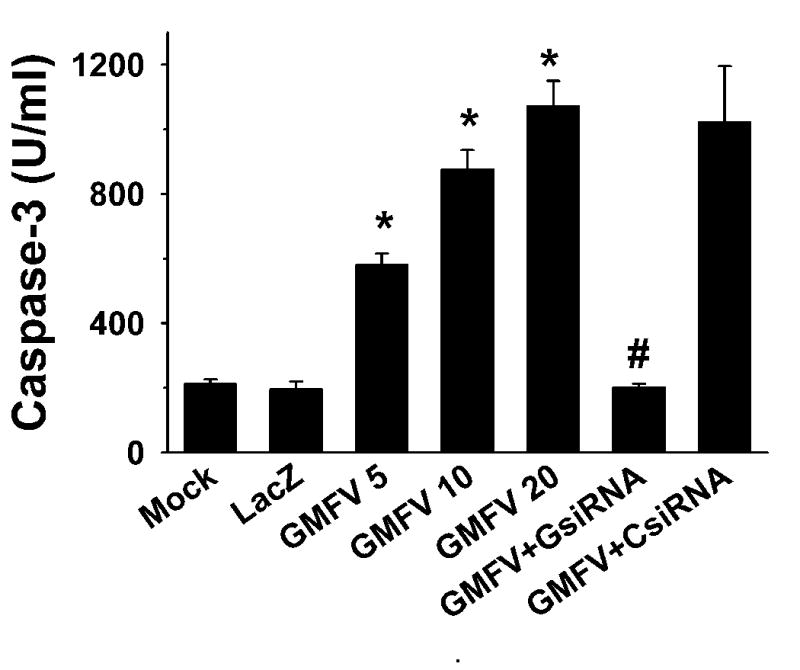
GMF dose-dependent activation of caspase-3 and significant blocking of GMF-induced activation by GsiRNA. Control scrambled CsiRNA had no such effect. Data expressed are the mean ±S.D. of three separate experiments. p< 0.001, *GMF-V versus control mock and LacZ); # GMF-V versus GsiRNA.
Fig. 8.

GMF overexpression up-regulate caspase expression. The mRNA expression levels were determined by quantitative real-time RT-PCR in total RNA isolated from GMF-V or LacZ transfected N18 cells. mRNA levels were estimated by determining ΔCT, the difference between the CT of the caspase of interest and the CT of the endogenous 18S rRNA control gene. * p< 0.05 (GMF-V versus control LacZ).
2. 7. Inhibitors of GSK-3β prevented GMF-dependent activation of caspase-3
Results in Fig. 9 show dose-dependent inhibition of cappase-3 activity by specific inhibitors of GSK-3β. A cell-permeable peptide (CPP) inhibitor, substrate-specific, competitive inhibitor of GSK-3β (Calbiochem) (Fig. 9A) and LiCl (Fig. 9B) could prevent GMF-dependent caspase-3 activation. These results suggest that the inhibition of GSK-3β activity was directly related to suppressing caspase-3 activity and that GSK-3β is upstream of caspase-3.
Fig. 9.
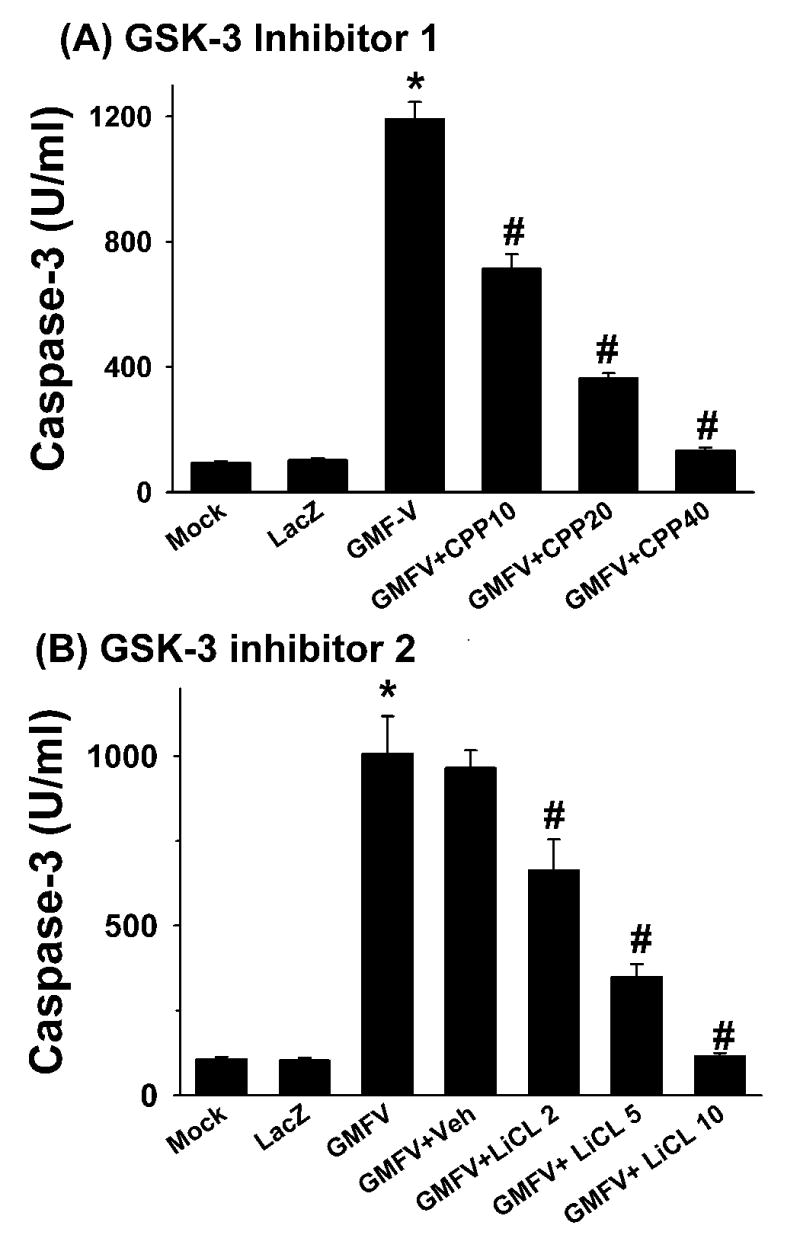
Inhibitors of GSK-3β prevented GMF-dependent activation of caspase-3. Cells were infected with GMF-V or LacZ at 20 MOI in the presence of a specific cell-permeable peptide (CPP) inhibitor of GSK-3 β at 10, 20, and 40 μM concentration (A), or with LiCl at 2, 5, and 10 mM (B). Inhibitors were added 4 h before infection. Cells were harvested after 48 h of incubation and caspase-3 activity was measured as described in Methods. The data shown are mean ± SD of three separate experiments. p< 0.001 (*GMF-V versus control mock and LacZ, # GMF-V versus inhibitors)
2. 8. Selective inhibitors of GMF, GSK-3β and caspase-3 significantly increased cell viability
To examine the involvement of GSK-3β and caspase-3 in GMF-dependent loss of N18 cell viability, we used selective cell-permeable inhibitors of GSK-3β and caspase-3. Results show that GMF-specific siRNA (GsiRNA) and CPP inhibitors of GSK-3β and caspase-3 significantly increased cell viability (Fig. 10). These results demonstrate that the loss of cell viability by the overexpression of GMF in N18 cells is through activation of the GSK-caspase-mediated signaling mechanism.
Fig. 10.
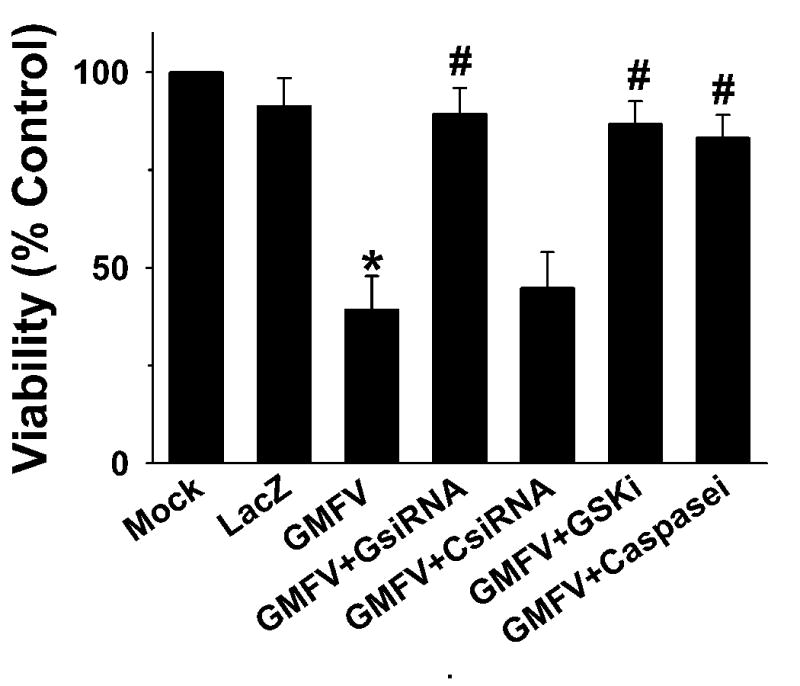
The effects of GMF, GSK-3β, and caspase-3 inhibitors on GMF-induced loss of cell viability. N18 cells (1 × 105) were plated on 24-well plates. After 24 h, the cells were infected with mock, LacZ or GMF-V at 20 MOI. Selective inhibitors of GMF, GsiRNA (20 nM), cell-permeable peptide GSK-3 β inhibitor (20 μM), or cell-permeable CPP32 caspase-3 inhibitor (0.25 nM) were added 4 h before infection. Cell viability was assessed after 96 h by MTT assay as described in methods. The data shown are mean ± SD of three separate experiments. p< 0.001 (*GMF-V versus control mock and LacZ, # GMF-V versus inhibitors)
3. Discussion
Glia maturation factor (GMF), a 17 kDa highly conserved protein, with 99% homology between humans and rodents, was isolated, sequenced and cloned in our laboratory (Kaplan et al., 1991; Lim et al., 1989; Zaheer et al., 1993). The GMF gene is localized to the long arm of human chromosome 14 (Lander et al., 2001). Previously, we established GMF as an intracellular signal transduction regulator. In cell-free assays, GMF is phosphorylated by protein kinase C (PKC), protein kinase A (PKA), casein kinase II (CKII), and ribosomal S6 kinase (RSK) (Zaheer and Lim, 1996; Zaheer and Lim, 1997); and PKA-phosphorylated GMF is an inhibitor of the ERK isoform of MAP kinase (Zaheer and Lim, 1996; Zaheer and Lim, 1997) while at the same time a stimulator of the p38 isoform (Lim and Zaheer, 1996), which implicates GMF protein in stress-activated responses. In C6 as well as in normal astrocytes, GMF overexpression stimulates p38 MAP kinase activity, and activates the redox enzyme CuZn superoxide dismutase (CuZnSOD) and the transcription factors NF-κB (Lim et al., 1998; Lim et al., 2000). Overexpression of GMF in PC12 pheochromocytoma cells activates p38 MAP kinase and its downstream MAPKAP-kinase 2 (Zaheer and Lim, 1998). Kaimori et al. (Kaimori et al., 2003) demonstrated oxidative stress enhancing ability of GMF in the pathophysiology of a renal disease. Overexpression of GMF in proximal tubular cells leads to vulnerability to oxidative injury through p38 pathway and changes in antioxidant enzyme activities (Kaimori et al., 2003). A recent study by Baldwin et al. (Baldwin et al., 2006) demonstrated that the transient overexpression of GMF in glioblastoma cells activated p38 MAPK signaling and also increased susceptibility of the tumor cells to cytotoxicity of cisplatin, a chemotherapeutic agent. More recently, we studied pro inflammatory cytokine profiles of primary glial cells following GMF overexpression. The data demonstrate that overexpression of GMF is necessary for the induction of GM-CSF in astrocytes and that GM-CSF is the key molecule for GMF-dependent production of proinflammatory cytokines in microglial cells. The results also demonstrate that the increased release of GMF-dependent microglial proinflammatory cytokines is cytotoxic to oligodendrocytes, the myelin-forming cells of the central nervous system (Zaheer et al., 2007b). We described novel interactions of brain cells to produce the mediators of neurodegenerative diseases such as multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE) (Zaheer et al., 2007a; Zaheer et al., 2007b; Zaheer et al., 2007c).
Glycogen synthase kinase 3 (GSK-3) is discovered as an enzyme mainly involved in glucose metabolism. GSK-3 consists of two isoforms, GSK-3α and GSK-3β. GSK-3β is expressed in neuronal tissue. The abnormal increases in GSK-3 levels and activity have been associated with neuronal apoptosis, hyperphosphorylation of microtubule associated protein tau, and as well as a decline in cognitive performance (Hetman et al., 2002; Mookherjee and Johnson, 2001; Schubert et al., 2004; Stoothoff and Johnson, 2001). Moreover, transgenic mice overexpressing GSK-3β reported to show abnormal tau hyper phosphorylation and increased neuronal apoptosis (Lucas et al., 2001). Recent reports suggest that phosphatidylinositol-3 kinase/protein kinase B (Akt) signaling pathway in the survival of neurons that leads to the inhibition of GSK-3 by increasing Ser9 phosphorylation (Cross et al., 1995). GSK3 is also reported as an important negative regulator in the insulin-signaling pathway and play a significant role in insulin resistance in diabetes. Thus, the identification of novel inhibitors of GSK3 may lead to potential therapeutic intervention for diseases such as Alzheimer's disease and diabetes.
In the present study we report that a replication-defective adenovirus construct of GMF cDNA (GMF-V) induced overexpression of GMF protein in neuroblastoma (N18) cells. Overexpression of GMF in N18 cells is correlated with the increase in toxicity and loss of cell viability. A significant increase in GSK-3β activity occurred after infection with GMF-V compared to the no infection (mock) and lacZ controls. Overexpression of GMF also increased caspase-3 activity, an early marker of apoptosis. Depletion of GMF mRNA by introducing GMF-specific siRNA completely blocked GSK-3β and caspase-3 activation whereas control scrambled siRNA had no effect. A cell-permeable specific inhibitor of GSK-3β and lithium completely prevented GMF-dependent activation of caspase-3. Our results demonstrate that GMF is involved in the signaling leading to the activation of GSK-3β, caspase-3 and inactivation of Akt in neuroblastoma cells and strongly suggest the involvement of GMF in neuro degeneration. This is the first study to demonstrate that GMF overexpression causes apoptosis through activation of the GSK-caspase-mediated signaling mechanism.
The hallmark of neurodegenerative disorder, Alzheimer's disease, is characterized by the presence of β-amyloid plaques and hyperphosphorylated tau neurofibrillary tangles, in addition to loss of neurons. The pathological activity of tau protein in AD brain is dependent on the level of its phosphorylation. A number of kinases, including GSK-3 and cdk5, can aberrantly phosphorylate tau. It appears that the GMF-GSK-3β-caspase pathway of apoptosis also includes hyperphosphorylated tau in GMF over expressed neuroblastoma cells and both, intrinsic and extrinsic pathways contributed to the apoptosis by the execution caspase-3 since, caspase-1 was also elevated in these cells. In this investigation, we did not measure the activities of p38 and NFκB and thus their participation and hierarchy in GMF overexpressing neuroblastoma is unknown. Future examination in normal neuronal cells will be needed to confirm the relevance of our results in understanding the neurodegenerative diseases. Never the less results of our studies demonstrate that overexpression of GMF in N18 cells contribute to aberrant increases in tau phosphorylation in a site-specific manner and remained elevated at least for 48 h. These findings provide new insights into the role and signaling mechanisms mediated by GMF in activation of GSK-3β, tau phosphorylation and apoptosis and may have significance in the development of molecular targets for future therapeutic application.
4. Experimental Procedure
4. 1. Reagents
Adenovirus constructs were prepared at the University of Iowa Gene Transfer Vector Core as described earlier (Davidson et al., 1993; Lim et al., 1998; Zaheer and Lim, 1998; Zaheer et al., 2002; Zaheer et al., 2001), using a replication-defective adenovirus (serotype 5) vector. The constructs contained either a full-length GMF cDNA (Ad5CMVGMF), or a cytoplasmic lacZ cDNA (Ad5CMVcytolacZ). G2-09 was a monoclonal antibody against GMF and was affinity-purified with protein-A. Affinity purified polyclonal antibody against GSK-3β, caspase-3, and β-actin were obtained from Cell Signaling Technology (Danvers, MA). Rabbit polyclonal antibodies against phospho-tau (pSer396 and pSer404), non-phosphorylated forms (total) of tau and HRP-conjugated IgG secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Enhanced chemluminescence (ECL) Western blot detection reagents were purchased from Amersham Pharmacia Biotech, Inc. (Piscataway, NJ). Glycogen synthase peptide-2 was from Millipore-Upstate Biotechnology, Inc.(Billerica, MA). [32P]-ATP (3000 Ci/mmol) was purchased from NEN Life Science Products, Inc. (Boston, MA). A cell-permeable myristoylated form of GSK-3β peptide inhibitor (Myr-N-Gly-Lys-Glu-Ala-Pro-Pro-Ala-Pro-Pro-Gln-Ser(PO3H)-Pro-NH2 and a cell-permeable (CPP32) inhibitor of caspase-3 (Ac-Ala-Ala-Val-Ala-Leu-Leu-Pro-Ala-Val-Leu-Leu-Ala-Leu-Leu-Ala-Pro-Asp-Glu-Val-Asp-CHO were purchased from EMD-Calbiochem (La Jolla, CA). Trizol® reagent, protein-G Agarose and ThermoScript™ RT-PCR system for first-strand cDNA synthesis were purchased from Invitrogen Corporation, CA. PCR primers were synthesized at Integrated DNA Thechnologies (Coralville, IA).
4. 2. Cell culture
Mouse neuroblastoma cells, N18, were obtained from American Type Culture Collection, Rockville, MD and maintained in our laboratory. The N18 cells were grown in DMEM/F12 medium supplemented with 5% fetal bovine serum, 2 mM L-glutamine, and antibioics penicillin (100 units/ml) and streptomycin (100 μg/ml) at 37°C in a humidified atmosphere of 5% CO2.
4. 3. Overexpression of GMF in N18 cells
Transient transfection with adenoviral vector to overexpress GMF in N18 cells were carried out as described earlier (Zaheer et al., 2002; Zaheer et al., 2007b). Briefly, the replication-defective human adenovirus vector containing either full length GMF cDNA (Ad5CMVGMF) or a cytoplasmic lacZ cDNA (Ad5CMVcytolacZ) were added to cells at 20 MOI (multiplicity of infectivity) or as noted, in serum-free and antibiotic-free DMEM/F12 medium for 4 h. The efficiency of infection was estimated to be over 95% as determined by X-gal staining. At the end of this infection period, cells were gently rinsed once with DMEM/F12 containing 5% FBS and allowed to grow in the same medium for additional time. The cells were harvested at the time indicated for each experiment. In mock-transfected controls, the procedure was carried out in the absence of virus.
4. 4. si RNA-mediated down-regulation of GMF expression
The small interfering RNA (siRNA) for GMF was designed based upon established characteristic of siRNA targeting constructs (Elbashir et al., 2001; Zaheer et al., 2007b). The sequences for GMF (accession no. Z11558) were as follows:
Sense strand GMF siRNA 5′-GAUUGACAAGGAUAAACGCUU-3′; and antisense 5′-GCGUUUAUCCUUGUCAAUCUU-3′. A scrambled sequence was designed as a mismatch control: sense 5′-AGAUAGACACGAUGGUACA UU-3′; and antisense 5′-CGUUCUACUAGUCUAUUCGUU-3′. We blast-search (NCBI database) selected GMF siRNA (GsiRNA) sequence to ensure that only GMF gene is targeted. Specific and control scrambled siRNA (CsiRNA) duplexes were formed as described earlier (Elbashir et al., 2001; Zaheer et al., 2007b). For experiments, N18 cells were seeded at 1×105 in 24-well plates at least 24 h prior to siRNA treatment. Transfections with siRNA (20 nM, or as noted) were carried out essentially as described earlier (Zaheer et al., 2007b). For co-transfection experiments, cells were first transfected with siRNA 4 h prior to GMF-V (20 MOI) infection. Cells were harvested 24 h or as noted after transfection.
4. 5. Caspase-3 Activity Assays
Cells were pulverized under liquid nitrogen and homogenized in ice-cold lysis buffer. Caspase-3 activity in lysates was determined using a colorimetric caspase-3 assay kit according to the manufacturer's instructions (Clontech). Samples were read at 405 nm in a microplate reader.
4. 6. GSK-3 β Immune-kinase assay
Cells were homogenized in a lysis buffer containing protease and phosphatase inhibitors, and then briefly sonicated. After 15 min on ice, the samples were centrifuged at 100,000 g for 1 h at 4°C. Cell lysates were incubated with anti-GSK-3β for 2 h, and then complexes were collected with protein G–Sepharose beads for an additional 1 h. Beads were washed four times in lysis buffer followed by final wash in assay buffer, and then were incubated for 20 min at 30°C with glycogen synthase peptide-2 (25 μM) and 200 μM γ[32P]ATP (3,000–4,000 cpm/pmol) in the presence of 10 mM MgCl2 and with or without 10 mM LiCl. Contents of the assays were spotted onto P81 phosphocellulose papers that were washed and then subjected to liquid scintillation counting. Kinase activity was reduced to background levels in the presence of LiCl suggesting the activity measured was specific for GSK-3β.
4. 7. Cell viability assay
The effect of H2O2 on cell survival was measured using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Hansen et al., 1989). N18 cells were grown in 24-well plates and treated with variable concentrations of H2O2 in fresh culture medium. Twenty-four hours after the treatments, MTT was added and the cells were incubated for 4 h at 37°C. The blue formazine formed in viable cells was dissolved by adding an equal volume of lysis buffer made of 20% sodium dodecylsulphate dissolved in a 1:1 mixture of water and N,N-dimethylformamide, pH 4.7. Two hundred microliter of the homogenous lysate from each well of 24-well plates was transferred into 96-well plates and the optical density measured at 570 nm.
4. 8. Analysis of DNA fragmentation
Apoptosis was determined by a DNA fragmentation assay. For this purpose, cells were scraped after 72 h of transfection in PBS and collected by centrifugation. The cells were then lysed using a buffer containing 9 mM Tris (pH 8.0), 35 mM sucrose and 1 mM EDTA, followed by incubation at 37° C overnight with 0.125 mL of 25% SDS. RNase A was added at a concentration of 1 μg/mL for 1 h at 37° C. Potassium acetate (8 M) was added to obtain a concentration of 1.33 M and the lysate was extracted twice with an equal volume of chloroform/isoamyl alcohol (24:1, v/v). The aqueous layer was taken and the DNA was precipitated in 90% ethanol at -20° C overnight. Samples were centrifuged and the pellets suspended in 10 mM Tris (pH 8.0) and 1 mM EDTA. DNA samples were electrophoresed in a 2% agarose gel along with DNA markers.
4. 9. PI staining
The occurrence of apoptosis was also analyzed by propidium iodide (PI) staining of cells. Apoptotic cells were identified by the appearance of pyknotic nuclei. To identify these features, N18 cells are seeded on a coverslip into the wells of a plate and allow to attach and grow before treating with desired agent. At the end of the 72 h transfection, cells were fixed with ethanol and treated with 0.1 volume of 1 mg/ml RNase A followed by incubation for 1 hr at 37°C. After washing with PBS, PI was added at a concentration of 1 μg/mL in PBS and allowed to incubate with the cells for 20 min at room temperature. Cells were washed again with PBS, mounted and visualized under a fluorescence microscope.
4. 10. Western Immunoblotting
Cells were extracted with a lysis buffer consisting of 1% Triton X-100, 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 50 mM NaF, 0.1 mM sodium vanadate, 1 mM benzamidine, 1 mM PMSF (phenylmethanesulfonylflouride), and 10 μg/ml each of aprotinin, leupeptin, chymostatin, pepstatin A and antipain. Equal amount of protein samples were separated on 4-20% gradient gels by SDS-polyacrylamide gel electrophoresis, and electroblotted onto nitrocellulose membranes. Protein blots were probed with specific primary antibodies (1:1000 v/v) and developed by using the appropriate secondary antibody conjugated to horse radish peroxidase (HRP) (1:2000 v/v), and by using the enhanced chemiluminescence (ECL) method as recommended by the manufacturer.
4. 11. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
RT-PCR was carried out as described earlier (21,22). For this purpose total RNA was isolated from cultured cells by the acid guanidinium thiocyanate-phenol-chloroform method (Chomczynski and Sacchi, 1987), using the RNAzol B reagent (Tel-Test, Inc., Friendswood, TX). The first strand cDNA synthesis was carried out, using a ThermoScript RT-PCR system kit (GibcoBRL, Life Technologies). PCR was carried out essentially as described earlier (Zaheer et al., 1995a; Zaheer et al., 1995b), using a Perkin-Elmer Thermal Cycler. PCR products were electrophoretically analyzed on a 1.8% agarose gel containing ethidium bromide. Highly specific oligonucleotide primers of 20-22 nucleotides were designed as described earlier in detail. The following primers were used: GMF, 5′-ACG CTG GGA GTA AGA ACA AGC T-3′ (sense) and 5′-GGT CCT CGG TGT TTC TTA TTT CAA A-3′ (antisense); caspase-1, 5′-CAC AGC TCT GGA GAT GGT GA -3′ (sense) and 5′-TCT TTC AAG CTT GGG CAC TT-3′ (antisense); caspase-2, 5′-CAG CTC CAA GAG GTT TTT CG-3′ (sense) and 5′-CCA GCA TCA CTC TCC TCA CA-3′ (antisense); caspase-6, 5′-TTC AGA CGT TGA CTG GCT TG-3′ (sense) and 5′-TCC AGC TTG TCT GTC TGG TG-3′ (antisense); caspase-9, 5′-TGC CCT TGC CTC TGA GTA GT-3′ (sense) and 5′-AAC AAA GAA ACG CCC ACA AC-3′ (antisense) and 18S, 5′-TAA GTC CCT GCC CTT TGT ACA-3′ (sense) and 5′-GAT CCG AGG GCC TCA CTA AAC-3′ (antisense).
4. 12. Statistical analysis
Results presented as mean values ± standard deviations. Statistical significance was assessed with one-way ANOVA followed by Tukey's procedure using SigmaStat software (SPP, Chicago, IL). A value of p< 0.05 was considered statistically significant.
Acknowledgments
We thank Yanghong Wu, Ramasamy Thangavel, and Satya Mathur for excellent technical help. This work was supported by the Department of Veterans Affairs Merit Review award (to A.Z.) and by the National Institute of Neurological Disorders and Stroke grant NS-47145 (to A.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baldwin RM, et al. Protection of glioblastoma cells from cisplatin cytotoxicity via protein kinase Ciota-mediated attenuation of p38 MAP kinase signaling. Oncogene. 2006;25:2909–19. doi: 10.1038/sj.onc.1209312. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cross DA, et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Davidson BL, et al. A model system for in vivo gene transfer into the central nervous system using an adenoviral vector. Nat Genet. 1993;3:219–23. doi: 10.1038/ng0393-219. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Hansen MB, et al. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–10. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Hetman M, et al. ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. J Biol Chem. 2002;277:49577–84. doi: 10.1074/jbc.M111227200. [DOI] [PubMed] [Google Scholar]
- Kaimori JY, et al. Induction of glia maturation factor-beta in proximal tubular cells leads to vulnerability to oxidative injury through the p38 pathway and changes in antioxidant enzyme activities. J Biol Chem. 2003;278:33519–27. doi: 10.1074/jbc.M301552200. [DOI] [PubMed] [Google Scholar]
- Kaplan R, et al. Molecular cloning and expression of biologically active human glia maturation factor-beta. J Neurochem. 1991;57:483–90. doi: 10.1111/j.1471-4159.1991.tb03777.x. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lim R, et al. Purification and characterization of glia maturation factor beta: a growth regulator for neurons and glia. Proc Natl Acad Sci U S A. 1989;86:3901–5. doi: 10.1073/pnas.86.10.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R, Zaheer A. In vitro enhancement of p38 mitogen-activated protein kinase activity by phosphorylated glia maturation factor. J Biol Chem. 1996;271:22953–6. doi: 10.1074/jbc.271.38.22953. [DOI] [PubMed] [Google Scholar]
- Lim R, et al. Overexpression of glia maturation factor in C6 cells promotes differentiation and activates superoxide dismutase. Neurochem Res. 1998;23:1445–51. doi: 10.1023/a:1020715126326. [DOI] [PubMed] [Google Scholar]
- Lim R, et al. Complete amino acid sequence of bovine glia maturation factor beta. Proc Natl Acad Sci U S A. 1990;87:5233–7. doi: 10.1073/pnas.87.14.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R, et al. Activation of nuclear factor-kappaB in C6 rat glioma cells after transfection with glia maturation factor. J Neurochem. 2000;74:596–602. doi: 10.1046/j.1471-4159.2000.740596.x. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, et al. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. Embo J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon K, et al. Diminished degradation of myelin basic protein by anti-sulfatide antibody and interferon-gamma in myelin from glia maturation factor-deficient mice. Neurosci Res. 2007;58:156–63. doi: 10.1016/j.neures.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookherjee P, Johnson GV. Tau phosphorylation during apoptosis of human SH-SY5Y neuroblastoma cells. Brain Res. 2001;921:31–43. doi: 10.1016/s0006-8993(01)03074-8. [DOI] [PubMed] [Google Scholar]
- Schubert M, et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A. 2004;101:3100–5. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV. Hyperosmotic stress-induced apoptosis and tau phosphorylation in human neuroblastoma cells. J Neurosci Res. 2001;65:573–82. doi: 10.1002/jnr.1187. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Expression of glia maturation factor beta mRNA and protein in rat organs and cells. J Neurochem. 1993;60:914–20. doi: 10.1111/j.1471-4159.1993.tb03237.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Lim R. In vitro inhibition of MAP kinase (ERK1/ERK2) activity by phosphorylated glia maturation factor (GMF) Biochemistry. 1996;35:6283–8. doi: 10.1021/bi960034c. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Lim R. Protein kinase A (PKA)- and protein kinase C-phosphorylated glia maturation factor promotes the catalytic activity of PKA. J Biol Chem. 1997;272:5183–6. doi: 10.1074/jbc.272.8.5183. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Lim R. Overexpression of glia maturation factor (GMF) in PC12 pheochromocytoma cells activates p38 MAP kinase, MAPKAP kinase-2, and tyrosine hydroxylase. Biochem Biophys Res Commun. 1998;250:278–82. doi: 10.1006/bbrc.1998.9301. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Overexpression of glia maturation factor in astrocytes leads to immune activation of microglia through secretion of granulocyte-macrophage-colony stimulating factor. Biochem Biophys Res Commun. 2002;294:238–44. doi: 10.1016/S0006-291X(02)00467-9. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Diminished cytokine and chemokine expression in the central nervous system of GMF-deficient mice with experimental autoimmune encephalomyelitis. Brain Res. 2007a;1144:239–47. doi: 10.1016/j.brainres.2007.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer A, et al. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochem Res. 2001;26:1293–9. doi: 10.1023/a:1014241300179. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J Neurochem. 2007b;101:364–76. doi: 10.1111/j.1471-4159.2006.04385.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Reduced severity of experimental autoimmune encephalomyelitis in GMF-deficient mice. Neurochem Res. 2007c;32:39–47. doi: 10.1007/s11064-006-9220-x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Expression of mRNAs of multiple growth factors and receptors by neuronal cell lines: detection with RT-PCR. Neurochem Res. 1995a;20:1457–63. doi: 10.1007/BF00970594. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Expression of mRNAs of multiple growth factors and receptors by astrocytes and glioma cells: detection with reverse transcription-polymerase chain reaction. Cell Mol Neurobiol. 1995b;15:221–37. doi: 10.1007/BF02073330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer S, et al. Glia Maturation Factor Regulation of STAT Expression: A Novel Mechanism in Experimental Autoimmune Encephalomyelitis. Neurochem Res. 2007d doi: 10.1007/s11064-007-9383-0. [DOI] [PubMed] [Google Scholar]