

Abstract
Mutations in KCNE2, which encodes the minK-related protein 1 (MiRP1), are associated with an increased risk of arrhythmias; however, the underlying mechanisms are unknown. MiRP1 is thought to associate with many K+ channel α-subunits, including HERG K+ channels, which have a major role in suppressing arrhythmias initiated by premature beats. In this study we have investigated in chinese hamster ovary (CHO) cells at 37 °C the effects of co-expressing HERG K+ channels with either wild-type (WT) MiRP1 or one of three mutant MiRP1 subunits, T8A, Q9E and M54T. The most significant effects of MiRP1 subunits on HERG channels were a more negative steady-state activation for HERG + T8A MiRP1 and a more positive steady-state activation for HERG + M54T MiRP1 compared to either HERG + WT MiRP1 or HERG alone. All three mutants caused a significant slowing of deactivation at depolarised potentials. T8A MiRP1 also caused an acceleration of inactivation and recovery from inactivation compared to HERG + WT MiRP1. During ventricular action potential clamp experiments there was a significant decrease in current in the early phases of the action potential for HERG + WT MiRP1 channels compared to HERG alone. This effect was not as prominent for the mutant MiRP1 subunits. During premature action potential clamp protocols, the T8A and Q9E mutants, but not the M54T mutant, resulted in significantly larger current spikes during closely coupled premature beats, compared to HERG + WT MiRP1. At longer coupling intervals, all three mutants resulted in larger current spikes than HERG alone or HERG + WT MiRP1 channels. It is therefore possible that augmentation of HERG currents in the early diastolic period may be pro-arrhythmic.
MinK-related protein 1 (MiRP1) is a 123 amino acid protein with a single membrane-spanning domain, encoded by the KCNE2 gene (Abbott et al. 1999). MiRP1 has been shown to associate with multiple cardiac ion channel α-subunits, including HERG K+ channels (Abbott et al. 1999), Kv4.2 channels (Zhang et al. 2001), HCN2 channels (Yu et al. 2001) and KCNQ1 channels (Tinel et al. 2000). Furthermore, the expression of MiRP1 in the heart has recently been definitively shown at the protein level (Jiang et al. 2003), confirming that it is likely to be an important regulator of cardiac electrophysiology.
Mutations in KCNE2 are associated with congenital long QT syndrome (LQTS; Abbott et al. 1999), a condition characterised by prolongation of the ventricular action potential (AP) and a markedly increased risk of lethal ventricular arrhythmias. There are at least six genetic subtypes of LQTS (Keating & Sanguinetti, 2001). KCNE2 was the sixth genetic locus found to be associated with LQTS, hence these cases are referred to as LQTS type 6. The mechanism(s) by which mutations in MiRP1 increase the risk of ventricular arrhythmias, however, is uncertain. Loss of function (Curran et al. 1995), as well as gain of function (Lees-Miller et al. 2000), mutations in HERG K+ channels are a well established cause of LQTS. Additionally, HERG K+ channels are the molecular targets for a wide range of drugs that cause drug-induced LQTS (Vandenberg et al. 2001). Therefore most work on the possible mechanisms of pro-arrhythmia of MiRP1 mutations has focused on the modulation of HERG K+ channels.
MiRP1 co-immunoprecipitates with HERG when the two subunits are co-expressed in CHO cells (Abbott et al. 1999). Furthermore, co-expression of MiRP1 with HERG in Xenopus oocytes results in an acceleration of deactivation, a reduction in single channel conductance compared to HERG channels expressed alone (Abbott et al. 1999). Co-expression of MiRP1 also alters the HERG K+ channel sensitivity to drugs (Abbott et al. 1999). The hypothesis that increased drug sensitivity could explain the pro-arrhythmic effect of MiRP1 mutations (Abbott et al. 1999) was subsequently supported by the finding that a subgroup of patients with drug-induced LQTS had a polymorphism in the KCNE2 gene (Sesti et al. 2000). Weepura et al. (2002), however have questioned the role of MiRP1 as a β-subunit for HERG K+ channels. They also found that co-expression of MiRP1 did not alter the drug sensitivity of HERG K+ channels when expressed in a mammalian cell line (Weerapura et al. 2002). More recently, Isbrandt et al. (2002) identified a new MiRP1 mutation, V65M, associated with congenital LQTS that results in acceleration of inactivation of HERG K+ channels, further supporting the possible role of MiRP1 in modulating the kinetics of HERG K+ channels. In this study we have therefore re-examined the possible role of mutant MiRP1 subunits in modulating the kinetics of HERG K+ channels.
The voltage dependence of ion channel activation and inactivation kinetics is most easily studied in transfected cell lines or Xenopus oocytes using square pulse voltage clamp techniques. The behaviour of channels during physiological voltage waveforms, such as ventricular APs, has then been reconstructed by mathematical modelling (Noble et al. 1998). An alternative, and more direct approach, has been the use of AP clamp protocols to record ion channel activity in response to voltage clamp waveforms resembling in vivo APs. This approach has the advantage that it is independent of any assumptions that are inherent in any modelling approach (Noble et al. 1998). This technique has been particularly useful for studying HERG K+ channels (Hancox et al. 1998; Zhou et al. 1998; Lu et al. 2001).
In this study we have used AP clamp techniques to characterise at 37 °C the effects of WT and mutant MiRP1 subunits co-expressed with HERG K+ channels in CHO cells. We show that three MiRP1 mutations, T8A, Q9E and M54T, identified in patients with LQTS (Abbott et al. 1999), have variable effects on the kinetics of HERG K+ channels. Furthermore, the mutant MiRP1 subunits have significant effects on the currents recorded during closely coupled premature beats, and this may contribute to the increased risk of arrhythmias seen in patients with these mutant MiRP1 subunits.
METHODS
Cloning and cell culture
A CHO cell line stably expressing HERG (CHOHERG+) was created by transfecting cells with HERG cDNA subcloned into the pIRES-Neo vector (a kind gift from Gail Robertson, Department of Physiology, University of Wisconsin, USA) and selecting G418 (Life Technologies, CA, USA)-resistant colonies. hMiRP1 cDNA was cloned by RT-PCR from RNA extracted from human ventricular tissue (obtained from the Papworth Hospital Human Tissue Bank, Papworth, UK). The T8A, Q9E and M54T mutants (Abbott et al. 1999) were introduced via PCR using mismatched primers (see Table 1) and the Expand high fidelity PCR system (Roche, Switzerland). All clones were verified by sequencing and subcloned into the pTSV40 vector (Invitrogen, UK) so that the MiRP1 subunit would be co-expressed with enhanced green fluorescent protein (eGFP) from the same plasmid, thereby ensuring that the cells expressing eGFP were also expressing MiRP1. CHOHERG+ cells were transiently transfected with 1.8 μg of WT or mutant hMiRP1 and 5 μl of LipofectAMINE (Life Technology, CA, USA). Transfected cells were identified by detection of eGFP fluorescence prior to electrophysiological recordings as previously described (Lu et al. 2001). In HERG alone experiments, an empty pTSV40 vector was transfected into the CHOHERG+ cells.
Table 1.
Primers used for sequential PCR reactions
| Primers | Sequence |
|---|---|
| Flanking primers | F ATTTAGGTGACACTATAGAATA |
| R GAATGTAATACGACTCACTATA | |
| MiRP1 T8A primers | R GTCTGTGCGAAATTGGATAAAG |
| F AATTTCGCACAGACGCTGGAAG | |
| MiRP1 Q9E primers | R GCGTCTCTGTGAAATTGGATAA |
| F TTCACAGAGACGCTGGAAGACG | |
| MiRP1 M54T primers | R ATCACCGTGAGGTACAGGATGA |
| F TACCTCACGGTGATGATTGGAA |
Mutant nucleotides are underlined. First round PCRs were performed using the forward flanking primer-reverse mutant primer and the reverse flanking primer-forward mutant primer. The resultant ‘megaprimers’ were used in the second round PCR. The final PCR products were cloned into the pTSV40 vector and sequenced. F, forward; R, reverse.
Electrophysiology
Electrophysiological experiments were performed as previously described (Lu et al. 2001). Briefly, cells were superfused with Normal Tyrode solution (mM): 129 NaCl, 5 sodium pyruvate, 5 sodium acetate, 4 KCl, 1 MgCl2, 1.8 CaCl2, 11.1 glucose and 5 Hepes (titrated to pH 7.4 with NaOH) at 37 °C. The patch pipette solution contained (mM): 140 KCl, 1 MgCl2, 5 MgATP, 10 EGTA and 5 Hepes (pH 7.4 with KOH). The measured liquid : liquid junction potential between the internal and external solutions was -3 mV, which has not been corrected for. All experiments were conducted at 37 ± 1 °C. Current recordings were made using an Axopatch 200B amplifier (Axon Instruments, CA, USA). Capacitance current transients were electronically subtracted. Series resistance was typically 3-4 MΩ, which was compensated by ≈70 %. The largest currents were ≈5 nA, which would have resulted in a maximum voltage error of ≈5 mV, although for most experiments this error was < 2 mV. Acquisition and analysis of data were performed using pClamp6 software (Axon Instruments). All summary data were analysed using Microsoft Excel and values are expressed as means ± S.E.M.
Electrophysiology protocols
Steady-state activation was measured from tail currents recorded at -50 mV following 3 s steps to voltages in the range -70 mV to +40 mV. Data were fitted with a Boltzmann function of the form:
where g is conductance, gmax is the maximum conductance, Vt is the test voltage, V1/2 is the mid-point of activation and k is the slope factor
Steady-state inactivation was measured from instantaneous tail currents recorded at potentials in the range -120 mV to +60 mV following a 3 s depolarisation step to +20 mV using the ‘rectification factor’ method (Sanguinetti et al. 1995). Rectification factors, R, were calculated according to the formula:
where G is the maximal conductance of HERG, n is the activation variable at +20 mV (which was assumed to be 1), Vt is the test voltage and Vrev is the reversal potential. R values were then fitted to a Boltzmann distribution (see above).
Rates of activation were measured using an envelope of tails protocol. Briefly, cells were depolarised to potentials in the range 0 to +60 mV for variable durations and then stepped to -50 mV. Rate constants were obtained by fitting a single exponential to the envelope of peak tail currents for each voltage, as previously described (Lu et al. 2001). Rates of deactivation were obtained by fitting double exponential curves to the decaying phase of tail currents in the range -40 to -100 mV. Rates of inactivation were obtained by fitting a single exponential to currents recorded during double pulse protocols consisting of a depolarisation to +20 mv for 3 s, repolarisation to -80 mV for 10 ms then stepping to voltages in the range -20 to +60 mV. Rates of recovery from inactivation were obtained from the same pulse protocol used to measure deactivation (see above) and fitting a single exponential to the initial portion of the tail current (see Fig. 3).
Figure 3. Effect of T8A MiRP1 subunits on inactivation of HERG.
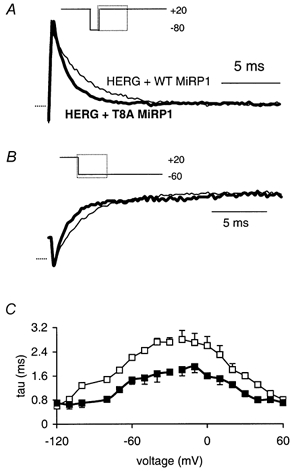
A, inactivation at +20 mV for HERG + WT MiRP1 (thin trace) and HERG + T8A MiRP1 (thick trace). B, recovery from inactivation at -60 mV for HERG + WT MiRP1 (thin trace) and HERG + T8A MiRP1 (thick trace). C, summary of voltage dependence of rates of inactivation for HERG + WT (□) compared to HERG + MiRP1 T8A (▪).
Action potential clamp protocols
Ventricular AP waveforms were derived from Oxsoft Heart 4.8 (Noble, 1999). APs were scaled to give a resting membrane potential of -85 mV, a maximum overshoot potential of +40 mV, a maximum upstroke velocity of 125 V s−1 and a duration at 90 % repolarisation (APD90) of 350 ms, similar to that for human ventricular endocardial APs (Peeters et al. 1995). The digitised AP waveforms were converted to pClamp6 format for use as voltage clamp waveforms using the DacFile command in pClamp6.
RESULTS
Steady-state activation and inactivation
The voltage dependence of steady-state activation was measured from tail currents recorded at -50 mV following 3 s steps to voltages in the range -60 to +40 mV and the data fitted using a Boltzmann function. The midpoint of the steady-state activation curve (V1/2) was significantly shifted in a negative direction from -32.3 ± 1.4 mV for HERG alone (n = 6) to -36 ± 1.0 mV for HERG + WT MiRP1 (n = 5, P < 0.05, ANOVA; Fig. 1A). The V1/2 of steady-state activation was also significantly shifted to the left by T8A MiRP1 (-43 ± 2.4 mV, n = 4) or to the right for M54T MiRP1 (-26 ± 2.3 mV, n = 6), compared to HERG alone or HERG + WT MiRP1. The V1/2 for Q9E MiRP1 (-34 ± 0.7 mV, n = 5) was not significantly different from either HERG alone or HERG + WT MiRP1.
Figure 1. Effect of mutant MiRP1 subunits on steady-state activation of HERG.
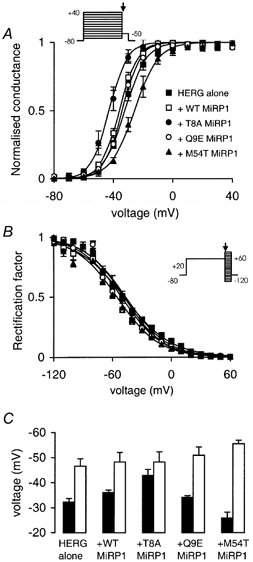
Steady-state activation (A) and steady-state inactivation (B) curves for HERG alone (▪) and HERG co-expressed with WT (□), T8A (•), Q9E (○) or M54T (▴) MiRP1 subunits. The protocols used to record currents for activation and inactivation are illustrated in the insets (see text for further details). C, summary of the V1/2 of activation (▪) and inactivation (□) for HERG alone and HERG expressed with WT or mutant MiRP1 subunits. Data are means ± S.E.M. (n = 4-7).
The voltage dependence of steady-state inactivation was measured from instantaneous tail currents recorded at potentials in the range -120 to +60 mV following a 3 s depolarisation step to +20 mV using the ‘rectification factor’ method (Sanguinetti et al. 1995). The co-expression of WT, T8A, or Q9E MiRP1 with HERG did not significantly alter the voltage dependence of steady-state inactivation (Fig. 1B). M54T MiRP1 did, however, cause a small but significant negative shift in the V1/2 for inactivation (from -45.6 ± 2.9 mV, n = 4, for HERG alone to -55.4 ± 1.4 mV, n = 6, for HERG + M54T MiRP1, P < 0.05). The V1/2 values for activation and inactivation are summarised in Fig. 1C. For HERG alone, as well as HERG + WT or mutant MiRP1 subunits, the V1/2 of activation was less negative than the V1/2 of inactivation. Thus the voltage range for which channels will be open at steady state, i.e. not inactive and not closed, is small. In addition, as the V1/2 values for activation and inactivation get further apart the voltage range for which channels will be open at steady state, the so-called window current gets smaller. Thus, from Fig. 1C it can be seen that the window current will be smallest for HERG + M54T MiRP1 and largest for HERG + T8A MiRP1.
Effects of MiRP1 on kinetics of HERG K+ channels
The presence of WT or mutant MiRP1 subunits did not affect the rates of activation of HERG K+ channels for voltages in the range 0 to +60 mV, measured using an envelope of tails protocol (Lu et al. 2001; as previously described). The co-expression of the different MiRP1 isoforms, however, had small but significant effects on the rates of deactivation, inactivation and recovery from inactivation (see below).
Deactivation of HERG is voltage dependent and shows two clearly defined components, a rapid phase with a time constant of 10-100 ms and a slower phase with a time constant of up to many seconds (Zhou et al. 1998). Deactivation was significantly slower for the HERG + M54T MiRP1 channels compared to HERG alone or HERG + WT MiRP1 (Fig. 2). Furthermore, a significantly larger fraction of HERG + M54T MiRP1 current deactivation was represented by the slow component at depolarised potentials compared to HERG alone (Table 2). WT MiRP1, T8A MiRP1 and Q9E MiRP1 also had significant effects on the voltage dependence of the fraction of current deactivating by the fast pathway at depolarised potentials (Table 2) although the rate constants themselves were not altered (data not shown).
Figure 2. Effect of M54T MiRP1 subunits on deactivation of HERG.
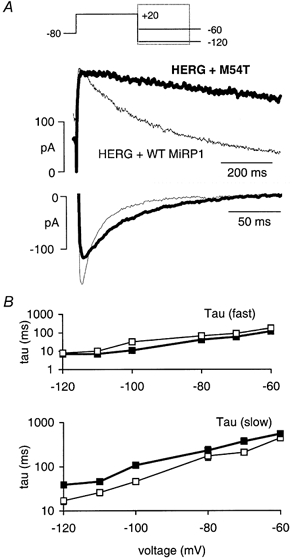
A, deactivation at -60 and -120 mV for HERG + WT MiRP1 (thin trace) and HERG + M54T MiRP1 (thick trace). B, voltage dependence of the rate constants of deactivation (plotted on a semi-logarithmic scale) for HERG + WT and HERG + M54T MiRP1.
Table 2.
Voltage dependence of Af/(Af + As) of deactivation
| −120 mV | −110 mV | −100 mV | −80 mV | −70 mV | −60 mV | |
|---|---|---|---|---|---|---|
| HERG | 0.65 ± 0.13 | 0.61 ± 0.09 | 0.76 ± 0.09 | 0.78 ± 0.02 | 0.53 ± 0.02 | 0.78 ± 0.11 |
| WT | 0.63 ± 0.08 | 0.67 ± 0.04 | 0.67 ± 0.11 | 0.62 ± 0.05† | 0.44 ± 0.02† | 0.29 ± 0.03† |
| T8A | 0.80 ± 0.04* | 0.71 ± 0.05 | 0.70 ± 0.06 | 0.40 ± 0.11* | 0.18 ± 0.03* | 0.17 ± 0.01* |
| Q9E | 0.75 ± 0.01 | 0.80 ± 0.05* | 0.76 ± 0.03 | 0.47 ± 0.06* | 0.27 ± 0.05* | 0.24 ± 0.05 |
| M54T | 0.70 ± 0.03 | 0.65 ± 0.08 | 0.71 ± 0.10 | 0.40 ± 0.07* | 0.19 ± 0.02* | 0.17 ± 0.02* |
Af and As are the magnitudes of the fast and slow components of deactivation, respectively.
P < 0.05 (ANOVA) HERG + mutant MiRP1 compared to HERG + WT MiRP1.
P < 0.05 (ANOVA) HERG alone compared to HERG + WT MiRP1.
The most marked differences in the rates of inactivation and recovery from inactivation were between HERG + WT MiRP1 channels and HERG + T8A MiRP1 channels (Fig. 3). There were also significant, although smaller, differences in the rates of inactivation and recovery from inactivation for the other channels. For example, the rates of inactivation at +20 mV were 1.3 ± 0.1, 1.2 ± 0.2, 1.6 ± 0.3, 1.8 ± 0.2 and 2.1 ± 0.1 ms (all n ≥ 4) for HERG + T8A, HERG alone, HERG + Q9E, HERG + WT and HERG + M54T, respectively, and the rates of recovery from inactivation at -60 mV were 1.5 ± 0.1, 1.3 ± 0.1, 1.7 ± 0.2, 2.2 ± 0.1 and 1.8 ± 0.1 ms. The rates of inactivation and recovery from inactivation were significantly faster for HERG + T8A MiRP1 and HERG alone compared to HERG + WT MiRP1 at all voltages tested. The rates of inactivation and recovery from inactivation for HERG + Q9E MiRP1 were generally faster than for HERG + WT MiRP1 and HERG + M54T MiRP1, although these differences were only significant in the voltage range -10 to -30 mV.
Action potential clamp studies
A typical example of a current trace recorded from a CHOHERG+ cell in response to a ventricular AP waveform is shown (grey line) in Fig. 4. The increase in current with progressive repolarisation reflects the recovery of channels from the inactive into the open state during repolarisation (Lu et al. 2001), with the subsequent decrease representing the net effect of a decrease in electrochemical driving force for K+ efflux and channel deactivation as well as continuing recovery from inactivation. Transient transfection of CHOHERG+ cells with WT and mutant MiRP1 subunits had subtle effects on the current changes during ventricular AP waveforms (black traces in each of the four panels in Fig. 4). Due to different levels of channel density in different experiments, currents were normalised relative to the maximum current in each cell. The traces in Fig. 4 therefore give an indication of the profile of current change during a ventricular AP waveform for each channel combination. The most significant change seen was during the early stages of the AP for HERG + WT MiRP1 compared to HERG alone (Fig. 4A). Due to the overlapping effects of recovery from inactivation, deactivation and the decrease in electrochemical driving force which results in smaller currents during terminal repolarisation it is difficult to discern significant differences in currents between HERG alone and HERG + WT or mutant MiRP1 subunits during the later phases of the AP. Subtle differences in current during terminal repolarisation can however be very important and so we probed this further using double AP pulse protocols (Lu et al. 2001).
Figure 4. Effect of mutant MiRP1 subunits on HERG currents recorded during ventricular AP waveforms.
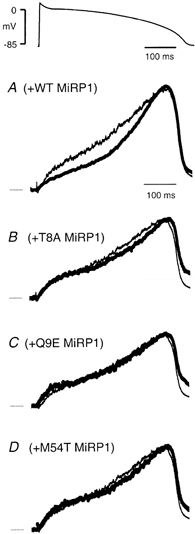
Typical current trace recorded during a ventricular AP waveform for HERG alone (thin trace) compared to: HERG + WT MiRP1 (A), HERG + T8A MiRP1 (B), HERG + Q9E MiRP1 (C), and HERG + M54T MiRP1 (D). All currents were normalised relative to the maximum current recorded during the AP waveform. Dotted lines indicate zero current line.
Effects of premature stimulation on MiRP1 mutants
During double pulse stimulation protocols there is a large transient increase in outward current at the onset of the second depolarisation (Fig. 5A). This large increase in current occurs because the HERG channels have recovered from inactivation into the open state during the repolarisation phase but have not yet deactivated, and thus a second depolarisation, which results in a large increase in the electrochemical driving force for K+ efflux, results in a large increase in current. The channels, however, rapidly inactivate again, hence this large outward current is only transient. The variation of the magnitude of the transient increase in outward current as the coupling interval between the two depolarisations is changed is determined by the rates of recovery from inactivation and subsequent deactivation of the channels (Lu et al. 2001). As different MiRP1 isoforms have different effects on inactivation and deactivation, we investigated how these differences would affect the response of HERG K+ channels to premature AP stimuli. Typical families of current traces for HERG, with and without WT or T8A MiRP1 subunits, recorded during successive coupled pairs of APs separated by progressively longer time intervals are illustrated in Fig. 5A. At the shortest coupling intervals shown, APD90 - 20 ms, the size of the transient increase in outward current during the second pulse is relatively small. As the coupling interval is lengthened, the magnitude of the current spike increases, with the largest currents occurring for coupling intervals of APD90 or APD90 + 10 ms. For coupling intervals > APD90 + 10 ms the magnitude of the current spike declines as the coupling interval increases.
Figure 5. Effects of mutant MiRP1 subunits on HERG currents recorded during premature ventricular AP waveforms.
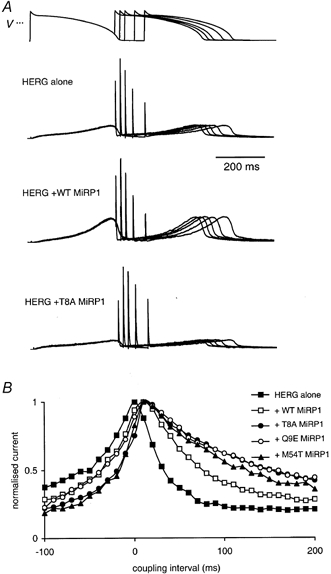
A, typical examples of currents recorded from HERG alone, HERG + WT MiRP1 and HERG + T8A MiRP1, during premature ventricular AP clamp protocols where the premature stimulus was introduced at the point of APD90 - 20 ms, APD90, APD90 + 20 ms, APD90 + 50 ms and APD90 + 100 ms (voltage waveform shown above). All current traces have been normalised to the maximum current during the premature depolarisation. B, the magnitude of the transient outward current recorded during premature ventricular AP clamp protocols plotted versus the coupling interval for the premature beat for HERG alone (▪, n = 4), and HERG co-expressed with WT (□, n = 3), T8A (•, n = 3), Q9E (○, n = 3) or M54T (▴, n = 3) MiRP1 subunits. Current magnitude has been normalised to the maximum transient outward current response. The coupling interval was defined relative to APD90 of the ventricular AP waveform, which in these experiments was 300 ms. For purposes of clarity, error bars are not shown.
The mean data for the magnitude of the outward current spikes as the coupling interval was increased from APD90 - 100 ms to APD90 + 200 ms are illustrated in Fig. 5B. Co-expression of WT or mutant MiRP1 resulted in the coupling interval for the peak current spike occurring at APD90 + 10 ms (n ≥ 3) compared to APD90 for HERG alone (n = 7, P < 0.05). The magnitude of the current spike was reduced at longer coupling intervals but this decay was less marked in the case of the HERG + mutant MiRP1 channels compared to either HERG + WT MiRP1 or HERG alone.
It is difficult to compare current densities from one experiment to another and from one channel subunit combination to another due to difficulties in controlling transfection efficiency between experiments. Furthermore, differences in activation, inactivation and deactivation will all influence peak current densities measured during AP waveforms (see Discussion). Nevertheless, it is important to consider the effects that mutant MiRP1 subunits may have on HERG current density under voltage clamp conditions similar to those that the channels will experience in vivo. We therefore looked at relative peak current densities during premature AP protocols by normalising the peak transient current during the second depolarisation (Iprem) to the peak current recorded during the preceeding AP (IAP; Fig. 6). The value of the peak ratio Iprem/IAP for all subunit combinations is plotted in Fig. 6A. The peak Iprem/IAP was significantly larger for HERG + T8A MiRP1 and HERG + Q9E MiRP1 compared to the value for HERG alone or HERG + WT MiRP1 (P < 0.05, ANOVA). Conversely, the value of the peak ratio Iprem/IAP for HERG + M54T MiRP1 was not significantly different to that for either HERG alone or HERG + WT MiRP1.
Figure 6. Summary of effects of mutant MiRP1 subunits on HERG currents recorded during premature ventricular AP waveforms.
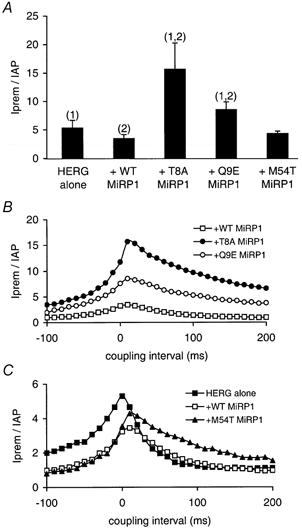
A, the magnitude of the peak outward current spike recorded during premature ventricular AP clamp protocols (Iprem) normalised to the peak current recorded during the preceding AP waveform (IAP). Numbers above the bars indicate (1) significantly different from HERG + WT MiRP1 (ANOVA, P < 0.05) and (2) significantly different from HERG alone (ANOVA, P < 0.05). B and C, the normalised values of the peak current (Iprem/IAP) recorded during premature ventricular AP clamp protocols plotted versus the coupling interval for the premature beat: HERG co-expressed with WT (□, n = 3), T8A (•, n = 3) or Q9E (○, n = 3)MiRP1 subunits (B); and HERG co-expressed with WT (□, n = 3), HERG alone (▪, n = 4) or M54T (▴, n = 3) MiRP1 subunits (C). The coupling interval was defined relative to APD90 of the ventricular AP waveform, which in these experiments was 300 ms. For purposes of clarity, error bars are not shown.
The values of the ratio Iprem/IAP recorded at all coupling intervals for HERG alone and HERG + WT or mutant MiRP1 subunits are plotted in Fig. 6B and C. From Fig. 6B it is apparent that the magnitude of the ratio Iprem/IAP was significantly larger for HERG + T8A MiRP1 and HERG + Q9E MiRP1 compared to HERG + WT MiRP1 at all coupling intervals. The ratio Iprem/IAP was significantly larger for HERG alone compared to HERG + WT MiRP1 only at short coupling intervals (Fig. 6C) whereas the ratio Iprem/IAP for HERG + M54T MiRP1 was only larger than that for HERG + WT MiRP1 at long coupling intervals (Fig. 6C).
DISCUSSION
Mutations in MiRP1 are associated with an increased risk of cardiac arrhythmias (Abbott et al. 1999). The mechanisms underlying the increased risk of arrhythmias are not fully resolved although most interest has focused on the possible modification of HERG K+ channels (Abbott et al. 1999, 2001; Cui et al. 2000; Sesti et al. 2000; Mazhari et al. 2001). In this study we report that three LQTS6-associated MiRP1 mutations (T8A, Q9E and M54T; Abbott et al. 1999) have variable effects on HERG K+ channel kinetics but they all affect the response of HERG K+ channels to premature AP stimuli, a known pro-arrhythmic stimulus in patients with LQTS (Benhorin & Medina, 1997).
Kinetics of channel gating
Previous reports on the effects of MiRP1 on the voltage dependence of activation are quite variable. Abbott et al. (1999) reported a positive shift in activation V1/2 following co-expression of WT MiRP1 (studied in Xenopus oocytes), whilst Weepura et al. (2002) reported a negative shift (studied in CHO cells at 35-37 °C) and Mazhari et al. (2001) and Cui et al. (2000) found no change (studied in HEK 293 and CHO cells, respectively, at room temperature). Our results are most similar to those of Weepura et al. (2002), although we found a smaller shift with co-expression of MiRP1, -3.7 mV, compared to the -10 mV in the previous study. Similar to Abbott et al. (1999), we found that the T8A mutant MiRP1 caused a significant negative shift in the V1/2 of activation. However, we found that M54T caused a significant positive shift in activation V1/2, whereas they reported a significant positive shift for Q9E but not M54T. The reasons for these discrepancies are uncertain.
WT MiRP1 does not affect the voltage dependence of steady-state inactivation (Fig. 1B; Abbott et al. 1999; Mazhari et al. 2001). In this study we report for the first time the effects of mutant MiRP1 subunits on the voltage dependence of steady-state inactivation. Neither T8A nor Q9E MiRP1 subunits affected the voltage dependence of steady-state inactivation; however, M54T MiRP1 subunits did cause a small but significant negative shift in the V1/2 of steady-state inactivation (Fig. 1B).
Mazhari et al. (2001) reported that WT MiRP1 accelerated the rates of inactivation and recovery from inactivation. This contrasts with the data presented here, where we found that co-expression of WT MiRP1 resulted in a small but significant slowing of inactivation and recovery from inactivation. The most significant differences between the experimental conditions used here and in the study by Mazhari et al. (2001) are: temperature, 37 °C compared to room temperature; and cell lines used, CHO cells compared to HEK 293 cells, respectively. Temperature certainly has profound effects on the rates of HERG channel gating (Zhou et al. 1998). Conversely, it is possible that there may be different proteins expressed in CHO cells compared to HEK 293 cells that could affect the results. We were not able to detect expression of any MinK-related peptides in CHO cells using RT-PCR with degenerate primers. Such an approach cannot definitively exclude the presence of MiRPs in the CHO cell line but it is unlikely. Therefore we suspect the most likely explanation for the difference between our study and that of Mazhari et al. (2001) is the difference in temperature.
WT MiRP1 has been reported to cause an acceleration of deactivation at potentials in the range -120 to -90 mV (Abbott et al. 1999; Mazhari et al. 2001; Weerapura et al. 2002). Furthermore, in the case of the M54T mutant the rate constant for the fast deactivation component was reduced even further (Abbott et al. 1999). Both Weepura et al. (2002) and Mazhari et al. (2001), however, reported that there was a slowing of deactivation at more depolarised potentials, although the differences did not reach statistical significance. We found that both WT and mutant MiRP1 subunits had a tendency towards faster deactivation at very negative potentials but a slowing of deactivation at more positive potentials, and this was primarily due to a greater proportion of channels deactivating via the slow pathway at depolarised potentials (Table 2). Thus our results are broadly similar to those reported by others, i.e. in the physiologically relevant voltage range, approximately -80 to -60 mV, deactivation of HERG K+ channels is slowed by co-expression with WT, T8A, Q9E or M54T MiRP1 subunits.
It is difficult to resolve all the discrepancies between the different studies on the effects of MiRP1 subunits on HERG K+ channels, but a few general themes are apparent. Firstly, MiRP1 appears to cause acceleration of deactivation at very negative potentials (<-90 mV) but a slowing of deactivation at less negative potentials (>-85 mV). MiRP1 has modest effects on activation and inactivation, although the effects are more marked with mutant MiRP1 subunits. In particular the T8A mutant appears to cause a significant negative shift in the activation V1/2.
Currents recorded during action potential waveforms
In mammalian cell lines it is difficult to control absolute levels of expression during transient transfection experiments. There is also cell-to-cell variation in the level of expression of HERG channels in the stable cell line. It is also possible that MiRP1 or its mutants may increase or decrease channel protein expression and thereby current density. Therefore, to compare current density from cell to cell during AP voltage clamp protocols we normalised the currents to the maximum current observed during the AP (Fig. 4).
There were relatively small changes in the profile of the current recorded during ventricular AP waveforms in the presence of WT or mutant MiRP1 subunits. WT MiRP1 resulted in a decrease in the initial current phase but during the terminal repolarisation phase, where HERG currents normally make a significant contribution to AP repolarisation (Hancox et al. 1998), it was difficult to discern any significant differences in the current profile between HERG alone and HERG + WT or mutant MiRP1 subunits (Fig. 4). Small differences during this phase, however, can be crucially important for how cells will respond to early after-depolarisations and ectopic beats (Lu et al. 2001).
The variation in the magnitude of the current spike with changes in coupling interval for premature beats reflects the rates of recovery from inactivation and rates of deactivation (discussed in more detail in Lu et al. 2001). Two notable features are apparent from the premature stimulation protocols in this study. Firstly, when the magnitudes of the current spikes were normalised relative to the maximum current spike it is apparent that the decay in the magnitude of the current spike at longer coupling intervals was slowest for channels formed with mutant MiRP1 subunits (Fig. 5B). The decay in the magnitude of the current spike was also slower for HERG + WT MiRP1 compared to HERG alone. These differences reflect the different overall rates of deactivation of the different channel types at -85 mV. Secondly, when we normalised the peak current spikes during the second depolarisation to the peak current recorded during the preceding AP, the current spikes were significantly larger for HERG + T8A MiRP1 and HERG + Q9E MiRP1 compared to HERG alone, HERG + WT MiRP1 or HERG + M54T MiRP1. This normalisation procedure is predicated on the assumption that for a similar channel density the MiRP1 subunits would all result in similar levels of current during a ventricular AP waveform. Given that the duration of the plateau of a ventricular AP (200-300 ms) is significantly longer than the activation rate constants at 37 °C for HERG ± MiRP1 subunits at 0 to +20 mV (≈100 ms) it is likely that they will all activate to a similar extent. However during the repolarisation phase they will recover from inactivation at different rates and so one might expect that the peak current recorded for HERG alone and HERG + T8A MiRP1 (i.e. channels with the fastest rates of recovery from inactivation) would be larger than for the other channel combinations. It is therefore likely that in Fig. 6 we have underestimated the magnitude of the peak currents for HERG + T8A MiRP1 (the largest) and HERG alone. Conversely, we have probably overestimated the magnitude of the peak currents for HERG + WT MiRP1 (the smallest) during premature pulses, as these channels have the slowest recovery from inactivation. Therefore, if anything, it is likely that we have underestimated the differences with the normalisation procedure that we used to obtain the data in Fig. 6
Pro-arrhythmic mechanisms in LQTS6
Mutations in MiRP1 are thought to be the cause of LQTS type 6 (Abbott et al. 1999). MiRP1 can associate with a number of ion channel α-subunits (including HERG (Abbott et al. 1999), KvLQT1 (Tinel et al. 2000), Kv4.2 (Yu et al. 2001) and HCN2 (Yu et al. 2001)). The mechanism(s) underlying LQTS and an increased risk of arrhythmias in patients with LQTS6, however, has not been fully elucidated. Mutations in HERG are clearly associated with LQTS (Curran et al. 1995). Furthermore, HERG K+ channels are thought to be particularly important for suppression of arrhythmias initiated by premature beats (Miller, 1996; Smith et al. 1996; Lu et al. 2001). Altered function of HERG subunits is therefore a plausible mechanism for increased arrhythmic risk in patients with MiRP1 mutations. If we assume that in normal patients, the rapid component of the delayed rectifier currents is mediated by channels formed by co-assembly of HERG + WT MiRP1, then what is the effect of different MiRP1 mutations? The most significant finding in our study is that replacement of WT with T8A or Q9E MiRP1 results in a significantly larger peak current during closely coupled AP stimuli (Fig. 6B). Furthermore, all three mutant subunits result in larger currents at longer coupling intervals (Fig. 5B). The current spikes in response to a premature beat are thought to protect the heart from arrhythmias by suppressing the propagation of premature beats. One might expect that an increase in the HERG current spikes would enhance the protective effect. However, the integrated response of a heart to a premature beat will also be determined by the response of other channels and, most notably, the recovery from inactivation of voltage-gated sodium channels. It is well known that subtle changes in sodium channel function that result in apparent ‘gain of function’ or ‘loss of function’ can both result in an increased risk of arrhythmia (Balser, 1999). Thus, it is possible that increases in HERG conductance during a premature stimulus, by antagonising the depolarisation caused by activation of sodium channels, would result in an apparent ‘loss of function’ in sodium channel activity and so be pro-arrhythmic (Balser, 1999).
Acknowledgments
We thank Gea-Ny Tseng for helpful discussions and comments on the manuscript and providing us with preliminary data for the expression of MiRP1 protein in the heart. M.P.M.-S. and J.I.V. were recipients of British Heart Foundation Basic Science Awards (BS/10 and BS/95002). Y.L. was a recipient of a Cambridge Overseas Trust Studentship and an Overseas Research Student Award. This work was also funded in part by a British Heart Foundation Project Grant (PG/2000108 to J.I.V., M.P.M.-S. and C.L.-H.H.) and an NH&MRC Career Development Award (NHMRC no. 213415 to J.I.V.).
REFERENCES
- Abbott GW, Goldstein SA, Sesti F. Do all voltage-gated potassium channels use MiRPs? Circ Res. 2001;88:981–983. doi: 10.1161/hh1001.091869. [DOI] [PubMed] [Google Scholar]
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Balser JR. Sodium ‘channelopathies’ and sudden death: must you be so sensitive? Circ Res. 1999;85:872–874. doi: 10.1161/01.res.85.9.872. [DOI] [PubMed] [Google Scholar]
- Cui J, Melman Y, Palma E, Fishman Gi, McDonald TV. Cyclic AMP regulates the HERG K(+) channel by dual pathways. Curr Biol. 2000;10:671–674. doi: 10.1016/s0960-9822(00)00516-9. [DOI] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Hancox JC, Levi AJ, Witchel HJ. Time course and voltage dependence of expressed HERG current compared with native ‘rapid’ delayed rectifier K current during the cardiac ventricular action potential. Pflugers Arch. 1998;436:843–853. doi: 10.1007/s004240050713. [DOI] [PubMed] [Google Scholar]
- Isbrandt D, Friederich P, Solth A, Haverkamp W, Ebneth A, Borggrefe M, Funke H, Sauter K, Breithardt G, Pongs O, Schulze-Bahr E. Identification and functional characterization of a novel KCNE2 (MiRP1) mutation that alters HERG channel kinetics. J Mol Med. 2002;80:524–532. doi: 10.1007/s00109-002-0364-0. [DOI] [PubMed] [Google Scholar]
- Jiang M, Liu J, Zhang M, Tseng G-N. KCNE2 protein is expressed in human heart and LQT6-related mutations in KCNE2 can cause a gain or loss of function. Biophys J. 2003;84:413a. [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Duan Y, Teng GQ, Thorstad K, Duff HJ. Novel gain-of-function mechanism in K+ channel-related long-QT syndrome: altered gating and selectivity in the HERG N629D mutant. Circ Res. 2000;86:507–513. doi: 10.1161/01.res.86.5.507. [DOI] [PubMed] [Google Scholar]
- Lu Y, Mahaut-Smith MP, Varghese A, Huang CL, Kemp Pr, Vandenberg JI. Effects of premature stimulation on HERG K+ channels. J Physiol. 2001;537:843–851. doi: 10.1111/j.1469-7793.2001.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhari R, Greenstein JL, Winslow RL, Marban E, Nuss HB. Molecular interactions between two long-QT syndrome gene products, HERG and KCNE2, rationalized by in vitro and in silico analysis. Circ Res. 2001;89:33–38. doi: 10.1161/hh1301.093633. [DOI] [PubMed] [Google Scholar]
- Miller C. The inconstancy of the human heart. Nature. 1996;379:767–768. doi: 10.1038/379767a0. [DOI] [PubMed] [Google Scholar]
- Noble D. Oxsoft Heart Program Manual. V4.8. Oxford: Oxsoft Ltd; 1999. [Google Scholar]
- Noble D, Varghese A, Kohl P, Noble P. Improved guinea-pig ventricular cell model incorporating a diadic space, IKr and IKs, and length- and tension-dependent processes. Can J Cardiol. 1998;14:123–134. [PubMed] [Google Scholar]
- Peeters GA, Sanguinetti MC, Eki Y, Konarzewska H, Renlund DG, Karwande SV, Barry WH. Method for isolation of human ventricular myocytes from single endocardial and epicardial biopsies. Am J Physiol. 1995;268:H1757–1764. doi: 10.1152/ajpheart.1995.268.4.H1757. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, Priori SG, Roden DM, George AL, Jr, Goldstein SA. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci U S A. 2000;97:10613–10618. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 2000;19:6326–6330. doi: 10.1093/emboj/19.23.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg J, Walker B, Campbell T. HERG K+ channels: friend and foe. Trends Pharmacol Sci. 2001;22:240–246. doi: 10.1016/s0165-6147(00)01662-x. [DOI] [PubMed] [Google Scholar]
- Weerapura M, Nattel S, Chartier D, Caballero R, Hebert TE. A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J Physiol. 2002;540:15–27. doi: 10.1113/jphysiol.2001.013296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Wu J, Potapova I, Wymore RT, Holmes B, Zuckerman J, Pan Z, Wang H, Shi W, Robinson RB, El-Maghrabi MR, Benjamin W, Dixon J, McKinnon D, Cohen IS, Wymore R. MinK-related peptide 1: a beta subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ Res. 2001;88:E84–87. doi: 10.1161/hh1201.093511. [DOI] [PubMed] [Google Scholar]
- Zhang M, Jiang M, Tseng GN. minK-related peptide 1 associates with Kv4. 2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circ Res. 2001;88:1012–1019. doi: 10.1161/hh1001.090839. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]