

Abstract
NifH (dinitrogenase reductase) has three important roles in the nitrogenase enzyme system. In addition to its role as the obligate electron donor to dinitrogenase, NifH is required for the iron–molybdenum cofactor (FeMo-co) synthesis and apodinitrogenase maturation. We have investigated the requirement of the Fe–S cluster of NifH for these processes by preparing apoNifH. The 4Fe–4S cluster of NifH was removed by chelation of the cluster with α, α′-bipyridyl. The resulting apoNifH was tested in in vitro FeMo-co synthesis and apodinitrogenase maturation reactions and was found to function in both these processes. Thus, the presence of a redox active 4Fe–4S cluster in NifH is not required for its function in FeMo-co synthesis and in apodinitrogenase maturation. This, in turn, implies that the role of NifH in these processes is not one of electron transfer or of iron or sulfur donation.
Keywords: Azotobacter vinelandii, nitrogenase, dinitrogenase reductase, nitrogen fixation, molybdenum, iron–sulfur cluster
Nitrogenase is composed of two protein components, NifKD (dinitrogenase or MoFe protein) and NifH (dinitrogenase reductase or Fe protein). Dinitrogenase is an α2β2 tetramer encoded by nifD and nifK genes, respectively (1, 2) and it contains two distinct types of metal clusters: the P-clusters and the iron-molybdenum cofactor (FeMo-co) (3–6). NifH, an α2 dimer (1, 2), contains a single 4Fe–4S cluster which is ligated to both α-subunits through Cys-97 and Cys-132 (7). The enzyme also contains two binding sites for MgATP, one on each subunit (8). NifH has at least three distinct functions in the nitrogenase enzyme system (9). First, it serves as the electron donor to dinitrogenase. During catalysis, reduced NifH transfers electrons, one at a time, to dinitrogenase, with the concomitant hydrolysis of two molecules of MgATP (10).
The second role of NifH is as a required participant for the synthesis of the FeMo-co (11–13). It is known that the products of at least six nif genes, nifQ, nifV, nifB, nifH, nifN, and nifE (but not nifK or nifD), are involved in the biosynthesis of FeMo-co (2, 14, 15). Klebsiella pneumoniae and Azotobacter vinelandii strains with mutations in nifB, nifN or nifE produce a cofactorless apodinitrogenase that can be activated in vitro by the addition of isolated FeMo-co (2, 5).
The third role of NifH involves its participation in the maturation of apodinitrogenase (16). Apodinitrogenase refers to dinitrogenase lacking FeMo-co (but containing the P-clusters). The subunit composition of apodinitrogenase from different A. vinelandii mutants depends on the presence of NifH in those strains. The apodinitrogenase in extracts of A. vinelandii mutants carrying a deletion of nifH and in certain nifH mutants is tetrameric (α2β2) and requires the addition of NifH and MgATP for its activation by purified FeMo-co (16, 17). NifH mediates the association of the α2β2 form of apodinitrogenase with a protein referred to as γ to yield a hexameric (α2β2γ2) FeMo-co-activatable species (16). This NifH- and MgATP-dependent association of γ2 with α2β2 apodinitrogenase is termed as apodinitrogenase maturation. γ is a non-nif protein and has been shown to function as a chaperone-insertase during the biosynthesis of dinitrogenase (18).
The features of NifH that enable it to function in FeMo-co synthesis and in apodinitrogenase maturation are largely unknown. Mutations in nifH that render the protein inactive in substrate reduction do not necessarily affect its ability to function in cofactor synthesis and/or apodinitrogenase maturation. For example, an altered form of NifH in which Ala-157 is substituted by Ser, has been shown to be impaired in substrate reduction but not in FeMo-co synthesis or apodinitrogenase maturation (19, 20). Similarly, the NifH from K. pneumoniae strain UN1041 where Arg-101 is replaced by His has been shown to be greatly diminished in electron transfer to dinitrogenase but fully active in FeMo-co synthesis and apodinitrogenase maturation (21, 22). On the other hand, the NifH from A. vinelandii mutant UW97, in which Phe replaces Ser-44, is not only impaired in catalysis, but also in FeMo-co synthesis and in apodinitrogenase maturation (16, 23).
The Ala-157 → Ser form of NifH (UW91) has been shown to bind MgATP but unable to undergo the nucleotide-induced conformational change, unable to transfer electrons to dinitrogenase, and unable to hydrolyze MgATP, yet able to synthesize catalytically active dinitrogenase. This result suggests that a form of NifH that is able to bind nucleotide may be sufficient for its function in FeMo-co synthesis and apodinitrogenase maturation (20). Similarly, A. vinelandii strain DJ576 (24) produces a NifH (Asp-125 → Glu) that is impaired in MgATP hydrolysis, but contains an active dinitrogenase. The ability of these forms of NifH, impaired in catalysis (electron transfer) and with altered MgATP reactivities, to function in FeMo-co synthesis and apodinitrogenase maturation strongly suggests a role(s) for NifH independent of that in substrate reduction.
In an attempt to understand the role(s) of NifH, the requirement of the 4Fe–4S cluster of the protein for FeMo-co synthesis and apodinitrogenase maturation was investigated. Previously Howard and coworkers (24) investigated the activities of mutant A. vinelandii strains with Cys → Ser substitutions at the sites of Fe–S cluster ligation to NifH. These strains failed to accumulate active NifH or dinitrogenase, perhaps suggesting a role for the Fe–S cluster of NifH in FeMo-co synthesis or apodinitrogenase maturation. Here, we report the ability of apoNifH (NifH without the 4Fe–4S cluster) to function in both FeMo-co synthesis and apodinitrogenase maturation in vitro.
MATERIALS AND METHODS
Strains.
A. vinelandii strains DJ54 (ΔnifH; (17), CA11.1 [ΔnifHDKΔvnfDGK::spc (25)], UW45 [nifB (26)], and DJ91 [nifH C97S (27)] have been described. Growth, derepression and cell breakage have been described (28). All strains were grown in the presence of Mo and were nif derepressed.
Materials.
Sodium dithionite (DTH) was from Fluka. α,α′-bipyridyl, leupeptin, phenylmethylsulfonyl fluoride, phosphocreatine, creatine phosphokinase, homocitrate lactone, and reactive red 120 were from Sigma. ATP was purchased as a disodium salt from Sigma and was of the highest purity available. Tris base and glycine were from Fisher Scientific. N-methyl formamide was obtained from Aldrich. DEAE-cellulose was Whatman DE-52. Sephadex G-25 was a Pharmacia Biotech product. Nitrocellulose membrane was from Millipore. Acrylamide/bis acrylamide solution (37.1:1%) and the equipment for SDS/PAGE were from Bio-Rad. Ammonium tetrathiomolybdate [(NH4)2MoS4] was a gift from D. Coucouvanis (University of Michigan, Ann Arbor).
Buffers.
Tris⋅HCl (pH 7.4) (25 mM) was used through out this work. Buffers were sparged with purified N2 for at least 30 min and were evacuated and flushed with purified argon on a gassing manifold repeatedly. All buffers contained 1.7 mM DTH, unless stated otherwise. All column chromatography buffers contained the protease inhibitors, phenylmethylsulfonyl fluoride (0.2 mM) and leupeptin (0.5 μg/ml).
In Vitro Preparation of apoNifH.
ApoNifH was prepared in vitro according to a published procedure (29). Purified NifH (2 mg of protein) was incubated with 20 μmol α,α′-bipyridyl in the presence of 2.5 mM MgATP and 2 mM DTH in a total volume of 0.5 ml at 25°C for 30 min. The apoNifH was purified using a 1 cm × 10 cm Sephadex G-25 column equilibrated with 25 mM Tris⋅HCl buffer (pH 7.4) containing 2 mM DTH.
Partial Purification of Apodinitrogenase from DJ54.
Column chromatography was performed at 4°C. A 30-ml extract (≈12 mg of protein/ml) of strain of DJ54 (ΔnifH) in 25 mM Tris⋅HCl (pH 7.4) was applied to a reactive red agarose column (1 cm × 21 cm) that had been equilibrated with 25 mM Tris⋅HCl. The column was further washed with two column volumes of equilibration buffer, and apodinitrogenase was eluted using 400 mM NaCl in 25 mM Tris⋅HCl buffer containing 20% glycerol. The fractions containing apodinitrogenase were collected anaerobically, and FeMo-co insertion assays were performed as described below.
FeMo-co Insertion into Apodinitrogenase and Isolation of Dinitrogenase.
A 6-ml of extract (≈12 mg of protein/ml) of strain DJ54 (ΔnifH) served as a source of α2β2 apodinitrogenase and was incubated with purified NifH or apoNifH (0.2 mg of protein), 1.5 ml of an ATP-regenerating mixture (containing 3.6 mM ATP/6.3 mM MgCl2/51 mM phosphocreatine/20 units/ml creatine phosphokinase/6.3 mM DTH) and 300 μl of a solution containing FeMo-co (equivalent to 13 nmol of Mo) for 30 min at 30°C. The mixtures were then applied to two identical DEAE cellulose columns (1 cm × 10 cm) that had been equilibrated with 100 mM NaCl in 25 mM Tris⋅HCl buffer containing 20% glycerol. The columns were further washed with 12 ml of column buffer to remove any unbound FeMo-co, and dinitrogenase was eluted with 200 mM NaCl in 25 mM Tris⋅HCl buffer containing 20% glycerol. The fractions were collected anaerobically and 25-μl aliquots were assayed for dinitrogenase activity by addition of excess NifH (0.1 mg of protein) and 0.8 ml of an ATP-regenerating mixture (as described above). Nitrogenase activity was quantitated by acetylene reduction as described (30).
NifH- and MgATP-Dependent Maturation of Tetrameric (α2β2) Apodinitrogenase.
Nine-milliliter serum vials were evacuated and flushed repeatedly with purified argon and rinsed with anaerobic Tris⋅HCl. The following were then added to appropriate vials: 100 μl of anaerobic Tris⋅HCl or 100 μl of an ATP-regenerating mixture (as described above); a source of apodinitrogenase and purified NifH (45 μg of protein) or apoNifH (25 μg of protein) in a total volume of 500 μl. The mixtures were incubated at 30°C for 30 min, after which they were placed on ice. Aliquots (20 μl) were subjected to anoxic native gel electrophoresis followed by immunoblotting using anti-γ antibody, as described below.
Activation of Apodinitrogenase with Purified FeMo-co (FeMo-co Insertion Assay).
FeMo-co was prepared in N-methyl formamide as described (5). The amount of FeMo-co used was in excess of the amount of apodinitrogenase in the assays as determined previously by titration. Nine-milliliter serum vials were evacuated and flushed repeatedly with purified argon and rinsed with anaerobic Tris⋅HCl. The following were then added to appropriate vials: 100 μl of Tris⋅HCl or 100 μl of ATP-regenerating mixture (as described above), 10 μl of a solution containing FeMo-co (equivalent to 0.43 nmol of Mo), 200 μl of desalted extract (≈1.8 mg of protein) of strain DJ54 (ΔnifH) as a source of α2β2 apodinitrogenase and 45 μg of purified NifH or 25 μg of apoNifH in a total volume of 510 μl. The mixtures were incubated for 30 min at 30°C after which 40 nmol of (NH4)2MoS4 were added to prevent further insertion of FeMo-co into apodinitrogenase (31). The vials were incubated for 10 min at room temperature, and 0.8 ml of ATP-regenerating mixture and an excess of purified NifH (0.1 mg of protein) was added to the assay mixtures. Nitrogenase activity was then quantitated by acetylene reduction as described (30).
In Vitro Synthesis of FeMo-co (FeMo-co Synthesis Assay).
FeMo-co synthesis reactions were carried out as described by Shah et al. (30). To 9-ml serum vials flushed with purified argon and rinsed with anaerobic Tris⋅HCl were added the following: 100 μl of anaerobic Tris⋅HCl, 20 μl of 5 mM homocitrate that had been treated with base to cleave the lactone (pH 8.0), 10 μl of 1 mM sodium molybdate, 200 μl of Tris⋅HCl or 200 μl of ATP-regenerating mixture (as described above), 200 μl of extract (≈1.8 mg of protein) of strain DJ54 (ΔnifH, as a source of NifNE, NifB-co, α2β2 apodinitrogenase, and γ2), and purified NifH (45 μg of protein) or apoNifH (25 μg of protein). The vials were incubated at 30°C for 30 min. A total of 10 nmol of (NH4)2MoS4 were added to the assay mixtures to prevent further synthesis of FeMo-co and its insertion into apodinitrogenase and the mixtures were incubated for 10 min at room temperature. An excess of NifH (0.1 mg of protein), 0.8 ml of the ATP-regenerating mixture (as previously described) and 0.5 ml of C2H2 were then added to the vials. The acetylene reduction reactions were stopped by the addition of 0.1 ml of 4 M NaOH after an incubation of 30 min at 30°C and C2H4 formed was quantitated by gas chromatography.
Visualization of γ Monomerization on Anaerobic Native Gels as an Indication of FeMo-co Synthesis.
FeMo-co synthesis assays were carried out as described above, except that 200 μl of desalted extract (≈2.5 mg of protein) of strain CA11.1 (ΔnifHDKΔvnfDGK::spc) was used as a source of the components required for FeMo-co synthesis. The vials were incubated for 30 min at 30°C, after which they were placed on ice. Aliquots (20-μl) of the reactions were subjected to anoxic native gel electrophoresis followed by immunoblotting using anti-γ antibody, as described below.
Anaerobic Native Gel Electrophoresis.
Proteins were separated on anaerobic native gels with a 7–14% acrylamide and 0–20% sucrose gradient as described (16).
Antibodies and Immunoblot Analysis.
Antibody to γ was a gift from Mary Homer and Gary P. Roberts (University of Wisconsin, Madison). Antibody to NifH from A. vinelandii was raised in rabbits. The protocols for immunoblotting and developing with modifications by Brandner et al. (32) have been described.
Protein Assays.
Protein concentrations were determined by the bicinchoninic acid method using BSA as standard (33).
RESULTS AND DISCUSSION
FeMo-co Activation of Tetrameric (α2β2) Apodinitrogenase in Extracts of A. vinelandii Strain DJ54 by apoNifH.
It has been previously reported that both NifH and MgATP are required for the insertion of FeMo-co into apodinitrogenase obtained from nifH mutants (16, 17). One role of NifH in the FeMo-co insertion process is in the formation of the hexameric apodinitrogenase containing γ2 (16). Both FeMo-co insertion assays and anoxic native PAGE were used to test if apoNifH prepared in vitro could function in the association of the free γ2 with the α2β2 apodinitrogenase in extracts of strain DJ54 (ΔnifH). The extracts of strain DJ54 were incubated with purified FeMo-co in the presence of NifH or apoNifH and in the presence or absence of MgATP, as described in Materials and Methods. The results of FeMo-co insertion assays are shown in Table 1. The data indicate that the apodinitrogenase in extracts of strain DJ54 can be activated by apoNifH and that this activation is MgATP-dependent. The reactions that contained apoNifH in the FeMo-co insertion phase showed activity comparable to those that contained NifH, indicating the competence of the apo-enzyme in the maturation process.
To confirm the formation of the α2β2γ2 apodinitrogenase species, anoxic, native PAGE was employed to monitor the actual association of the free γ2 with α2β2 apodinitrogenase in the presence and absence of nucleotide and in the presence of either purified NifH or apoNifH. A fraction enriched in α2β2 apodinitrogenase and γ2 was prepared from an extract of A. vinelandii strain DJ54 by chromatography on reactive red 120. This fraction was used in apodinitrogenase maturation reactions as described in Materials and Methods. Fig. 1 is the immunoblot of an anoxic native gel, developed with antibody to γ. The figure illustrates that apoNifH functions in the processing of α2β2 apodinitrogenase to a α2β2γ2 species. The reaction containing apoNifH shows similarity to the one containing NifH in that the reactions are dependent on the presence of MgATP (compare lanes 3 and 4). Similar results were obtained when the same set of reactions were repeated with extracts of DJ54 (data not shown). The level of γ associated with apodinitrogenase in reactions containing no nucleotide and/or NifH represents the background levels of cross reactivity at the apodinitrogenase position. Previous studies have shown the presence of multiple bands at the position of α2β2γ2 apodinitrogenase. The role of nucleotide in apodinitrogenase maturation is unclear. The functional binding site for MgATP during apodinitrogenase maturation might be on NifH, γ, or apodinitrogenase. Although the presence of nucleotide binding sites in NifH and dinitrogenase has been documented (8, 34), the ability of γ to bind nucleotide has not yet been tested.
Figure 1.
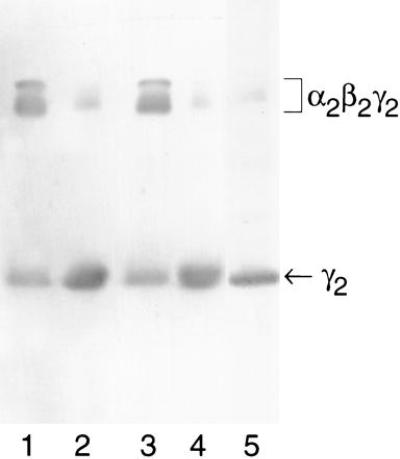
Immunoblot of an anoxic native gel developed with antibody to γ, illustrating the apoNifH- and MgATP-dependent association of γ2 with the α2β2 apodinitrogenase. FeMo-co insertion reactions containing a fraction enriched in the α2β2 apodinitrogenase and γ2 from DJ54 were subjected to anoxic native PAGE as described. Lanes: 1, reaction including NifH and MgATP; 2, reaction including NifH and excluding MgATP; 3, reaction including apoNifH and MgATP; 4, reaction including apoNifH and excluding MgATP; 5, reaction excluding NifH and MgATP.
Maturation of Tetrameric (α2β2) Apodinitrogenase to a FeMo-co Activatable Species (α2β2γ2) by apoNifH.
Another approach to investigate if apoNifH could function in the maturation of α2β2 apodinitrogenase was to test for dinitrogenase formed by the insertion of FeMo-co into apodinitrogenase. The α2β2 apodinitrogenase is not FeMo-co activatable, whereas the α2β2γ2 form is amenable to FeMo-co insertion. The formation of the α2β2γ2 apodinitrogenase species was tested in an indirect manner: by the isolation of catalytically active dinitrogenase formed by FeMo-co insertion into the newly matured apodinitrogenase. Purified NifH or apoNifH was used in apodinitrogenase maturation reactions containing an extract of strain DJ54 (as a source of α2β2 apodinitrogenase and γ2), MgATP, and purified FeMo-co, as described in Materials and Methods. After a 30-min incubation each mixture was applied to a DE-52 column. Fractions containing dinitrogenase were collected anaerobically from each column and were assayed for acetylene reduction activity following the addition of an excess of NifH and an ATP-regenerating mixture. The results of these assays are presented in Table 2. These data clearly indicate the synthesis of catalytically active dinitrogenase upon incubation of the extract with either NifH or apoNifH in presence of MgATP and purified FeMo-co. The amount of dinitrogenase synthesized in presence of apoNifH is comparable to that synthesized in presence of NifH, indicating the competence of apo-enzyme in the apodinitrogenase maturation process.
The actual step(s) requiring NifH during the process of apodinitrogenase maturation remains unknown. A complex formation between NifH and apodinitrogenase or between NifH and γ could be envisioned as a prerequisite for this process. However, the present study indicates that the presence of the 4Fe–4S cluster of NifH is not required for its function in the maturation of apodinitrogenase. Thus, a redox role for NifH during apodinitrogenase maturation can be ruled out.
Ability of apoNifH to Function in FeMo-co Synthesis.
Another function of NifH in the nitrogenase enzyme system is its indispensable role in the synthesis of FeMo-co. In vitro FeMo-co synthesis assays and anoxic PAGE were used to test the requirement of the 4Fe–4S cluster of NifH for its function in this process. The results of this study are shown in Table 3 and Fig. 2. ApoNifH was used in the in vitro FeMo-co synthesis assay in the place of NifH. The in vitro FeMo-co synthesis reaction contained the extract of strain DJ54, homocitrate, molybdate, MgATP, and a source of NifH as described in Materials and Methods. The results in Table 3 indicate the ability of apoNifH to function in the process of FeMo-co synthesis. The reactions containing apoNifH in the FeMo-co synthesis phase of the assay typically exhibited ≈50–60% of the activity exhibited by reactions containing NifH during the synthesis phase. The reason for this lower activity is unknown. The removal of the 4Fe–4S cluster might render the protein less reactive or less stable and thereby less efficient in cofactor synthesis.
Figure 2.
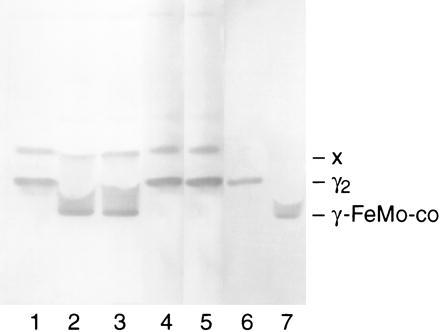
Immunoblot of an anoxic native gel developed with antibody to γ, illustrating γ monomerization upon binding FeMo-co. FeMo-co synthesis reactions were performed as described and subjected to anoxic native PAGE. Lanes: 1, reaction excluding NifH; 2, reaction including NifH; 3, reaction including apoNifH; 4, reaction including NifH and excluding MgATP; 5, reaction including apoNifH and excluding MgATP; 6, purified γ2; 7, purified γ2 plus excess purified FeMo-co.
FeMo-co synthesis can also be detected by monitoring the migration of γ on anoxic native gels. It has been reported by Homer et al. (18) that γ exists as a dimer (γ2) in cell extracts of mutants interrupted in the FeMo-co biosynthetic pathway. Upon in vitro synthesis of the cofactor or upon addition of purified FeMo-co to such extracts that also lack the structural genes for apodinitrogenase (nifK and nifD), γ has been shown to bind FeMo-co and monomerize (γ-FeMo-co) and consequently migrate at a different position on anoxic, native gels than the γ2. This provides a convenient, alternate assay for the synthesis of FeMo-co, whereby the shift in the position of γ correlates with FeMo-co synthesis; the position of γ in the gel is detected using anti-γ antibodies. FeMo-co synthesis assays were performed using desalted extracts of strain CA11.1 (ΔnifHDKΔvnfDGK::spc) in presence and absence of MgATP and in presence of either NifH or apoNifH, as described in Materials and Methods. The results of this study are presented in Fig. 2. γ in the extract of strain CA11.1 is a dimer (γ2) because the strain is impaired in FeMo-co synthesis (Fig. 2, lane 1). Upon addition of NifH to the reaction mixture (containing all components required for FeMo-co synthesis), FeMo-co is synthesized and accumulates on γ. γ bound to FeMo-co (γ-FeMo-co) electrophoreses as a faster migrating band on anoxic native gels and can be clearly distinguished from the slower migrating γ2 species (Fig. 2, compare lanes 1 and 2). γ in reactions that contained apoNifH also electrophoreses as the faster migrating band on anoxic native gels, indicating the presence of newly synthesized FeMo-co (Fig. 2, lane 3). Also, the reactions containing apoNifH show similarity to those containing NifH in that, MgATP is required for FeMo-co synthesis and consequently for the monomerization of γ (Fig. 2, lanes 4 and 5). A slower migrating species of γ (designated as “X”) is observed in extracts of CA11.1 which is yet uncharacterized.
The results of the above study demonstrate clearly the ability of apoNifH to function in the process of FeMo-co synthesis. Though the exact reaction(s) catalyzed by NifH during FeMo-co synthesis remains unknown, the experiments described here show that electron transfer by NifH is not required during FeMo-co synthesis because the 4Fe–4S deficient form is competent in this process. This rules out a redox role for NifH during FeMo-co synthesis. However, a role for NifH involving its binding to other protein components during the cofactor biosynthetic process, for example, NifNE or γ, cannot be ruled out.
Confirmation That holoNifH Is Not Reconstituted During in Vitro Assays.
Because we wish to demonstrate that apoNifH possesses the ability to function in FeMo-co synthesis and apodinitrogenase maturation, it is critical to show that there is no holoNifH present in our apo-enzyme preparation either before or during the course of the in vitro assays. At least three lines of evidence indicate that there are neither traces of NifH in the apo-enzyme preparation nor is there a regeneration of the 4Fe–4S cluster when apoNifH is added into extracts of strains DJ54 (ΔnifH) and CA11.1 (ΔnifHDKΔvnfDGK::spc).
First, the removal of the 4Fe–4S cluster to entirety is evidenced by a complete loss of substrate reduction activity (Table 4). To test if reconstitution of a catalytically active Fe–S center occurs upon addition of the apo-enzyme to cell extracts, apoNifH was incubated with extracts of either strain DJ54 or strain CA11.1, as described in Materials and Methods. Acetylene reduction activity of apoNifH was also tested in the presence of low levels of dinitrogenase in the reaction mixture because C2H2 reduction is inhibited at high ratios of dinitrogenase to NifH (35). The results presented in Table 4 clearly indicate that the fraction containing apoNifH does not contain catalytic amounts of NifH. Moreover, the inability of apoNifH, preincubated with extracts of strains DJ54 and CA11.1, to reduce C2H2 indicates that no regeneration of a catalytically active 4Fe–4S cluster in the apo-enzyme has taken place. The acetylene reduction assay is extremely sensitive and we would have easily detected 0.5 μg of active NifH in our assays. These results are compelling evidence for the absence of a catalytically active Fe–S cluster in apoNifH even after incubation of the apo-enzyme with extracts of strains containing deletions of the nifH gene.
Second, the apoNifH preparation was tested for the presence of iron by subjecting increasing concentrations of both purified NifH and the apoNifH to native, anaerobic gel electrophoresis, and staining the gel for iron (data not shown) according to a published procedure (36). Lanes containing apoNifH did not show the presence of iron while the lanes containing NifH showed the presence of iron-stained bands. The apoNifH preparation was also tested for iron content by inductively coupled plasma emission spectrometry and was found to contain less than 0.02 ppm of Fe, which was the background level of Fe found in the column equilibration buffer.
The third line of evidence showing that traces of NifH are not present in the apo-enzyme preparation is the absence of ADP ribosylated form of NifH by dinitrogenase reductase ADP ribosyl transferase (DRAT). ApoNifH is not a substrate for DRAT, whereas NifH is a substrate for the enzyme (37, 38). ADP ribosylation reactions of dinitrogenase reductase, apoNifH, and aliquots of apoNifH incubated with extracts of strains DJ54 and CA11.1 were carried out as described by Lowery and Ludden (38) and were analyzed for the presence of the ADP ribosylated species (data not shown). The apoNifH, by itself or upon incubation with extracts of strains DJ54 and CA11.1, did not show the presence of the modified species. This further confirms the absence of catalytic amounts of NifH in the apo-enzyme preparation and is evidence for the absence of regeneration of a calytically active Fe–S cluster upon incubation of the apo-enzyme with cell extracts of DJ54 and CA11.1.
The other concern we would like to address here is the potential for regeneration of the 4Fe–4S cluster by a NifS-catalyzed reaction. The A. vinelandii nifS gene product, NifS, has been shown to be involved in the activation of apoNifH by catalyzing the reconstitution of the 4Fe–4S cluster into the apo-protein in vitro in the presence of l-cysteine, ferrous ion, DTT, and MgATP (29). The reconstitution of the cluster into the in vitro formed apoNifH when the apo-enzyme is incubated with cell extracts during FeMo-co synthesis or apodinitrogenase maturation assays by a NifS-catalyzed reaction is highly improbable since the activity of NifS is inhibited by DTH (29), and all manipulations were carried out in presence of at least 1.7 mM DTH and in the absence of any added cysteine.
Because apoNifH, prepared in vitro was competent in the processes of apodinitrogenase maturation and FeMo-co synthesis, it was of considerable interest to investigate if a strain containing the apo-form of NifH could accumulate catalytically active dinitrogenase. The A. vinelandii strain DJ91 in which Cys-97 that ligates the 4Fe–4S cluster in NifH is substituted by serine (27) has been reported to lack a functional 4Fe–4S cluster. Thus the strain DJ91 contains an apo-form of NifH in vivo due to its inability to incorporate a functional 4Fe–4S cluster into the enzyme. However, it has been reported that the Cys-97 → Ser form of NifH cannot participate in FeMo-co synthesis (27). Our results with the strain DJ91 also showed that this strain of A. vinelandii did not accumulate mature dinitrogenase and thus we agree with the observations made by Howard et al. (27). Several reasons could exist for the inability of strain DJ91 to perform FeMo-co synthesis. To test if NifH in DJ91 was in its native dimeric state, we subjected the extract of strain DJ91 to anoxic native PAGE, followed by immunoblotting and developing with anti-NifH antibody (data not shown). The immunoblot showed no cross reactivity to the antibody, suggesting that this form of NifH may be unstable or more susceptible to degradation. On the other hand, the apo-form of NifH prepared in vitro by the action of Fe chelators like α,α′-bipyridyl and o-bathophenanthrolinedisulfonate is believed to be a dimer on the basis of gel filtration experiments and is believed to retain its ability to bind two molecules of MgATP, like the holo form (27). This ability of the in vitro formed apoNifH to retain its dimeric nature could be an important factor that enables it to function in FeMo-co synthesis and apodinitrogenase maturation.
CONCLUSION
The action of NifH during FeMo-co synthesis has long been suspected to be one of either electron transfer or one of Fe or S donation (13, 39). The high degree of similarity between the nifN and nifK sequences and the nifE and nifD sequences (40) and thus between NifNE and dinitrogenase, has lead to suggestions that the role of NifH in FeMo-co synthesis may be that of electron donation to NifNE, similar to its role in catalysis. Studies using 35S-labeled NifH suggest that the enzyme does not donate acid-labile sulfide to FeMo-co synthesized in vitro (13). Further, Allen et al. (41) have shown the metabolic product of NifB, NifB-co, serves as a specific Fe and S donor for FeMo-co synthesis. In this study, we have shown that the apo-form of NifH, though completely inactive in substrate reduction, is capable of functioning in the synthesis of FeMo-co and in the maturation of apodinitrogenase. Identification of specific regions in the protein important in these processes will be made possible by a more thorough study involving site specifically altered forms of NifH. The use of purified FeMo-co activation and FeMo-co synthesis systems should help elucidate the roles of dinitrogenase reductase, MgATP and γ in these processes.
Table 1.
Activation of apodinitrogenase in extracts of DJ54 by apoNifH
| System | Activity* |
|---|---|
| Complete† | 13.1 |
| Minus MgATP‡ | 0.8 |
| Minus NifH§ plus apoNifH | 11.6 |
| Minus NifH plus apoNifH minus MgATP | 1.0 |
Expressed as nmol of C2H4 formed per min/mg cell-extract protein. Values are the averages of duplicate assays performed separately.
FeMo-co insertion assays were performed as described. A complete system contained 0.2 ml of extract of strain DJ54 (1.8 mg of protein), purified NifH (45 μg of protein), MgATP-regenerating mixture, and an excess of purified FeMo-co in NMF (equivalent to 13 nmol of Mo).
A “minus MgATP” system contained all the components of the FeMo-co insertion assay, except that the MgATP-regenerating mixture was replaced by 0.1 ml of anaerobic Tris⋅HCl buffer.
The reaction contained apoNifH (25 μg of protein) during the FeMo-co insertion phase.
Table 2.
Synthesis of dinitrogenase in extracts of strain DJ54 incubated with NifH or apoNifH
The reaction mixture contained 6 ml of extract (≈12 mg of protein/ml) of strain DJ54 preincubated with an excess of purified FeMo-co N-methyl-formamide (equivalent to 13 nmol of Mo), 1.5 ml of an ATP-regenerating mixture, and purified NifH or apoNifH, as described.
A 25-μl aliquot of the fraction (0.21 mg of protein) was tested for dinitrogenase activity by the addition of excess purified NifH (0.1 mg of protein), ATP-regenerating mixture, and 0.5 ml C2H2. Activity is expressed as nmol of C2H4 formed per min/mg protein.
The reaction mixture was similar to the one described above except that it contained no source of NifH in the preincubation phase.
Table 3.
Ability of ApoNifH to function in in vitro FeMo-co synthesis
Expressed as nmol C2H4 formed per min/mg protein. Values are the averages of duplicate assays performed separately.
In vitro FeMo-co synthesis assays were performed as described. A complete system contained 0.1 ml anaerobic Tris⋅HCl buffer, 20 μl of 5 mM homocitrate, 10 μl of 1 mM sodium molybdate, 0.2 ml of an ATP-regenerating mixture, 0.2 ml extract of strain DJ54, and purified NifH (45 μg of protein). At the end of the 30-min incubation, 10 nmol of (NH4)2MoS4 was added and the substrate reduction phase of the assay was initiated by the addition of excess ATP-regenerating mixture, NifH (0.1 mg of protein), and C2H2.
The in vitro FeMo-co synthesis assay contained apoNifH (25 μg of protein) during the cofactor synthesis phase of the assay. After the 30-min incubation period, 10 nmol of (NH4)2MoS4 was added and the substrate reduction phase was carried out as described above.
Table 4.
Activities of dinitrogenase complemented with NifH or apoNifH
| Form of NifH* | Pretreatment† | Concentration of dinitrogenase, μg of protein | Activity‡ |
|---|---|---|---|
| NifH | None | 0.0 | <0.01 |
| NifH | None | 50.0 | 50.3 |
| NifH | None | 5.0 | 6.3 |
| ApoNifH | None | 50.0 | <0.01 |
| 5.0 | <0.01 | ||
| ApoNifH | Plus extract of | 50.0 | <0.01 |
| strain CA11.1 | 5.0 | <0.01 | |
| ApoNifH | Plus extract of | 50.0 | <0.01 |
| strain DJ54 | 5.0 | <0.01 |
Assays contained purified NifH or apoNifH (0.1 mg of protein) with high (50.0 μg of protein) or low (5.0 μg of protein) concentrations of purified dinitrogenase.
ApoNifH (0.1 mg of protein) was preincubated with 0.2 ml of extracts of either DJ54 or CA11.1 for 30 min at 30°C, after which the appropriate concentration of purified dinitrogenase was added.
Expressed as nmol C2H4 formed per minute.
Acknowledgments
We thank Gary Roberts, Tracey Foulger, and Carmen Rüttimann-Johnson for critically reading this manuscript and for helpful discussions. We also thank Sandra Grunwald for providing purified DRAT. Special thanks to Gary Roberts and Mary Homer for providing γ antibody to us. We also thank the University of Wisconsin Soil and Plant Analysis lab for the inductively coupled plasma analyses. This research was supported by National Institutes of Health Grant GM35332-11 to P.W.L.
ABBREVIATIONS
- DTH
sodium dithionite
- FeMo-co
iron-molybdenum cofactor
- DRAT
dinitrogenase reductase ADP ribosyl transferase
- NifH
dinitrogenase reductase
References
- 1.Burris R H. J Biol Chem. 1991;266:9339–9342. [PubMed] [Google Scholar]
- 2.Roberts G P, MacNeil T, MacNeil D, Brill W J. J Bacteriol. 1978;136:267–279. doi: 10.1128/jb.136.1.267-279.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Madden M S, Kindon N D, Ludden P W, Shah V K. Proc Natl Acad Sci USA. 1990;87:6517–6521. doi: 10.1073/pnas.87.17.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imperial J, Hoover T R, Madden M S, Ludden P W, Shah V K. Biochemistry. 1989;28:7796–7799. doi: 10.1021/bi00445a040. [DOI] [PubMed] [Google Scholar]
- 5.Shah V K, Brill W J. Proc Natl Acad Sci USA. 1977;74:3249–3253. doi: 10.1073/pnas.74.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawkes T R, McLean P A, Smith B E. Biochem J. 1984;217:317–321. doi: 10.1042/bj2170317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hausinger R P, Howard J. J Biol Chem. 1983;258:13486–13492. [PubMed] [Google Scholar]
- 8.Tso M Y W, Burris R H. Biochim Biophys Acta. 1973;309:263–270. doi: 10.1016/0005-2744(73)90024-7. [DOI] [PubMed] [Google Scholar]
- 9.Allen R M, Chatterjee R, Madden M S, Ludden P W, Shah V K. Crit Rev Biotechnol. 1994;14:225–249. doi: 10.3109/07388554409079834. [DOI] [PubMed] [Google Scholar]
- 10.Ljones T, Burris R H. Biochim Biophys Acta. 1972;275:93–101. doi: 10.1016/0005-2728(72)90027-8. [DOI] [PubMed] [Google Scholar]
- 11.Robinson A C, Dean D R, Burgess B K. J Biol Chem. 1987;262:14327–14332. [PubMed] [Google Scholar]
- 12.Filler W A, Kemp R M, Ng J C, Hawkes T R, Dixon R A, Smith B E. Eur J Biochem. 1986;160:371–377. doi: 10.1111/j.1432-1033.1986.tb09981.x. [DOI] [PubMed] [Google Scholar]
- 13.Shah V K, Hoover T R, Imperial J, Paustian T D, Roberts G P, Ludden P W. In: Role of nif Gene Products and Homocitrate in the Biosynthesis of the Iron-Molybdenum Cofactor. Bothe H, de Bruijn F J, Newton W E, editors; Bothe H, de Bruijn F J, Newton W E, editors. Cologne: Gustav Fischer; 1988. pp. 115–120. [Google Scholar]
- 14.Imperial J, Ugalde R A, Shah V K, Brill W J. J Bacteriol. 1984;158:187–194. doi: 10.1128/jb.158.1.187-194.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoover T R, Shah V K, Roberts G P, Ludden P W. J Bacteriol. 1986;167:999–1003. doi: 10.1128/jb.167.3.999-1003.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen R M, Homer M J, Chatterjee R, Ludden P W, Roberts G P, Shah V K. J Biol Chem. 1993;268:23670–23674. [PubMed] [Google Scholar]
- 17.Robinson A C, Chun T W, Li J-G, Burgess B K. J Biol Chem. 1989;264:10088–10095. [PubMed] [Google Scholar]
- 18.Homer M J, Dean D R, Roberts G P. J Biol Chem. 1995;270:24745–24752. doi: 10.1074/jbc.270.42.24745. [DOI] [PubMed] [Google Scholar]
- 19.Shah V K, Davis L C, Gordon J K, Orme-Johnson W H, Brill W J. Biochim Biophys Acta. 1973;292:246–255. doi: 10.1016/0005-2728(73)90269-7. [DOI] [PubMed] [Google Scholar]
- 20.Gavini N, Burgess B K. J Biol Chem. 1992;267:21179–21186. [PubMed] [Google Scholar]
- 21.Wolle D, Kim C, Dean D, Howard J B. J Biol Chem. 1992;267:3667–3673. [PubMed] [Google Scholar]
- 22.Lowery R G, Chang C L, Davis L C, McKenna M C, Stephens P J, Ludden P W. Biochemistry. 1989;28:1206–1212. doi: 10.1021/bi00429a038. [DOI] [PubMed] [Google Scholar]
- 23.Pulakat L, Hausman B S, Gavini N. J Biol Chem. 1996;271:1884–1889. doi: 10.1074/jbc.271.4.1884. [DOI] [PubMed] [Google Scholar]
- 24.Wolle D, Dean D R, Howard J B. Science. 1992;258:992–995. doi: 10.1126/science.1359643. [DOI] [PubMed] [Google Scholar]
- 25.Waugh S I, Paulsen D M, Mylona P V, Maynard R H, Premakumar R, Bishop P E. J Bacteriol. 1995;177:15505–15510. doi: 10.1128/jb.177.6.1505-1510.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joerger R D, Bishop P E. J Bacteriol. 1988;170:1475–1487. doi: 10.1128/jb.170.4.1475-1487.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howard J B, Davis R, Mouldenhauer B, Cash V L, Dean D R. J Biol Chem. 1989;264:11270–11274. [PubMed] [Google Scholar]
- 28.Shah V K, Davis L C, Brill W J. Biochim Biophys Acta. 1972;256:498–511. doi: 10.1016/0005-2728(72)90078-3. [DOI] [PubMed] [Google Scholar]
- 29.Zheng L, Dean D R. J Biol Chem. 1994;269:18723–18726. [PubMed] [Google Scholar]
- 30.Shah V K, Imperial J, Ugalde R A, Ludden P W, Brill W J. Proc Natl Acad Sci USA. 1985;83:1636–1640. doi: 10.1073/pnas.83.6.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah V K, Ugalde R A, Imperial J, Brill W J. J Biol Chem. 1985;260:3891–3894. [PubMed] [Google Scholar]
- 32.Brandner J P, McEwan A G, Kaplan S, Donohue T. J Bacteriol. 1989;171:360–368. doi: 10.1128/jb.171.1.360-368.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith P K, Krohn R I, Hermanson A K, Mallia A K, Gartner F H, Provezano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk D C. Anal Biochem. 1985;150:175–179. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 34.Miller R W, Robson R L, Yates M G, Eady R R. Can J Biochem. 1980;58:542–548. doi: 10.1139/o80-074. [DOI] [PubMed] [Google Scholar]
- 35.Shah V K, Davis L C, Brill W J. Biochim Biophys Acta. 1971;256:498–511. doi: 10.1016/0005-2728(72)90078-3. [DOI] [PubMed] [Google Scholar]
- 36.Kuo C H, Fridovich I. Anal Biochem. 1988;170:183–185. doi: 10.1016/0003-2697(88)90106-6. [DOI] [PubMed] [Google Scholar]
- 37.Grunwald S G, Ludden P W. J Bacteriol. 1997;179:3277–3283. doi: 10.1128/jb.179.10.3277-3283.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowery R G, Ludden P W. J Biol Chem. 1988;263:16714–16719. [PubMed] [Google Scholar]
- 39.Burgess B K. Chem Rev. 1991;90:1377–1406. [Google Scholar]
- 40.Brigle K E, Setterquist R A, Dean D R, Cantwell J S, Weiss M C, Newton W E. Proc Natl Acad Sci USA. 1987;84:7066–7069. doi: 10.1073/pnas.84.20.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen R M, Chatterjee R, Ludden P W, Shah V K. J Biol Chem. 1995;270:26890–26896. doi: 10.1074/jbc.270.45.26890. [DOI] [PubMed] [Google Scholar]