

Abstract
The effects of extracellular pH (pHo) on calcium-sensing non-selective cation (csNSC) channels in cultured mouse hippocampal neurons were investigated using whole-cell voltage-clamp and current-clamp recordings. Decreasing extracellular Ca2+ concentrations ([Ca2+]o) activated slow and sustained inward currents through the csNSC channels. Decreasing pHo activated amiloride-sensitive transient proton-gated currents which decayed to baseline in several seconds. With proton-gated channels inactivated by pre-perfusion with low pH solution or blocked by amiloride, decreasing pHo to 6.5 inhibited the csNSC currents with a leftward shift of the Ca2+ dose–inhibition curve. Increasing pH to 8.5, on the other hand, caused a rightward shift of the Ca2+ dose–inhibition curve and potentiated the csNSC currents. Intracellular alkalinization following bath perfusion of quinine mimicked the potentiation of the csNSC currents by increasing pHo, while intracellular acidification by addition and subsequent withdrawal of NH4Cl mimicked the inhibition of the csNSC currents by decreasing pHo. Intracellular pH (pHi) imaging demonstrated that decreasing pHo induced a corresponding decrease in pHi. Including 30 mM Hepes in the pipette solution eliminated the effects of quinine and NH4Cl on the csNSC currents, but only partially reduced the effect of lowering pHo. In current-clamp recordings, decreasing [Ca2+]o induced sustained membrane depolarization and excitation of hippocampal neurons. Decreasing pHo to 6.5 inhibited the low [Ca2+]o-induced csNSC channel-mediated membrane depolarization and the excitation of neurons. Our results indicate that acidosis may inhibit low [Ca2+]o-induced neuronal excitation by inhibiting the activity of the csNSC channels. Both the extracellular and the intracellular sites are involved in the proton modulation of the csNSC channels.
Extracellular concentrations of calcium ([Ca2+]o) in the central nervous system fall substantially in both physiological and pathological conditions. Repetitive electrical stimulation or iontophoretic applications of excitatory amino acids, for example, can produce up to a 0.5 mM decrease in [Ca2+]o (Heinemann & Pumain, 1980; Somjen, 1980; Krnjevic et al. 1982; Heinemann & Louvel, 1983), while experimentally induced seizure activity can decrease [Ca2+]o by ≈1.0 mM (Heinemann et al. 1977; Heinemann & Louvel, 1983). More pronounced and sustained decreases in [Ca2+]o can be recorded during spreading depression and periods of brain ischaemia (Hansen & Zeuthen, 1981; Ekholm et al. 1995). For example, a decrease of [Ca2+]o to ≈0.2 mM is commonly observed in brain ischaemia (Hansen & Zeuthen, 1981; Ekholm et al. 1995).
Decreases in [Ca2+]o are known to increase neuronal excitability (Hille, 1992). The detailed mechanism underlying enhanced neuronal excitation is, however, not fully understood. We previously demonstrated that decreases in [Ca2+]o excite central neurons through activation of a novel non-selective cation channel (Xiong et al. 1997). This channel is not blocked by specific inhibitors of known voltage-gated or ligand-gated ion channels. Since the gating of this channel is closely controlled by [Ca2+]o, it was named the calcium-sensing non-selective cation (csNSC) channel (Xiong & MacDonald, 1999). A cation channel with similar electrophysiological and pharmacological properties has also been described in cardiac cells (Mubagwa et al. 1997). Activation of the csNSC channel may well explain a major component of the enhanced neuronal excitability associated with the decreases in [Ca2+]o during epileptic seizures (Xiong et al. 2001). It is also likely that activation of the csNSC channel and subsequent long-lasting membrane depolarization may contribute to excitotoxicity associated with brain ischaemia.
Along with the decrease in [Ca2+]o, extracellular pH (pHo) also decreases dramatically during both seizure activity (Simon et al. 1985, 1987; Xiong & Stringer, 2000) and brain ischaemia (Siemkowicz & Hansen, 1981; Kraig et al. 1985; Siesjo, 1988). During brain ischaemia, for example, pHo typically falls to 6.5 under normoglycaemic conditions (Siemkowicz & Hansen, 1981) and may fall to as low as 6.0 under hyperglycaemic conditions (Siemkowicz & Hansen, 1981; Kraig et al. 1985). Such changes in pHo can modulate the activity of a variety of membrane receptors and ion channels (Tang et al. 1990; Traynelis & Cull-Candy, 1990; Zhu et al. 1999; Claydon et al. 2000). For example, N-methyl-D-aspartate (NMDA) receptor-gated channels are strongly inhibited by decreases in pHo (Tang et al. 1990; Traynelis & Cull-Candy, 1990). On the other hand, a decrease in pHo can itself induce inward currents by activating acid-sensing ion channels (ASICs) in both peripheral sensory (Mironov & Lux, 1993; Waldmann et al. 1997b) and central neurons (Waldmann et al. 1997a; Varming, 1999; Escoubas et al. 2000). In the present study, we investigated the effects of acidosis on the csNSC channel-mediated responses in cultured mouse hippocampal neurons. Our results show that acidosis may reduce low [Ca2+]o-induced neuronal excitation through inhibition of the csNSC channels.
METHODS
Dissociation and culture of mouse hippocampal neurons
Cultures of mouse hippocampal neurons were prepared according to previously described techniques (Xiong et al. 2001). The use of mice for neuronal cultures was reviewed and approved by the Institutional Animal Care and Use Committee of the University of Toronto and Legacy Clinical Research and Technology Center. Briefly, time-pregnant (E17) mice were anaesthetized with halothane followed by cervical dislocation. Fetuses were rapidly removed and hippocampi were dissected and placed in Ca2+ and Mg2+-free cold Hank's solution. The hippocampi were then incubated with 0.05 % trypsin-EDTA for 10 min at 37 °C, triturated with fire-polished glass pipettes, and plated in 35 mm poly-L-ornithine-coated culture dishes or 25 mm coverslips at densities of ≈1 × 106 per dish and 0.9 × 106 per coverslip. The culture medium consisted of Eagle's minimum essential medium (MEM) supplemented with 10 % heat-inactivated horse serum. After 3 days in vitro, growth of non-neuronal cells was inhibited by a 48 h exposure to medium containing 5 μM uridine and 5 μM (+)-5-fluor-2′-deoxyuridine. Cultures were fed twice a week and used for electrophysiological recordings 14–21 days after plating.
Electrophysiology
Whole-cell patch-clamp recordings were performed and analysed as described previously (Xiong et al. 1997). Patch electrodes were constructed from thin-walled borosilicate glass (1.5 mm diameter, WPI, Sarasota, FL, USA) on a two-stage puller (PP83, Narishige, Tokyo). The tips of the electrodes were heat-polished on a microforge (Model MF-83, Narishige, Tokyo) to a final diameter of 1–2 μm. The patch electrodes had resistances between 3 and 5 MΩ when filled with intracellular solution. Whole-cell currents were recorded using Axopatch 1-D amplifiers (Axon Instruments, Foster City, CA, USA). Data were filtered at 2 kHz and digitized at 5 kHz using Digidata 1200 DAC units (Axon Instruments). The on-line acquisition was done using pCLAMP software (version 8, Axon Instruments). During each experiment, a voltage step of −10 mV was applied periodically to monitor the cell capacitance and the access resistance. Recordings in which the access resistance or the capacitance changed by more than 10 % during the recordings were not included in data analysis (Xiong et al. 1998). Perforated-patch recording was performed as described previously (Valenzuela et al. 1996). Nystatin (300 μg ml−1) was included in the pipette solution to achieve low access resistance without a physical disruption of the patch membrane. Whole-cell current was recorded 5–10 min after the formation of high resistance seal (>5 GΩ) where the access resistance is below 20 MΩ. For cell-attached loose-patch recording, glass pipettes (impedance, 1–3 MΩ) were filled with regular bath solution. The pipette potential was held at 0 mV.
pHi measurement
Intracellular pH was assessed using the pH-sensitive probe BCECF, as described in detail previously (Boyarsky et al. 1988). Cultured mouse hippocampal neurons grown on 25 mm × 25 mm coverslips were incubated in the extracellular solution containing 2 μM BCECF-AM (Molecular Probes, Eugene, OR, USA) for 20 min at room temperature, washed 3 times and then incubated with BCECF-free solution for 30 min. Coverslips with BCECF-loaded cells were then transferred to a perfusion chamber on an inverted microscope (Nikon TE300). Cells were illuminated using a xenon lamp (75 W) and observed with a × 40 UV fluor oil-immersion objective lens. Video images were obtained using a cooled CCD camera (Sensys KAF 1401, Photometrics). Digitized images were acquired, stored and analysed in a personal computer controlled by Axon Imaging Workbench software (AIW2.1, Axon Instruments). The shutter and filter wheel (Lambda 10–2) were also controlled by AIW to allow for timed illumination of cells at either 440 nm (pH insensitive) or 490 nm (pH sensitive) excitation wavelengths. Background-subtracted BCECF fluorescence was detected at an emission wavelength of 535 nm. 490 nm/440 nm ratio images were acquired every 10 or 20 s and analysed by averaging pixel ratio values in circumscribed regions of cells in the field of view. Analysis was restricted to those neurons able to retain BCECF throughout the course of an experiment. The values were exported from AIW to SigmaPlot 2000 for statistical analysis and plotting.
Fluorescence ratios were calibrated in situ using the nigericin- high K+ method (Boyarsky et al. 1988) with the calibration solutions at five pH values: 6.5, 7.0, 7.5, 8.0 and 8.5.
Solutions and chemicals
Standard extracellular solution contained (mM): 140 NaCl, 5.4 KCl, 2 CaCl2, 25 Hepes, 33 glucose (pH 7.4 with NaOH; 320–330 mosmol l−1). For solutions with pH < 7.0, Mes was used as pH buffer instead of Hepes. MgCl2 (1 mM) was present in the current-clamp experiment but was excluded for some voltage-clamp experiments in order to maximize the activation of the csNSC channel. Tetrodotoxin (TTX, 0.5 μM) was added for all voltage-clamp experiments but excluded for some current-clamp recordings where firing of action potentials was studied.
For voltage-clamp experiments, the pipette solution contained (mM): 140 CsF or CsCl, 30 CsOH, 10 Hepes, 11 EGTA, 2 tetraethylammonium chloride (TEA), 1 CaCl2, 2 MgCl2 and 4 K2ATP (pH 7.3 with CsOH; 310 mosmol l−1). For current-clamp recordings the pipette solution contained (mM): 150 KF or KCl, 10 Hepes, 11 EGTA, 2 TEA, 1 CaCl2, 2 MgCl2 and 4 K2ATP (pH 7.3 with KOH; 310 mosmol l−1). For perforated- patch recording, the following solution was used in the pipette (mM): 136.5 potassium gluconate, 17.5 KCl, 9 NaCl, 1 MgCl2, 10 Hepes, 0.2 EGTA, 2 CaCl2, with 300 μg ml−1 nystatin. A multi-barrel perfusion system (SF-77, Warner Instrument Co., CT, USA) was employed to achieve a rapid exchange of solutions.
All experiments were performed at room temperature (22 -24 °C). Data are expressed as means ± s.e.m. Student's t test was employed for the analysis of statistical significance.
RESULTS
Lowering [Ca2+]o activates the csNSC currents independently of changes in the seal conductance
We have reported previously that, in cultured hippocampal neurons, lowering [Ca2+]o induced neuronal excitation by activating a slow inward current (Xiong et al. 1997). The current was sensitive to blockade by micromolar concentrations of Gd3+ but insensitive to known blockers of specific voltage-gated or ligand-gated channels. Since the activation of this current is closely controlled by the [Ca2+]o, it was tentatively named the calcium-sensing non-selective cation (csNSC) current (Xiong & MacDonald, 1999).
In acutely dissociated rat thalamic or sensory ganglion neurons, however, Formenti and De Simoni have recently shown that lowering [Ca2+]o can induce an inward current simply by increasing the seal conductance in the cell-attached configuration (Formenti & De Simoni, 2000). The change in seal conductance induced by lowering [Ca2+]o decreases with higher resistance seals and becomes very low with seals over 1 GΩ. For this reason, we have also performed cell-attached recordings in cultured hippocampal neurons to examine the effect of lowering [Ca2+]o on the seal resistance between the electrode and the cell. In a total of eight cells recorded, an average seal resistance of 8.3 ± 0.6 GΩ was achieved in the presence of 2.0 mM [Ca2+]o. Lowering [Ca2+]o to 0.1 mM induced only a minor and insignificant change in seal resistance to 8.2 ± 0.7 GΩ (n = 8, P = 0.63, Fig. 1A and B). With this change of seal resistance, the seal conductance is only changed from 0.120 to 0.122 nS. Such a minor shift in seal resistance is too small to account for the large amplitude of whole-cell current recorded in hippocampal neurons (Fig. 1C, left panel).
Figure 1. Low [Ca2+]o-activated current in cultured hippocampal neurons is not due to changes in the seal conductance.
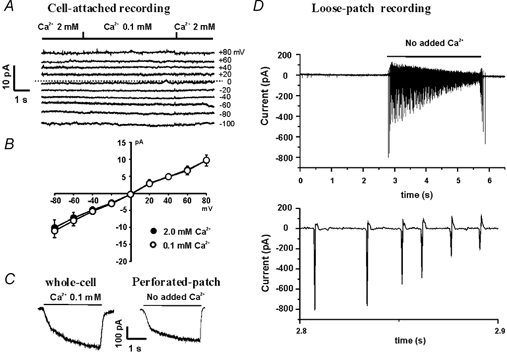
A, example traces showing the effect of low [Ca2+]o on the seal currents recorded at the potentials indicated to the right of each current trace. B, summary data showing the current–voltage relationship (I-V curve) in the presence of 2.0 mM [Ca2+]o (•) and 0.1 mM [Ca2+]o (○). The seal resistance, calculated from the slope of the I-V curve, was 8.3 ± 0.6 GΩ for 2.0 mM [Ca2+]o and 8.2 ± 0.7 GΩ for 0.1 mM [Ca2+]o (n = 8, P = 0.63). C, example traces showing the activation of the csNSC current in conventional whole-cell and perforated-patch configurations. For whole-cell recording, low access resistance (< 10 MΩ) was achieved by applying a brief pulse of suction to disrupt the patch membrane, while for perforated-patch recording, low access resistance (˜15 MΩ) was achieved by membrane perforation with 300 μg ml−1 nystatin. D, top panel, example traces showing the low [Ca2+]o-induced neuronal excitation in a cell-attached loose-patch recording. With the pipette potential held at 0 mV which eliminates the driving force across the seal between the pipette and the cell, lowering [Ca2+]o from 1.3 to 0 mM excites the neuron independently of changes in the seal conductance. D, bottom panel, a section of the trace above on an expanded time scale showing the membrane current corresponding to the firing of individual action potentials.
Since it is difficult to accurately measure the seal resistance in whole-cell recording, it may be argued that during the formation of the whole-cell patch configuration, the seal resistance is reduced due to a physical disruption of the patch membrane. This reduction in the seal resistance probably makes it more sensitive to the changes in [Ca2+]o. To address this possibility, we have also used the perforated-patch recording technique (Horn & Marty, 1988) to study the low [Ca2+]o-activated current following membrane perforation. With this recording technique, the patch membrane is not physically disrupted during the formation of a low access resistance seal for the recording of whole-cell currents, thus the seal resistance is expected to be the same as in the cell-attached configuration (i.e. before perforation). Nystatin (300 μg ml−1) was included in the pipette solution to achieve the perforated-patch recording as described previously (Valenzuela et al. 1996). Five minutes after the formation of a high resistance seal (>5 GΩ), the low [Ca2+]o-activated current was activated by step reduction of [Ca2+]o. As shown in Fig. 1C (right panel), lowering [Ca2+]o from 1.3 to 0 mM activated a large inward current in perforated-patch recordings (n = 6), similar to the current recorded in the conventional whole-cell recordings (Fig. 1C, left panel).
To further determine whether the low [Ca2+]o-mediated response is due to a change in seal conductance, we also employed the loose-patch technique to record responses to lowering [Ca2+]o with the pipette potential held at 0 mV. Cell-attached loose-patch recording can be used for detection of the firing of action potentials without any physical damage to the cell membrane (Smith & Otis, 2003). When the pipette potential is held at 0 mV, the loose-patch electrode can record the membrane current independently of changes in seal conductance. Taking advantage of this technique, we were able to record high frequency firing of action potentials induced by lowering [Ca2+]o (from 1.3 to 0 mM) while holding the pipette potential at 0 mV (Fig. 1D). Since the driving force crossing the seal resistance is 0 mV, low [Ca2+]o-induced neuronal excitation cannot be explained by changes in the seal conductance. The most likely explanation is that lowering [Ca2+] activates an inward current which causes membrane depolarization, resulting in the firing of action potentials. This effect of low [Ca2+]o is consistent with the low [Ca2+]o-induced membrane depolarization and excitation of hippocampal neurons seen in current-clamp recordings (Xiong et al. 1997; see also Fig. 9). Together these data strongly suggest that low [Ca2+]o can activate the csNSC channel, resulting in neuronal excitation that is independant of any changes in the seal conductance.
Figure 9. Effect of acidosis on the csNSC channel-mediated membrane depolarization and the excitation of hippocampal neurons.
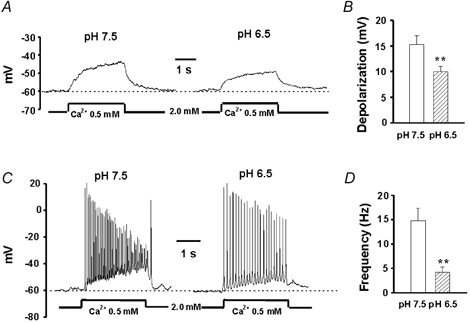
A, representative traces showing membrane depolarization in the presence of TTX (0.5 μM) by lowering [Ca2+]o from 2.0 to 0.5 mM at pH 7.5 and 6.5. B, summary data from 7 neurons showing the effect of acidosis on the amplitude of membrane depolarization induced by lowering [Ca2+]o. The amplitude of membrane depolarization induced by lowering [Ca2+]o to 0.5 mM was 15.3 ± 1.7 mV at pH 7.5 and 9.9 ± 1.0 mV at pH 6.5 (n = 6, P < 0.01). C, representative traces showing membrane depolarization and excitation of a hippocampal neuron by lowering [Ca2+]o from 2.0 to 0.5 mM at pH 7.5 or 6.5. D, summary data from 8 neurons showing the frequency of action potential firing at pH 7.5 and 6.5. The frequency of action potential firing in the presence of 0.5 mM [Ca2+]o was 14.8 ± 2.6 Hz at pH 7.5 and 4.3 ± 1.1 Hz at pH 6.5 (n = 8, P < 0.01). All solutions contained 1 mM Mg2+. ** P < 0.01 compared with the pH 7.5 group.
Transient synergistic interaction between low pH- and low Ca2+-induced responses
The effects of acidosis on the csNSC current were initially studied by comparing the peak amplitude of the inward current activated by decreasing the [Ca2+]o with or without a simultaneous decrease in pHo. At a holding potential of −60 mV, step reductions in [Ca2+]o from 2.0 mM to 0.1 mM without changing the pH (7.5) induced a slowly activated inward current in all cultured hippocampal neurons. The average amplitude of this current at 2 s after switching to 0.1 mM [Ca2+]o was −306 ± 45 pA (n = 7; Fig. 2A, left panel). In the same neurons, a step reduction in pHo from 7.5 to 6.5, without changing [Ca2+]o (2.0 mM), induced a transient inward current of −464 ± 76 pA (Fig. 2A, middle panel) probably mediated via proton-gated or acid-sensing ion channels (ASICs; Waldmann et al. 1999). When [Ca2+]o and pHo were simultaneously reduced, inward currents with an average amplitude of −2059 ± 421 pA (n = 6) were recorded (Fig. 2A, right panel). These currents were substantially larger than the simple sum of each separate current (P < 0.01), indicating a synergistic interaction between acidosis and low [Ca2+]o-induced responses. This enhancement of inward current by simultaneous decreases of [Ca2+]o with pH is probably due to removal of Ca2+ blockade of the ASICs, as described previously by Immke & McCleskey, who found that chelating [Ca2+]o by lactate potentiates the ASIC current in cardiac sensory neurons (Immke & McCleskey, 2001).
Figure 2. Synergistic interaction between low [Ca2+]o- and low pH-induced responses.
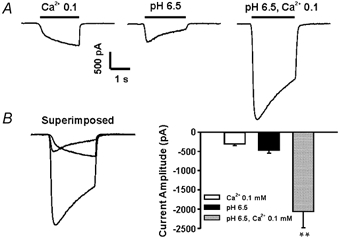
Cultured hippocampal neurons were initially perfused with bath solution containing 2.0 mM [Ca2+]o at pH 7.5. A, example traces showing the interaction between low pH- and low [Ca2+]o-mediated responses. At a holding potential of −60 mV, a step decrease of [Ca2+]0 from 2.0 to 0.1 mM activated a slow and sustained inward current through the csNSC channels (left panel), while a step decrease of pH from 7.5 to 6.5 activated a transient inward current, probably through proton-gated channels (middle panel). Simultaneous decreases in pH and [Ca2+]0 activated a much larger inward current (right panel). B, superimposed traces (left panel) and bar graph (right panel) showing the synergism between low pH- and low [Ca2+]0-induced responses. ** P < 0.01.
Decreasing pHo inhibits the csNSC current when proton-induced currents are inactivated or blocked
In order to study the effect of changing pH on the csNSC current without interference by proton-gated currents, neurons were pre-perfused with low pH solution (6.5) for at least 2 min prior to activating the csNSC channels. As shown in Fig.3A, proton-gated currents inactivate rapidly and completely within ≈5 s. This fast decay of the proton-gated current is in contrast to the same current recorded in cardiac sensory neurons and in dorsal root ganglion neurons where a non-inactivating and sustained component of the proton-gated current is also observed (Waldmann et al. 1997a; Benson et al. 1999). This difference is largely due to the different subunits of proton-gated channels expressed in hippocampal neurons and in sensory neurons. For example, ASIC3 (or DRASIC), which mediates a slow component of the proton-gated currents, specifically expresses in sensory neurons (Waldmann et al. 1997a), while in hippocampal neurons, ASIC1, a subunit mediating a transient component of the proton-gated current, predominates (Wemmie et al. 2002). The fast decay of the proton-gated current in cultured hippocampal neurons makes it possible to use pretreatment of the neurons with low pH solutions to avoid contamination of the csNSC currents by the current passing through the proton-gated channels. In addition, in some recordings, we have used amiloride to block the proton-gated currents (Waldmann & Lazdunski, 1998). As shown in Fig. 3B, amiloride caused a concentration-dependent inhibition of proton-gated currents in cultured hippocampal neurons (IC50 of ≈11 μM). To block these currents, 100 μM amiloride was included in the bath solution when recording the csNSC current at lowered pH. This concentration of amiloride had no direct effect on the csNSC current (Fig. 3C), though higher concentrations (>500 μM) slightly inhibited the current (not shown). Finally, further evidence that ASICs play no role in generating the csNSC responses comes from experiments we are carrying out on cells from ASIC1 knock-out mice, which lack the normal ASIC-mediated response to pH, but still show a response of normal amplitude to low [Ca2+]o (X. P. Chu et al., unublished observations).
Figure 3. Time-dependent inactivation and amiloride blockade of the currents through proton-gated channels.
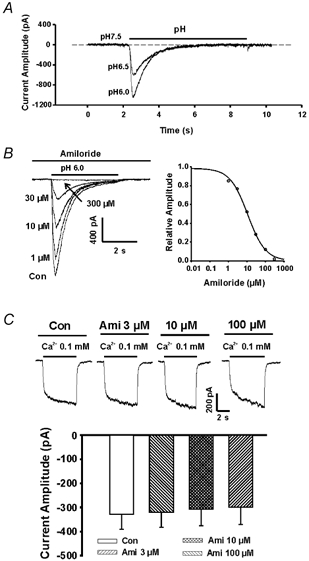
A, example traces showing the time course of the currents activated by decreasing pH from 7.5 to 6.5 and 6.0. The currents decayed to baseline within 5 s. B, left panel, representative traces showing the effect of amiloride on proton-gated currents activated by decreasing pH to 6.0; right panel,dose-dependent block of proton-gated currents by amiloride, with an IC50 of 11 μM. C, representative traces and summary bar graph showing the lack of blockade of the csNSC current by amiloride with concentrations up to 100 μM.
In contrast to the effect of co-applications of low pH and low [Ca2+]o, pre-treating the neurons at pH 6.5 inhibited the csNSC current, reducing the amplitude from a mean value of −336 ± 48 pA to −206 ± 35 pA (≈40 % decrease, n = 7, P < 0.01; Fig. 4A and B). Increasing the pH to 8.5, on the other hand, enhanced the csNSC currents, with the mean amplitude rising to −526 ± 76 pA (P < 0.05). These data indicate that increasing the proton concentration inhibits the csNSC channels.
Figure 4. pH dependence of the csNSC current.
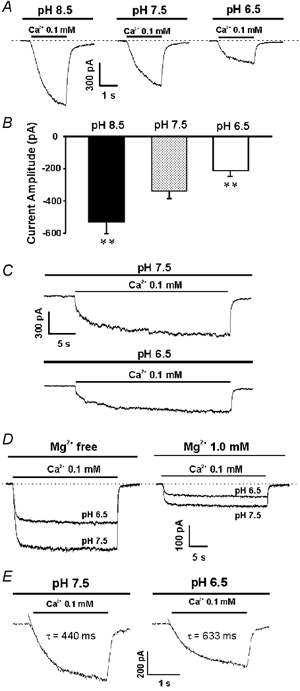
A, example traces showing the csNSC currents activated at pH 6.5, 7.5 and 8.5 in the same neuron. Neurons were perfused with different pH solutions for at least 2 min before activating the csNSC current. A decrease in pH to 6.5 inhibited the current while an increase in pH to 8.5 potentiated the current. B, summary data showing the amplitude of the csNSC currents activated at different pH values(397 ± 74 pA at pH 7.5; 206 ± 35 pA at pH 6.5; and 584 ± 87 pA at pH 8.5, n = 7). ** P < 0.01 when compared with pH 7.5 group. C, prolonged current traces demonstrate that both the rate of onset and the steady state of the csNSC current are affected by pHo. D, example traces showing the inhibition of the csNSC currents by low pH in the absence and presence of 1 mM Mg2+. E, example traces and exponential fits showing the effect of pH on the activation time constant (τ) of the csNSC currents.
To test whether changing the pH affects the steady state of the csNSC current, currents were also activated by lowering Ca2+ for a prolonged period of time, e.g. 30 s. As shown in Fig. 4C and D, both the rate of onset and the steady state of the csNSC current were affected by pHo.
To maximize the csNSC current, Mg2+ was not included in the extracellular solution in the above experiments, because it inhibits the csNSC channel (Xiong et al. 1997). To test whether the presence Mg2+ might influence the proton-induced inhibition of the csNSC channel, we also studied the effect of lowering pH on the csNSC current in the presence of 1 mM Mg2+. Although a higher concentration of Mg2+(e.g. 1.5 mM) may be considered more physiologically relevant, 1 mM was chosen since during intense synaptic excitation or in pathological conditions where [Ca2+]o decreases, a simultaneous decrease in the concentration of extracellular Mg2+ is observed (Krnjevic et al. 1982; Heinemann et al. 1990). Using 1 mM Mg2+ is therefore closer to the situations where the csNSC channels are expected to be activated.
Although the amplitude of the csNSC current decreases in the presence of Mg2+, a substantial amount of current remains. In three neurons tested, lowering the pH to 6.5 in the presence of 1 mM Mg2+ induced a 43.6 ± 4.1 % inhibition of the csNSC current (Fig. 4D), similar to the level of inhibition in the absence of Mg2+.
The effect of pH on the rate of activation of the csNSC channels was studied by comparing the time constant of the current activation at neutral (7.5) and low (6.5) pH. The time constants at both pH values were fitted by single exponentials. Although the time constant varies significantly in different cells recorded, lowering pHo in general slows down the activation of the csNSC channels. This is demonstrated by an increase in the activation time constant from 414 ± 41 ms at pH 7.5 to 679 ± 95 ms at pH 6.5 (n = 6, P < 0.05; Fig. 4E).
A concentration-response analysis demonstrated that the IC50 for proton inhibition of the csNSC current was at pH 6.2 ± 0.3 (Fig. 5, n = 4). At a physiological pH of 7.5, about 20 % of the csNSC current is inhibited by protons. This effect of protons on the csNSC channel is similar to what is observed for NMDA receptors where about 50 % of the current is inhibited at physiological pH (Traynelis & Cull-Candy, 1990).
Figure 5. Concentration-dependent inhibition of the csNSC currents by protons.
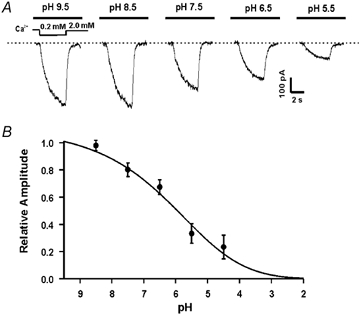
Currents were activated by a step decrease of [Ca2+]0 from 2.0 to 0.2 mM at the holding potential of −60 mV. A, representative traces taken from the same neuron demonstrate that the csNSC currents are inhibited by protons in a concentration-dependent manner. B, summary data showing the concentration-dependent effect of protons on the csNSC current. The mean IC50 for proton inhibition of the csNSC current is pH 6.2 ± 0.3 (n = 4).
Proton-induced inhibition of the csNSC currents is voltage independent
To determine whether protons act at a site within the transmembrane electric field (e.g. within the channel), we examined the voltage sensitivity of the proton-induced inhibition of the csNSC currents. Whole-cell currents activated by lowering Ca2+ from 2.0 to 0.1 mM were recorded over a range of holding potentials from −60 to +20 mV at normal or low pH. As shown in Fig. 6, whole-cell csNSC currents reversed near 0 mV at both pH 7.5 and 6.5, and the I-V relationships were near linear over the entire voltage range tested. Furthermore, decreasing pH to 6.5 inhibited the current to a similar extent at all potentials tested, illustrating a lack of voltage dependence (Fig. 6B and C). At pH 6.5, the amplitudes of the csNSC currents at −60, −40, −20 and +20 mV were decreased to 68 ± 5 %, 66 ± 6 %, 59 ± 8 % and 67 ± 9 % of the control values recorded at pH 7.5 (Fig. 6C, n = 5, P > 0.05).
Figure 6. Voltage-independent effect of pH on the csNSC current.
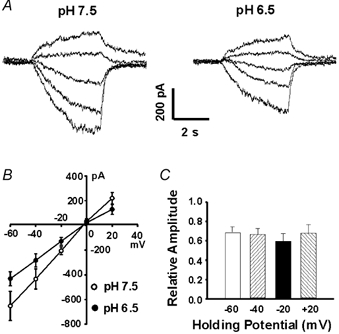
A, representative traces showing the effect of acidosis on the current–voltage relationship of the csNSC channel. Currents were activated by lowering Ca2+ from 2.0 to 0.1 mM over a range of holding potentials from −60 to +20 mV, at normal (7.5, left panel) and low (6.5, right panel) pH. B, I–V curves of the csNSC channel at pH 7.5 and 6.5. Both I–V curves display a linear relationship with reversal potentials near 0 mV. A decrease in pH to 6.5 inhibits the amplitude of the csNSC currents at all potentials. C, summary data showing the voltage-independent inhibition of the csNSC currents by pH 6.5. The amplitudes of the csNSC currents at −60, −40, −20 and +20 mV were decreased by pH 6.5 to 68 ± 5 %, 66 ± 6 %, 59 ± 8 % and 67 ± 9 % of the control currents recorded at pH 7.5, n = 5, P > 0.05.
Decreasing pH increases the potency of Ca2+ blockade
Ca2+ is an effective endogenous blocker of the csNSC channels (Xiong et al. 1997). To study the possible mechanism underlying the proton-induced inhibition of this current, we explored the possibility that changing pH may alter the potency of Ca2+ block. A concentration-inhibition curve was therefore constructed for the Ca2+ blockade of the csNSC current at physiological (7.5) or reduced pH. As shown in Fig. 7, decreasing the pH to 6.5 induced a leftward shift of the Ca2+ concentration-inhibition curve, indicating an increase in the potency of Ca2+ blockade. In addition to causing a leftward shift in the dose–inhibition curve, decreasing the pH to 6.5 also decreased the maximal current activated by lowering [Ca2+]o to 0 mM (Fig.7A), perhaps due to a direct block of the channel by protons. In contrast, increasing the pH to 8.5 induced a rightward shift in the dose–inhibition curve, consistent with a decrease in the potency of Ca2+ blockade (Fig. 7). The IC50 values for Ca2+ block of the csNSC currents were 0.12 ± 0.01 mM for pH 7.5 (n = 6), 0.08 ± 0.01 mM for pH 6.5 (n = 5) and 0.18 ± 0.02 mM for pH 8.5 (n = 7, P < 0.05 for both pH 6.5 and pH 8.5 when compared to pH 7.5). The Hill coefficients were 1.29 ± 0.12 (n = 7), 1.17 ± 0.09 (n = 6) and 0.96 ± 0.13 (n = 5) for pH 8.5, pH 7.5 and pH 6.5, respectively. Though there seems to be a trend that the Hill coefficient increases with pH increases, no significant differences were detected among different pH groups.
Figure 7. Effects of pH on the dose–inhibition relationship of Ca2+ blockade of csNSC current.
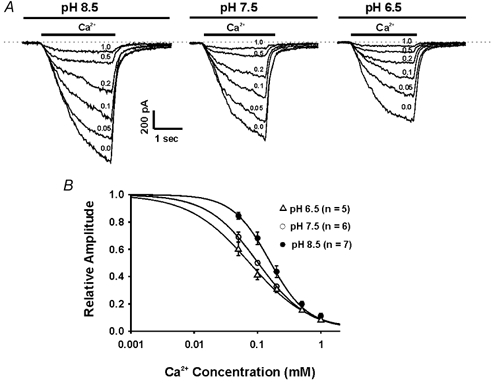
A, representative traces showing the dose-dependent Ca2+ block of the csNSC currents at pH 7.5, 6.5 and 8.5. B, summary data showing the dose–inhibition curves of Ca2+ blockade of the csNSC current at pH 6.5 (▵), 7.5 (○), and 8.5 (•). The mean IC50 values for Ca2+ blockade are 0.08 ± 0.01 (n = 5) at pH 6.5, 0.12 ± 0.01 (n = 6) at pH 7.5 and 0.16 ± 0.02 (n = 7) at pH 8.5.
Both extracellular and intracellular sites are involved in proton inhibition of the csNSC currents
Changes in extracellular pH may alter intracellular pH (Schlue & Dorner, 1992; Deitmer & Rose, 1996). To determine whether changes in intracellular pH are involved in the proton-induced modulation of the csNSC currents, we have studied the effect of quinine, an agent known to cause intracellular alkalinization (Dixon et al. 1996), on the amplitude of the csNSC currents. As shown in Fig. 8A and B, bath applications of 0.5 mM quinine for 2 min enhanced the csNSC currents by 60 ± 18 % (n = 6, P < 0.05).
Figure 8. Effects of changing intracellular pH on the csNSC current.
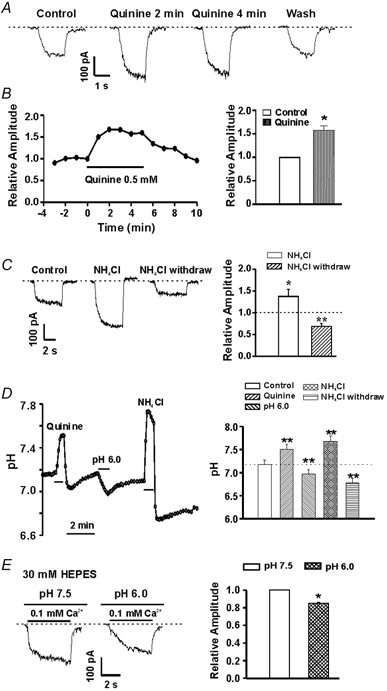
A, representative traces showing potentiation of the csNSC current by bath perfusion of quinine (0.5 mM). Currents were activated by a step decrease in [Ca2+]o from 2.0 to 0.2 mM at a holding potential of −60 mV. B, left, time course of quinine potentiation; right, summary data showing the effect of quinine on the csNSC current. Two minutes of perfusion with quinine increased the amplitude of the csNSC current by 60 ± 18 % (n = 6, P < 0.05). C, representative traces and bar graph illustrating the effect of bath perfusion of NH4Cl (15 mM) and subsequent withdrawal on amplitude of the csNSC current. NH4Cl was perfused for 3 min, followed by rapid washout of NH4Cl. The amplitude of the csNSC current was increased to 137 ± 16 % during the initial perfusion of NH4Cl and decreased to 68.8 ± 7.0 % of the control value after a 3 min washout of NH4Cl (n = 4–5, P < 0.05). D, effects of quinine, NH4Cl withdrawal and lowering pHo on intracellular pH (pHi) in cultured hippocampal neurons. pHi was measured using BCECF as pH indicator and calibrated with 10 μM nigericin–high K+ solutions (see Methods). E, example traces and summary bar graph showing the inhibition of the csNSC current by acidosis with intracellular solution containing 30 mM Hepes. * P < 0.05, ** P < 0.01, compared with the control group.
The effect of intracellular acidification was then examined using an NH4Cl pre-pulse protocol (Deitmer & Ellis, 1980). The addition and subsequent removal of NH4Cl is commonly used to alter intracellular pH (Deitmer & Ellis, 1980; Kupriyanov et al. 1999; Bonnet et al. 2000) based on the following principle: NH3, which is in equilibrium with NH4+, penetrates into the cell faster than NH4+, where it binds H+, resulting in transient alkalinization of the cytoplasm. Subsequent washout of the NH4Cl then results in a faster removal of NH3 from the cytoplasm and shifts the equilibrium (NH4+ → NH3 + H+) towards NH4+ dissociation and the release of H+, resulting in intracellular acidosis (Deitmer & Ellis, 1980). In guinea-pig hippocampal neurons, 3 min perfusion of 20 mM NH4Cl, followed by washout, induced up to a 0.5 unit decrease in intracellular pH (Bonnet et al. 2000). To study the effect of intracellular acidification on the csNSC current, 15 mM NH4Cl was perfused for 3 min and followed by washout. As shown in Fig. 8C, 3 min after the removal of NH4Cl, the amplitude of the csNSC current decreased to 68.8 ± 7.0 % of the control value (n = 5, P < 0.01). Together, these data suggest that the effect of changing pHo on the activity of the csNSC channels is probably due to a change in intracellular pH.
To gain direct evidence that bath perfusion of quinine, NH4Cl withdrawal, and changes in pHo indeed induce alterations in pHi, we have measured pHi using BCECF as a pH indicator. Neurons were loaded with the membrane-permeable form of BCECF (BCECF-AM, 2 μM) before imaging. As shown in Fig. 8D, in intact neurons, bath perfusion of quinine induced a rapid increase in pHi from 7.18 ± 0.09 to 7.51 ± 0.10 (n = 9, P < 0.01). On the other hand, lowering pHo to 6.0 caused a decrease in pHi to 6.97 ± 0.09 (n = 9, P < 0.01). Bath perfusion of NH4Cl (10 mM) and subsequent withdrawal induced a transient increase followed by a relatively long-lasting decrease in pHi to 6.78 ± 0.09 (n = 6, P < 0.01; Fig. 8D). This transient increase and subsequent decrease in pHi by NH4Cl application and withdrawal are consistent with their effect on the csNSC current (Fig. 8C). In ≈20 % of neurons, a transient post-quinine acidosis lasting for ≈1 min was observed (Fig. 8D). The reason for this acid rebound is not clear. Since the majority of cells did not show this response, the effect of this transient acidosis on the csNSC current was not further characterized.
To more closely mimic the conditions in which the csNSC currents were recorded, we also performed pHi measurements in neurons under whole-cell voltage-clamp conditions. Individual neurons were loaded with the membrane-impermeable dye BCECF (BCECF free acid, 100 μM) through the patch electrode. Hepes at 10 mM was also included in the pipette solution, as for current recording. Imaging was performed 10 min after the formation of the whole-cell configuration at a holding potential of −60 mV. As in intact neurons, addition of quinine, NH4Cl withdrawal or altering pHo still induced significant changes in pHi. In four cells tested, perfusion of quinine increased pHi from 7.28 ± 0.14 to 7.83 ± 0.20 (P < 0.01), while washout of NH4Cl decreased pHi to 7.01 ± 0.13 (P < 0.05). Similarly, changing pHo to 6.0 or 6.5 decreased pHi to 6.84 ± 0.08 and 6.93 ± 0.09 (P < 0.05), respectively. These findings indicate that 10 mM Hepes in the intracellular solution is not sufficient to eliminate the changes of pHi. Raising intracellular Hepes to 30 mM, however, largely eliminates the changes in pHi induced by quinine and NH4Cl, as well as their effects on the csNSC current (n = 3, not shown), indicating that changes in intracellular pH are involved in the modulation of the csNSC currents. Inclusion of 30 mM Hepes in the pipette solution, however, only partially reduced the inhibition of the csNSC current by low pH. In four neurons tested, lowering pHo from 7.5 to 6.0 inhibited the csNSC current from −183 ± 37 pA to −154 ± 32 pA (15 ± 8 % inhibition, n = 4, P < 0.05, Fig. 8E). This finding, together with the effects of quinine and NH4Cl, suggests that both the intracellular and extracellular site(s) are probably involved in the inhibition of the csNSC current by protons.
Decreasing pH inhibits the csNSC-mediated membrane depolarization and neuronal excitation
The effect of pH on the csNSC-mediated membrane depolarizations and the subsequent excitation of cultured hippocampal neurons was studied in the current-clamp configuration in the presence of 1 mM Mg2+. In the absence of the Na+ channel blocker TTX, the majority of cultured hippocampal neurons displayed spontaneous action potentials at a resting potential of ≈-55 mV. The frequency of these action potentials was largely dependent on the membrane potential: hyperpolarization reduced or eliminated firing while moderate depolarization increased the frequency of action potentials (not shown). The effects of acidosis on the csNSC channel-mediated responses were studied in either the absence or presence of TTX. At a holding potential of −60 mV and in the presence of TTX, lowering [Ca2+]o from 2 to 0.5 mM at pH 7.5 induced a sustained membrane depolarization with a mean value of 15.3 ± 1.7 mV (n = 6). However, when pH was lowered to 6.5, the same decrease in [Ca2+]o induced a membrane depolarization of only 9.9 ± 1.0 mV (P < 0.01; Fig. 9A and B). In the absence of TTX, decreasing Ca2+ from 2 to 0.5 mM at pH 7.5 not only induced membrane depolarization but also induced a high frequency (14.8 ± 2.6 Hz, n = 8) firing of action potentials (Fig. 9C). We previously demonstrated that this enhanced excitation is largely due to activation of the csNSC channels (Xiong et al. 1997, 2001). Decreasing pH to 6.5 not only decreased the amplitude of the membrane depolarization but also decreased the frequency of action potential firing in the presence of 0.5 Ca2+ to 4.3 ± 1.1 Hz (P < 0.01; Fig. 9C and D).
DISCUSSION
It is well known that decreases in [Ca2+]o enhance neuronal excitability (Hille, 1992). The exact mechanism underlying increased neuronal excitability is, however, not fully understood. Lowering [Ca2+]o reduces the shielding of negatively charged groups located at the membrane surface (Hille, 1992; Zhou & Jones, 1995). By this mechanism Ca2+ may influence the voltage-dependent activation of various ion channels (Hille, 1992). Calcium also alters the gating and the permeability of several voltage-gated ion channels (Zhou & Jones, 1995) and in some cases channel selectivity is lost when Ca2+ is reduced to extremely low levels. For example, Na+ will readily permeate L-type Ca2+ channels when [Ca2+]o is lowered to a nanomolar range (Almers & McCleskey, 1984; Fukushima & Hagiwara, 1985; Hess et al. 1986; Matsuda, 1986). Such low values of [Ca2+]o, however, are not expected under either physiological or pathological conditions. In contrast to these effects of extremely low concentrations of Ca2+, the csNSC channel, which is not voltage dependent, can be activated by very moderate decreases in [Ca2+]o (Xiong et al. 1997; Xiong & MacDonald, 1999). For example, in voltage-clamp recording, measurable inward currents can be detected by decreasing [Ca2+]o from 2 to 1 mM. In current-clamp recording, a threshold for membrane depolarization can be reached by a decrease in [Ca2+]o of as little as 0.1 mM (Xiong et al. 1997). These findings suggest that activation of the csNSC current and the subsequent long-lasting membrane depolarization may largely be responsible for the enhanced neuronal excitation observed in the presence of low [Ca2+]o, i.e. during seizures and periods of brain ischaemia.
In acutely dissociated rat thalamic and sensory ganglion neurons, Formenti and De Simoni have recently reported that lowering [Ca2+]o may induce inward current in cell-attached recordings by decreasing the seal resistance (Formenti & De Simoni, 2000). To test whether a similar current can be activated in cultured hippocampal neurons, we have also performed cell-attached recording and examined the effect of lowering [Ca2+]o on the seal resistance. In contrast to the study by Formenti & De Simoni (2000) using acutely dissociated rat thalamic and sensory ganglion neurons, our study in cultured hippocampal neurons demonstrated that lowering [Ca2+]o induced only a minor and insignificant change in seal conductance (e.g. from 0.120 to 0.122 nS). Such a minor shift in seal resistance is too small to account for the large amplitude of low [Ca2+]o-activated whole-cell current.
The reason for the difference between our results and those of Formenti and De Simoni is not clear. One possible explanation is that the seal resistance in our recording is much higher than the value reported by Formenti and De Simoni. In our recordings, an average seal resistance of over 8 GΩ was achieved in the presence of 2 mM CaCl2 in the bath solution, while in Formenti and De Simoni's recordings, a relatively low seal resistance of ≈1–2 GΩ was reported (Formenti & De Simoni, 2000). As described by Formenti and De Simoni, the change in seal conductance induced by lowering [Ca2+]o decreases with high resistance seals and becomes very low with seal resistances over 1GΩ. This probably explains why we do not see any obvious changes in the seal resistance on lowering [Ca2+]o in our recordings. The difference in seal resistance between our recordings and those of Formenti and De Simoni may be due to the different preparations used: cultured hippocampal neurons vs. acutely dissociated thalamic or sensory ganglion neurons. It is likely that the membrane of cultured neurons is cleaner and healthier than the acutely enzyme-dissociated neurons which would not have fully recovered from the injury caused by the dissociation process. Nevertheless, our seal resistance is closer to the values commonly recommended for high quality patch-clamp experiments.
Since it is difficult to accurately measure the seal resistance in the whole-cell configuration, it may be argued that during the formation of whole-cell patch clamps, the seal resistance is reduced by disruption of the patch-membrane. This reduction in seal resistance may make it more sensitive to changes in [Ca2+]o. To address this possibility, we have also performed perforated-patch recordings and studied the low [Ca2+]o-induced current after the membrane perforation. It is expected that if there is no physical disruption of the patch membrane seal resistance should not be affected during the formation of low access resistance by membrane perforation (Horn & Marty, 1988). Our data showing that a similar low [Ca2+]o-induced current can be activated during perforated-patch recording further suggests that the csNSC current is not due to changes in seal conductance.
In addition, the following arguments can also be made against the possibility that a change in the seal resistance is responsible for the whole-cell currents induced by lowering [Ca2+]o in our studies. (a) It is recognized that, even though lowering [Ca2+]o may induce a current by affecting the seal resistance in the cell-attached configuration, the effects of changing Ca2+ in the whole-cell configuration with a high resistance seal, where Rs >> Rm, is predominantly due to a change in membrane conductance (Formenti & De Simoni, 2000). In our recordings, seal resistances of > 5 GΩ were routinely achieved before patch excision, and the average input resistance (as a close estimation of membrane resistance) measured by a 10 mV hyperpolarization pulse from the holding potential of −60 mV is ≈200 MΩ. In this situation, the main effects observed on changing [Ca2+]o should be largely due to changes in membrane conductance, i.e. opening of ion channels. (b) Our study using the loose-patch recording technique also demonstrated that lowering [Ca2+]o can induce neuronal excitation independently of changes in the seal conductance. Together, our data strongly suggest that lowering [Ca2+]o can activate the csNSC channels independently of changes in seal conductance.
The brain normally depends on the complete oxidation of glucose, with the end-products being CO2 and H2O, for essentially all its energy requirements. During pathological conditions such as hypoxia/ischaemia, anaerobic glycolysis leads to lactic acid accumulation, causing a decrease in pH. Extracellular pH typically falls to 6.5 during ischaemia under normoglycaemic conditions, and it can fall below 6.0 during severe ischaemia or under hyperglycaemic conditions (Rehncrona, 1985; Nedergaard et al. 1991). Since acidosis co-exists with low Ca2+ in pathological conditions including epileptic seizures and brain ischaemia, it is important to know if the lowered pH influences the activity of the csNSC channels. Here we have demonstrated that the activity of the csNSC channel in cultured hippocampal neurons is indeed regulated by pHo: decreasing pHo inhibited, while increasing pHo potentiated, the responses mediated by the csNSC channels.
Since Ca2+ is an effective endogenous blocker of the csNSC channel (Xiong et al. 1997), we have explored the possibility that pH regulates the csNSC channel by modulating the potency of Ca2+. Consistent with an increase in the potency of the Ca2+ blockade, our data demonstrated that decreasing pHo caused a leftward shift in the concentration-inhibition curve. Increasing pHo, on the other hand, decreased the potency of the Ca2+ blockade. In addition to a leftward shift in the concentation-inhibition curve, our data also demonstrated that decreases in pHo caused a reduction in the maximal response mediated by the csNSC channels. This result indicates that an increase in the potency of the Ca2+ blockade may not be the only mechanism underlying proton inhibition of the csNSC channels. It is likely that protons may block the csNSC channel directly, as exemplified by proton blockade of the L-type Ca2+ channels (Chen et al. 1996) and the cardiac Kcnk3 channels (Lopes et al. 2000).
Changing extracellular pH is anticipated to alter intracellular pH (Schlue & Dorner, 1992; Deitmer & Rose, 1996). To determine whether changes in intracellular pH affect the activity of the csNSC channels, we tested the effect of quinine, an agent known to cause intracellular alkalinization (Dixon et al. 1996), and NH4Cl withdrawal, a protocol known to cause intracellular acidification (Deitmer & Ellis, 1980), on the amplitude of the csNSC-mediated currents. Our results demonstrated that an increase in intracellular pH by quinine and a decrease in intracellular pH by NH4Cl withdrawal had similar effects to an increase or decrease in pHo, respectively. pH imaging experiments clearly demonstrated that addition of quinine or removal of NH4Cl caused a corresponding increase or decrease in pHi. Similarly, decreasing pHo also induced a subsequent decrease in pHi. These results suggest that changes in pHi are involved in the modulation of the csNSC channels. Our finding that a high concentration of intracellular Hepes only partially reduced the effect of changing pHo indicates that both the intracellular and the extracellular site(s) are involved in the inhibition of the csNSC current by protons. This dual modulation of the csNSC channels by protons is similar to the proton-induced modulation of other ion channels, e.g. Kir2.3 K+ channel (Zhu et al. 1999) and a volume-sensitive K+ current (Hougaard et al. 2001).
The detailed physiological and pathological role of the csNSC channel activation is not clear at present time. It is likely that activation of the csNSC channel may play an important role in a positive feedback system during excessive neuronal depolarization, which probably contributes to the excitatory neuronal injury associated with epileptic seizures and brain ischaemia. Excitatory neuronal injury involves activation of glutamate receptors and excessive Ca2+ entry through NMDA receptor-gated channels (Choi, 1994). Membrane depolarization is an essential step in the activation of NMDA channels by relieving voltage-dependent Mg2+ blockade (Novelli et al. 1988; MacDonald & Nowak, 1990). It is likely that activation of the csNSC channels and the resultant long-lasting membrane depolarization may contribute to excitatory neuronal injury. During brain ischaemia, for example, energy deprivation initially causes dysfunction of ATP-dependent ionic pumps (e.g. Na+-K+ pump) (Siesjo, 1992). This deficiency of the Na+-K+ pump then results in an increase in [K+]o and [Na+]i. High [K+]o and [Na+]i depolarize neurons leading to activation of voltage-gated Ca2+ channels and enhanced release of glutamate which activates NMDA channels (Bittigau & Ikonomidou, 1997). Activation of voltage-gated Ca2+ channels and NMDA channels induces entry of Ca2+ into neurons leading to a decrease in [Ca2+]o. Activation of the csNSC channel by the fall in [Ca2+]o will in turn enhance membrane depolarization, further facilitating the activation of NMDA receptors and voltage-gated Ca2+ channels. Enhanced activation of NMDA receptors and voltage-gated Ca2+ channels will cause additional decreases in [Ca2+]o and greater activation of the csNSC channels. This positive feedback system may eventually contribute to loading of neurons with intolerable amounts of Ca2+, with resultant neuronal injury. Inhibition of the activity of the csNSC channels by acidosis may act to reduce this positive feedback, hence attenuating Ca2+ overload and excitatory neuronal injury.
There has been controversy as to whether the acidosis generated during ischaemia contributes to the pathology or provides beneficial actions (Tombaugh & Sapolsky, 1993). Most in vivo studies have demonstrated that acidosis aggravates ischaemic brain damage (Kristian et al. 1994; Siesjo et al. 1996). The deleterious effects of acidosis may be related to its influence on the synthesis and degradation of cellular constituents, mitochondrial function, post-ischaemic blood flow, and stimulation of pathologic free radical reactions (Rehncrona, 1985; Tombaugh & Sapolsky, 1993). In addition, profound acidosis is expected to inhibit the astrocyte glutamate uptake system which may contribute to excitatory neuronal injury (Swanson et al. 1995). Furthermore, activation of ASICs in neurons by acidosis, as shown by our own study and others, might contribute to overall membrane depolarization and neuronal injury (Zhu et al. 2001). On the other hand, several in vitro studies have suggested that acidosis may in fact be beneficial in protecting neurons from excitotoxic injury (Giffard et al. 1990; Kaku et al. 1993; Sapolsky et al. 1996). One explanation is that a decrease in extracellular pH inhibits NMDA receptor channel activity (Tang et al. 1990; Traynelis & Cull-Candy, 1990). Our finding that lowering pHo inhibits the csNSC channel with resulting attenuation of neuronal excitation may suggest a new protective role for acidosis during ischaemia and seizures. Such an effect may contribute to the neuroprotective effect of moderate acidosis on focal ischaemia (Simon et al. 1993) and epileptic brain injury (Sasahira et al. 1997). Although a simultaneous decrease in [Ca2+]o with pH may induce enhanced membrane depolarization due to potentiation of ASIC channels, this synergism is expected to last for only a few seconds due to the transient nature of the ASIC current. In contrast, the inhibition of the csNSC channels by acidosis and subsequent decrease in membrane depolarization should be long lasting and have a dominant role in reducing low [Ca2+]o-induced neuronal excitation.
The overall effect of acidosis on neuronal injury in a biological system will therefore depend on the balance between its beneficial and deleterious actions. One potential therapeutic strategy could be to minimize the detrimental effects of acidosis, e.g. activation of the proton-gated channels, while preserving its beneficial effects, e.g. inhibition of the csNSC channels.
Acknowledgments
This work was supported by the Medical Research Foundation of Oregon and the American Heart Association (Grant 0230280N), the Canadian Institutes of Health Research and the Heart and Stroke Foundation of Canada.
References
- Almers W, McCleskey EW. Non-selective conductance in calcium channels of frog muscle: calcium selectivity in a single-file pore. J Physiol. 1984;353:585–608. doi: 10.1113/jphysiol.1984.sp015352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson CJ, Eckert SP, McCleskey EW. Acid-evoked currents in cardiac sensory neurons: A possible mediator of myocardial ischemic sensation. Circ Res. 1999;84:921–928. doi: 10.1161/01.res.84.8.921. [DOI] [PubMed] [Google Scholar]
- Bittigau P, Ikonomidou C. Glutamate in neurologic diseases. J Child Neurol. 1997;12:471–485. doi: 10.1177/088307389701200802. [DOI] [PubMed] [Google Scholar]
- Bonnet U, Leniger T, Wiemann M. Moclobemide reduces intracellular pH and neuronal activity of CA3 neurones in guinea-pig hippocampal slices-implication for its neuroprotective properties. Neuropharmacol. 2000;39:2067–2074. doi: 10.1016/s0028-3908(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells. I. Acid extrusion in absence and presence of HCO3−. Am J Physiol. 1988;255:C844–856. doi: 10.1152/ajpcell.1988.255.6.C844. [DOI] [PubMed] [Google Scholar]
- Chen XH, Bezprozvanny I, Tsien RW. Molecular basis of proton block of L-type Ca2+ channels. J Gen Physiol. 1996;108:363–374. doi: 10.1085/jgp.108.5.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Calcium and excitotoxic neuronal injury. Ann NY Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- Claydon TW, Boyett MR, Sivaprasadarao A, Ishii K, Owen JM, O'Beirne HA, Leach R, Komukai K, Orchard CH. Inhibition of the K+ channel Kv1. 4 by acidosis: protonation of an extracellular histidine slows the recovery from N-type inactivation. J Physiol. 2000;526:253–264. doi: 10.1111/j.1469-7793.2000.00253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer JW, Ellis D. Interactions between the regulation of the intracellular pH and sodium activity of sheep cardiac Purkinje fibres. J Physiol. 1980;304:471–488. doi: 10.1113/jphysiol.1980.sp013337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer JW, Rose CR. pH regulation and proton signalling by glial cells. Prog Neurobiol. 1996;48:73–103. doi: 10.1016/0301-0082(95)00039-9. [DOI] [PubMed] [Google Scholar]
- Dixon DB, Takahashi K, Bieda M, Copenhagen DR. Quinine, intracellular pH and modulation of hemi-gap junctions in catfish horizontal cells. Vision Res. 1996;36:3925–3931. doi: 10.1016/s0042-6989(96)00129-0. [DOI] [PubMed] [Google Scholar]
- Ekholm A, Kristian T, Siesjo BK. Influence of hyperglycemia and of hypercapnia on cellular calcium transients during reversible brain ischemia. Exp Brain Res. 1995;104:462–466. doi: 10.1007/BF00231980. [DOI] [PubMed] [Google Scholar]
- Escoubas P, De Weille JR, Lecoq A, Diochot S, Waldmann R, Champigny G, Moinier D, Menez A, Lazdunski M. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J Biol Chem. 2000;275:25116–25121. doi: 10.1074/jbc.M003643200. [DOI] [PubMed] [Google Scholar]
- Formenti A, De Simoni A. Effects of extracellular Ca2+ on membrane and seal resistance in patch-clamped rat thalamic and sensory ganglion neurons. Neurosci Lett. 2000;279:49–52. doi: 10.1016/s0304-3940(99)00951-9. [DOI] [PubMed] [Google Scholar]
- Fukushima Y, Hagiwara S. Currents carried by monovalent cations through calcium channels in mouse neoplastic B lymphocytes. J Physiol. 1985;358:255–284. doi: 10.1113/jphysiol.1985.sp015550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffard RG, Monyer H, Christine CW, Choi DW. Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res. 1990;506:339–342. doi: 10.1016/0006-8993(90)91276-m. [DOI] [PubMed] [Google Scholar]
- Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand. 1981;113:437–445. doi: 10.1111/j.1748-1716.1981.tb06920.x. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Louvel J. Changes in [Ca2+]o and [K+]o during repetitive electrical stimulation and during pentetrazol induced seizure activity in the sensorimotor cortex of cats. Pflugers Arch. 1983;398:310–317. doi: 10.1007/BF00657240. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD, Gutnick MJ. Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res. 1977;27:237–243. doi: 10.1007/BF00235500. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Pumain R. Extracellular calcium activity changes in cat sensorimotor cortex induced by iontophoretic application of aminoacids. Exp Brain Res. 1980;40:247–250. doi: 10.1007/BF00237788. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Stabel J, Rausche G. Activity-dependent ionic changes and neuronal plasticity in rat hippocampus. Prog Brain Res. 1990;83:197–214. doi: 10.1016/s0079-6123(08)61250-9. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J Gen Physiol. 1986;88:293–319. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA, USA: Sinauer Associates, Inc.; 1992. [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougaard C, Jorgensen F, Hoffmann EK. Modulation of the volume-sensitive K+ current in Ehrlich ascites tumour cells by pH. Pflugers Arch. 2001;442:622–633. doi: 10.1007/s004240100585. [DOI] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat Neurosci. 2001;4:869–870. doi: 10.1038/nn0901-869. [DOI] [PubMed] [Google Scholar]
- Kaku DA, Giffard RG, Choi DW. Neuroprotective effects of glutamate antagonists and extracellular acidity. Science. 1993;260:1516–1518. doi: 10.1126/science.8389056. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Pulsinelli WA, Plum F. Hydrogen ion buffering during complete brain ischemia. Brain Res. 1985;342:281–290. doi: 10.1016/0006-8993(85)91127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristian T, Katsura K, Gido G, Siesjo BK. The influence of pH on cellular calcium influx during ischemia. Brain Res. 1994;641:295–302. doi: 10.1016/0006-8993(94)90158-9. [DOI] [PubMed] [Google Scholar]
- Krnjevic K, Morris ME, Reiffenstein RJ. Stimulation-evoked changes in extracellular K+ and Ca2+ in pyramidal layers of the rat's hippocampus. Can J Physiol Pharmacol. 1982;60:1643–1657. doi: 10.1139/y82-243. [DOI] [PubMed] [Google Scholar]
- Kupriyanov VV, Xiang B, Kuzio B, Deslauriers R. pH regulation of K+ efflux from myocytes in isolated rat hearts: 87Rb, 7Li, and 31P NMR studies. Am J Physiol. 1999;277:H279–289. doi: 10.1152/ajpheart.1999.277.1.H279. [DOI] [PubMed] [Google Scholar]
- Lopes CM, Gallagher PG, Buck ME, Butler MH, Goldstein SA. Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J Biol Chem. 2000;275:16969–16978. doi: 10.1074/jbc.M001948200. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Nowak LM. Mechanisms of blockade of excitatory amino acid receptor channels. Trends Pharmacol Sci. 1990;11:167–172. doi: 10.1016/0165-6147(90)90070-O. [DOI] [PubMed] [Google Scholar]
- Matsuda H. Sodium conductance in calcium channels of guinea-pig ventricular cells induced by removal of external calcium ions. Pflugers Arch. 1986;407:465–475. doi: 10.1007/BF00657502. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Lux HD. NH4Cl-induced inward currents and cytoplasmic Ca2+ transients in chick sensory neurones. Neuroreport. 1993;4:1055–1058. doi: 10.1097/00001756-199308000-00016. [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Stengl M, Flameng W. Extracellular divalent cations block a cation non-selective conductance unrelated to calcium channels in rat cardiac muscle. J Physiol. 1997;502:235–247. doi: 10.1111/j.1469-7793.1997.235bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am J Physiol. 1991;260:R581–588. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- Rehncrona S. Brain acidosis. Ann Emerg Med. 1985;14:770–776. doi: 10.1016/s0196-0644(85)80055-x. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Trafton J, Tombaugh GC. Excitotoxic neuron death, acidotic endangerment, and the paradox of acidotic protection. Adv Neurol. 1996;71:237–244. [PubMed] [Google Scholar]
- Sasahira M, Lowry T, Simon RP. Neuronal injury in experimental status epilepticus in the rat: role of acidosis. Neurosci Lett. 1997;224:177–180. doi: 10.1016/S0304-3940(97)00168-7. [DOI] [PubMed] [Google Scholar]
- Schlue WR, Dorner R. The regulation of pH in the central nervous system. Can J Physiol Pharmacol. 1992;70:S278–S285. doi: 10.1139/y92-273. [DOI] [PubMed] [Google Scholar]
- Siemkowicz E, Hansen AJ. Brain extracellular ion composition and EEG activity following 10 min ischemia in normo- and hyperglycemic rats. Stroke. 1981;12:236–240. doi: 10.1161/01.str.12.2.236. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Acidosis and ischemic brain damage. Neurochem Pathol. 1988;9:31–88. doi: 10.1007/BF03160355. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part I: Pathophysiology. J Neurosurg. 1992;77:169–184. doi: 10.3171/jns.1992.77.2.0169. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Katsura KI, Kristian T, Li PA, Siesjo P. Molecular mechanisms of acidosis-mediated damage. Acta Neurochir Suppl (Wien) 1996;66:8–14. doi: 10.1007/978-3-7091-9465-2_2. [DOI] [PubMed] [Google Scholar]
- Simon RP, Benowitz N, Hedlund R, Copeland J. Influence of the blood-brain pH gradient on brain phenobarbital uptake during status epilepticus. J Pharmacol Exp Ther. 1985;234:830–835. [PubMed] [Google Scholar]
- Simon RP, Copeland JR, Benowitz NL, Jacob P, III, Bronstein J. Brain phenobarbital uptake during prolonged status epilepticus. J Cereb Blood Flow Metab. 1987;7:783–788. doi: 10.1038/jcbfm.1987.134. [DOI] [PubMed] [Google Scholar]
- Simon RP, Niro M, Gwinn R. Brain acidosis induced by hypercarbic ventilation attenuates focal ischemic injury. J Pharmacol Exp Ther. 1993;267:1428–1431. [PubMed] [Google Scholar]
- Smith SL, Otis TS. Persistent changes in spontaneous firing of Purkinje neurons triggered by the nitric oxide signaling cascade. J Neurosci. 2003;23:367–372. doi: 10.1523/JNEUROSCI.23-02-00367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Stimulus-evoked and seizure-related responses of extracellular calcium activity in spinal cord compared to those in cerebral cortex. J Neurophysiol. 1980;44:617–632. doi: 10.1152/jn.1980.44.4.617. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Farrell K, Simon RP. Acidosis causes failure of astrocyte glutamate uptake during hypoxia. J Cereb Blood Flow Metab. 1995;15:417–424. doi: 10.1038/jcbfm.1995.52. [DOI] [PubMed] [Google Scholar]
- Tang CM, Dichter M, Morad M. Modulation of the N-methyl-D-aspartate channel by extracellular H+ Proc Natl Acad Sci U S A. 1990;87:6445–6449. doi: 10.1073/pnas.87.16.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Sapolsky RM. Evolving concepts about the role of acidosis in ischemic neuropathology. J Neurochem. 1993;61:793–803. doi: 10.1111/j.1471-4159.1993.tb03589.x. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature. 1990;345:347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Xiong Z, MacDonald JF, Weiner JL, Frazier CJ, Dunwiddie TV, Kazlauskas A, Whiting PJ, Harris RA. Platelet-derived growth factor induces a long-term inhibition of N-methyl-D-aspartate receptor function. J Biol Chem. 1996;271:16151–16159. doi: 10.1074/jbc.271.27.16151. [DOI] [PubMed] [Google Scholar]
- Varming T. Proton-gated ion channels in cultured mouse cortical neurons. Neuropharmacol. 1999;38:1875–1881. doi: 10.1016/s0028-3908(99)00079-9. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Bassilana F, De Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem. 1997a;272:20975–20978. doi: 10.1074/jbc.272.34.20975. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997b;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Lingueglia E, De Weille J, Heurteaux C, Lazdunski M. H(+)-gated cation channels. Ann NY Acad Sci. 1999;868:67–76. doi: 10.1111/j.1749-6632.1999.tb11274.x. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M. H(+)-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol. 1998;8:418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Lu W, MacDonald JF. Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc Natl Acad Sci USA. 1997;94:7012–7017. doi: 10.1073/pnas.94.13.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, Chu XP, MacDonald JF. Effect of lamotrigine on the Ca2+-sensing cation current in cultured hippocampal neurons. J Neurophysiol. 2001;86:2520–2526. doi: 10.1152/jn.2001.86.5.2520. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, MacDonald JF. Sensing of extracellular calcium by neurones. Can J Physiol Pharmacol. 1999;77:715–721. [PubMed] [Google Scholar]
- Xiong ZG, Raouf R, Lu WY, Wang LY, Orser BA, Dudek EM, Browning MD, MacDonald JF. Regulation of N-methyl-D-aspartate receptor function by constitutively active protein kinase C. Mol Pharmacol. 1998;54:1055–1063. [PubMed] [Google Scholar]
- Xiong ZQ, Stringer JL. Extracellular pH responses in CA1 and the dentate gyrus during electrical stimulation, seizure discharges, and spreading depression. J Neurophysiol. 2000;83:3519–3524. doi: 10.1152/jn.2000.83.6.3519. [DOI] [PubMed] [Google Scholar]
- Zhou W, Jones SW. Surface charge and calcium channel saturation in bullfrog sympathetic neurons. J Gen Physiol. 1995;105:441–462. doi: 10.1085/jgp.105.4.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Chanchevalap S, Cui N, Jiang C. Effects of intra- and extracellular acidifications on single channel Kir2. 3 currents. J Physiol. 1999;516:699–710. doi: 10.1111/j.1469-7793.1999.0699u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Chu X, Miesch J, Simon R, Xiong Z. Proton-gated channels are involved in acidosis-induced neuronal injury. Soc Neurosci Abstr. 2001;332:17. [Google Scholar]