

Abstract
While large, myelinated dorsal root ganglion (DRG) neurons are capable of firing at high frequencies, small unmyelinated DRG neurons typically display much lower maximum firing frequencies. However, the molecular basis for this difference has not been delineated. Because the sodium currents in large DRG neurons exhibit rapid repriming (recovery from inactivation) kinetics and the sodium currents in small DRG neurons exhibit predominantly slow repriming kinetics, it has been proposed that differences in sodium channels might contribute to the determination of repetitive firing properties in DRG neurons. A recent study demonstrated that Nav1.7 expression is negatively correlated with conduction velocity and DRG cell size, while the Nav1.6 voltage-gated sodium channel has been implicated as the predominant isoform present at nodes of Ranvier of myelinated fibres. Therefore we characterized and compared the functional properties, including repriming, of recombinant Nav1.6 and Nav1.7 channels expressed in mouse DRG neurons. Both Nav1.6 and Nav1.7 channels generated fast-activating and fast-inactivating currents. However recovery from inactivation was significantly faster (˜5-fold at −70 mV) for Nav1.6 currents than for Nav1.7 currents. The recovery from inactivation of Nav1.6 channels was also much faster than that of native tetrodotoxin-sensitive sodium currents recorded from small spinal sensory neurons, but similar to that of tetrodotoxin-sensitive sodium currents recorded from large spinal sensory neurons. Development of closed-state inactivation was also much faster for Nav1.6 currents than for Nav1.7 currents. Our results indicate that the firing properties of DRG neurons can be tuned by regulating expression of different sodium channel isoforms that have distinct repriming and closed-state inactivation kinetics.
Primary sensory neurons convey a broad range of sensory information from the periphery to the CNS and different subpopulations exhibit distinct electrophysiological properties. While large myelinated dorsal root ganglion (DRG) neurons are capable of firing at high frequencies, small unmyelinated DRG neurons typically exhibit much lower maximum firing frequencies. The molecular basis for this difference has not been delineated. Voltage-gated sodium channels play a critical role in excitable cells including DRG neurons, because they underlie the depolarizing phase of action potentials which these cells use to transmit information, and may contribute to subthreshold currents that influence action potential electrogenesis. As might be expected from the different firing properties of different types of neurons, the voltage-dependent and kinetic properties of sodium currents recorded from different neuronal populations can show substantial differences (Raman & Bean, 1997; Cummins et al. 2002). Recently we demonstrated that while the TTX-sensitive currents of large DRG neurons (which give rise to myelinated A-fibres (Harper & Lawson, 1985) and can follow firing frequencies of > 100 Hz (Waddell & Lawson, 1990)) exhibit rapid repriming, i.e. fast recovery from inactivation (Everill et al. 2001), the TTX-sensitive currents of small DRG neurons (which give rise to unmyelinated or lightly myelinated axons and can follow significantly lower frequencies, e.g. ˜16 Hz, of stimulation (Waddell & Lawson 1990)) exhibit slow recovery from inactivation (Elliott & Elliott, 1993; Cummins & Waxman, 1997; Black et al. 1999). This suggests that the repriming rate of sodium channels may be an important determinant of maximum following frequency.
At least nine distinct voltage-gated sodium channels have been cloned from mammals (Black & Waxman, 1996; Goldin et al. 2000). Many of these channels have specific developmental, tissue or cellular distributions (Felts et al. 1997), and expression of recombinant channels in Xenopus oocytes and mammalian cells indicates that the different channels can have distinct kinetic and voltage-dependent properties (Smith & Goldin, 1998; Cummins et al. 1998, 2001). Interestingly, the Nav1.7 sodium channel, which is highly expressed in small DRG neurons (Gould et al. 2000; Djouhri et al. 2003), shows slow repriming kinetics when expressed in HEK293 cells (Cummins et al. 1998). Nav1.7 also displays slow onset of closed-state inactivation, a feature which permits it to respond to small, slow depolarizations (Cummins et al. 1998) and which may be relevant to its deployment at or close to sensory nerve terminals (Toledo-Aral et al. 1997). Nav1.6 (previously termed NaCh6 or Scn8a) is highly expressed in large DRG neurons while small DRG neurons express low levels of message for this isoform (Black et al. 1996). Several studies have indicated that Nav1.6 is the predominant isoform located at mature nodes of Ranvier (Caldwell et al. 2000; Krzemien et al. 2000; Boiko et al. 2001) along myelinated axons, which are notable for not firing in response to slow depolarizations but do fire in response to rapid depolarizations and are capable of following high-frequency stimulation (Kocsis et al. 1983). However, the repriming kinetics of Nav1.6 channels have not thus far been characterized.
In the present study, we hypothesized that, in order to support conduction of high-frequency impulse trains along myelinated axons, Nav1.6 channels would display rapid repriming and, because of their complementary patterns of deployment in large and small DRG neurons, we compared Nav1.6 and Nav1.7 sodium currents. Because the properties of sodium currents can depend on the cells in which they are expressed (Cummins et al. 2001), we characterized Nav1.6 and Nav1.7 channels expressed within DRG neurons. Our results show that Nav1.6 and Nav1.7 TTX-sensitive sodium channels have distinct repriming and closed-state inactivation properties. These differences may be important determinants of the integrative and firing properties of DRG sensory neurons.
METHODS
Construction of mammalian expression vectors encoding neuronal rat Nav1.6 channel
These studies utilized a cDNA construct that encodes the mouse NaV1.6 open reading frame. This construct varies from the original published mouse sequence (Burgess et al. 1995) at several nucleotides as reported by Smith et al. (1998). The complete open reading frame of mouse NaV1.6, which was generously provided by Dr A. Goldin (University of California, Irvine), was excised from a pLCT1 oocyte expression vector by digestion first with AatII, then blunting the ends with T4 DNA polymerase, followed by digestion with XhoI to release the Nav1.6 insert. A mammalian expression vector pcDNA3.1 (Invitrogen), modified to render it a low copy vector (Klugbauer et al.1995), was digested with ApaI, the ends were blunted with T4 DNA polymerase, and it was then digested with XhoI. In a subsequent ligation reaction the two DNA pieces were joined to form a 13 kb plasmid. Isolates that displayed the expected digestion pattern using BamHI were verified by sequencing the entire NaV1.6 insert.
For expression within DRG neurons, a TTX-resistant (TTX-R) derivative of NaV1.6 (NaV1.6r) was produced by converting tyrosine 371 to serine (Y371S) as previously described for the Nav1.3 channel (Cummins et al. 2001). Similarly, a TTX-R derivative of NaV1.7 (NaV1.7r) was produced by converting tyrosine 362 to serine (Y362S). These point mutations were introduced into the channel sequence using the Quick Change XL mutagenesis kit (Stratagene) with two mutagenic primers designed according to the manufacturer's instructions. The mutant constructs were subsequently verified by sequencing.
Culture of DRG neurons
DRG neurons were cultured from mice following a protocol approved by the Yale Animal Care and Use Committee. Mice were rendered unconscious by exposure to CO2 and decapitated. DRG neurons were cultured as described previously (Caffrey et al. 1992). Briefly, the L4 and L5 DRGs were harvested from adult Nav1.8-null mice (Akopian et al. 1999). The DRGs were treated with collagenase A (1 mg ml−1) for 25 min, and collagenase D (1 mg ml−1) and papain (30 u ml−1) for 25 min, dissociated in Dulbeccos's modified Eagle's medium and Ham's F12 medium supplemented with 10 % fetal bovine serum, and plated on glass coverslips. Nav1.8-null neurons were kept under standard tissue culture conditions for 5–7 days before biolistic transfections. We previously showed that Nav1.8-null DRG neurons do not express fast- or slow-inactivating TTX-R sodium currents (Cummins et al. 1999, 2001). Some Nav1.8-null DRG neurons do express persistent TTX-R sodium currents (Cummins et al. 1999), but these currents typically display amplitudes < 1 nA after several days in culture and run down quickly in the whole-cell recording configuration; therefore these persistent TTX-R currents are not significant under the culture and recording conditions used in the present study.
Biolistic transfection of Nav1.8-null DRG neurons
The Helios Gene Gun System (Bio-Rad Laboratories) was used for biolistic transfection of neurons (Wellmann et al. 1999). Nav1.6r or Nav1.7r DNA (10 μg) was mixed with 5 μg green fluorescent protein (GFP) DNA and biolistic cartridges were made as described previously (Cummins et al. 2001) using 1.6 μm gold particles. Immediately prior to biolistic transfection, the culture medium was removed from the Petri dish. The gene gun was held ˜2 cm above the cells and a pressure of ˜120 p.s.i. (˜827 kPa) was used to discharge the gold particles. Within 24 h the cells usually showed expression of GFP, indicating a successful biolistic transfection. Electrophysiological studies were conducted 18–48 h after transfection and most of the cells that expressed GFP also expressed fast-inactivating TTX-resistant sodium currents. Since these currents are not observed in untransfected Nav1.8-null neurons or Nav1.8-null neurons transfected with GFP alone, this confirmed that most of the cells that expressed GFP had also been successfully cotransfected with the Nav1.6r or Nav1.7r DNA.
Whole-cell patch-clamp recordings
Whole-cell patch-clamp recordings were conducted at room temperature (˜21 °C) using a HEKA EPC-9 amplifier. Data were acquired on a Windows-based Pentium-III computer using the Pulse program (v 8.1, HEKA Electronic, Germany). Fire-polished electrodes (0.8–1.5 MΩ) were fabricated from 1.7 mm capillary glass using a Sutter P-97 puller (Novato, CA, USA). To optimize space clamp, only isolated cells with a soma diameter of less than 30 μm were selected for recording. Cells were not considered for analysis if the initial seal resistance was less than 2 GΩ, if they had high leakage currents (holding current > 0.5 nA for DRG neurons), or an access resistance greater than 4 MΩ. The average access resistance was 1.8 ± 0.6 MΩ (mean ± S.D., n = 82). Voltage errors were minimized using 80–90 % series resistance compensation and the capacitance artifact was cancelled using the computer-controlled circuitry of the patch-clamp amplifier. Linear leak subtraction, based on resistance estimates from either four or five hyperpolarizing pulses applied before the depolarizing test potential, was used for all voltage-clamp recordings. Membrane currents were usually filtered at 5 kHz and sampled at 20 kHz. The pipette solution contained (mM): 140 CsF, 1 EGTA, 10 NaCl and 10 Hepes (pH 7.3). The standard bathing solution was (mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 0.05 CdCl2, and 10 Hepes (pH 7.3). The liquid junction potential for these solutions was < 8 mV; data were not corrected to account for this offset. The osmolarity of all solutions was adjusted to 310 mosmol l−1 (Wescor 5500 osmometer, Logan, UT, USA). The offset potential was zeroed before patching the cells.
Data analysis
Data were analysed using the Pulsefit (HEKA Electronic, Germany) and Origin (OriginLab Corp., Northampton, MA, USA) software programs. Unless otherwise noted, statistical significance was determined by P < 0.05 using Student's unpaired t test. Results are presented as means ± S.E.M. and error bars in the figures represent the S.E.M. The curves in the figures are drawn by eye unless otherwise noted. Time course data were fitted with single exponential functions.
RESULTS
Robust expression of Nav1.6r and Nav1.7r currents in Nav1.8-null DRG neurons
Because we were interested in the properties of Nav1.6 and Nav1.7 currents in DRG neurons, we expressed Nav1.6 and Nav1.7 channels directly in DRG neurons. In order to identify the Nav1.6 and Nav1.7 currents against the background of other TTX-sensitive sodium currents in DRG neurons, Nav1.6 and Nav1.7 channels were made TTX resistant (TTX-R) by replacing the tyrosine at a critical position for TTX binding (Sivilotti et al. 1997; Cummins et al. 2001) with a serine, and these TTX-R channels (referred to as NaV1.6r and Nav1.7r) were expressed in cultured DRG neurons from Nav1.8-null mice. These neurons, which lack functional Nav1.8 slow-inactivating TTX-R sodium currents and express very low levels of persistent TTX-R sodium current after several days in culture, provide a mammalian neuronal expression system in which other sodium channels, mutated to become TTX-R, can be studied in isolation (Cummins et al. 2001). Nav1.8-null DRG neurons in culture for 5–7 days were transfected with GFP alone or in combination with Nav1.6r or Nav1.7r. Sodium currents were recorded in the presence of 500 nM TTX, 24–48 h after transfection. No TTX-R sodium currents were observed in Nav1.8-null neurons transfected with GFP alone (peak amplitude 0.22 ± 0.04 nA, n = 20). By contrast, the Nav1.6r and Nav1.7r channels produced large fast-inactivating TTX-R sodium currents (Fig. 1A and B). The average peak current amplitude was 29.9 ± 3.8 nA (n = 46) for Nav1.6r currents and 29.5 ± 8.3 nA (n = 12) for Nav1.7r currents. The majority of DRG neurons recorded from were small (soma diameter < 30 μm) and the average cell capacitance was 22 ± 1 pF.
Figure 1. Comparison of Nav1.6r and Nav1.7r current properties.
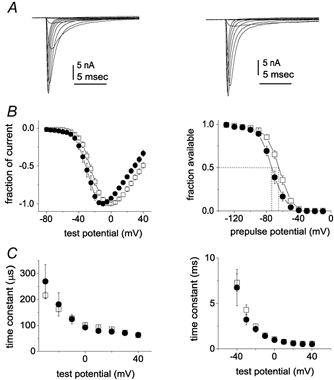
Representative traces from Nav1.8-null DRG neurons expressing Nav1.6r channels (A) and Nav1.7r channels (B) are shown. The currents were elicited by 50 ms test pulses to various potentials from −80 to 40 mV. Cells were held at −120 mV. C, normalized peak current-voltage relationship for Nav1.6r (▪; n = 16) and Nav1.7r channels (•; n = 10). D, voltage dependence of Nav1.6r (▪; n = 16) and Nav1.7r (•; n = 12) sodium current steady-state inactivation. Steady-state inactivation was estimated by measuring the peak current amplitude elicited by 20 ms test pulses to 0 mV after 500 ms prepulses to potentials over the range of −130 mV to −10 mV. Current is plotted as a fraction of the maximum peak current. E, activation time constants as a function of voltage are shown for Nav1.6r (▪; n = 14) and Nav1.7r (•; n = 7) currents in Nav1.8-null DRG neurons. F, inactivation time constants are shown for Nav1.6r (▪; n = 14) and Nav1.7r (•; n = 7) currents in Nav1.8-null DRG neurons as a function of voltage. The activation and inactivation time constants were estimated from Hodgkin-Huxley m3h fits to the currents elicited by 50 ms step depolarizations to voltages ranging from −40 to +40 mV.
Voltage dependence of activation and steady-state inactivation
The largest peak sodium current amplitude recorded from the Nav1.6r transfected DRG neurons was 101 nA. Endogenous TTX-sensitive sodium currents in DRG neurons can also be greater than 100 nA, but currents this large can be difficult to adequately voltage clamp. By using low resistance pipettes and 80–90 % series resistance compensation we were able to achieve reasonable voltage clamp of cells expressing even 30 nA currents (Fig. 1A and B). For our characterization of the voltage-dependent and kinetic properties we focused on data from cells expressing lower peak current amplitudes to minimize the voltage errors. For Fig. 1 we averaged data from 16 Nav1.6r transfected cells (8.1 ± 1.3 nA) and eight Nav1.7r transfected cells (13.9 ± 3.7 nA). The average access resistance in these cells was 1.8 ± 0.1 MΩ and the estimated maximum voltage error after series resistance compensation was 3.3 ± 0.6 mV. The current-voltage relationships for Nav1.6r and Nav1.7r currents are shown in Fig. 1C. The midpoint for voltage-dependent activation was −18.7 ± 1.4 mV and the slope value was 7.7 ± 0.3 for Nav1.6r. The midpoint for voltage-dependent activation was −27.3 ± 2.1 mV and the slope value was 6.2 ± 0.8 for Nav1.7r. The midpoint values were significantly different (P < 0.05). The steady-state inactivation curves, measured with 500 ms depolarizing prepulses, are shown in Fig. 1D. The midpoint of steady-state inactivation was −64.1 ± 1.7 mV and the slope factor was 7.0 ± 0.4 for Nav1.6r; and they were −71.3 ± 2.4 mV and 6.8 ± 0.4 for Nav1.7r. The midpoints of inactivation were significantly different (P < 0.05). The currents elicited by step depolarizations ranging from −40 to +40 mV were fitted using a m3h Hodgkin and Huxley (1952) model to estimate the time constants for activation (Fig. 1E) and fast inactivation (Fig. 1F). The kinetics of activation and fast inactivation were not significantly different (P > 0.05) for Nav1.6r and Nav1.7r currents.
Recovery from inactivation
Large and small cutaneous afferent DRG sensory neurons display dramatic differences in the repriming kinetics of their TTX-sensitive sodium currents (Everill et al. 2001). While the TTX-sensitive sodium currents in small DRG neurons reprime slowly (τ˜90 ms at −80 mV), the repriming of TTX-sensitive sodium currents in large DRG neurons is fast (τ˜12 ms at −80 mV). We were interested in comparing the repriming kinetics of Nav1.6, which is expressed at higher levels in large, compared to small, DRG neurons (Black et al. 1996), and Nav1.7, which is more highly expressed in small DRG neurons (Gould et al. 2000; Djouhri et al. 2003).
The repriming kinetics of Nav1.6r channels expressed in DRG neurons were fast (τ˜8 ms at −80 mV; Fig. 2A and B, left). The repriming kinetics of Nav1.6r currents were measured at recovery voltages from −140 to −60 mV and single exponential fits were used to estimate the repriming time constants that are shown in Fig. 2C (left). By contrast the repriming kinetics of Nav1.7r channels expressed in DRG neurons were slow (τ˜ 70 ms at −80 mV; Fig. 2A and B, right). The repriming time constants of Nav1.7r currents estimated at recovery voltages from −140 to −60 mV are shown in Fig. 2C (right).
Figure 2. Repriming (recovery from inactivation) kinetics are much faster for Nav1.6r than Nav1.7r channels in Nav1.8-null DRG neurons.
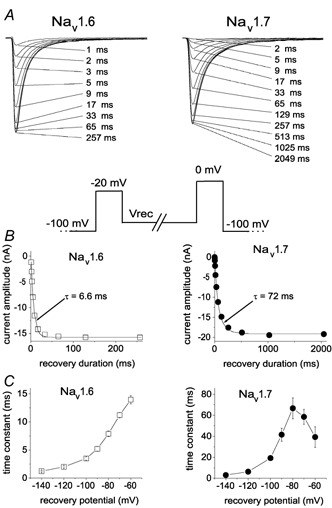
A, family of current traces from representative neurons expressing Nav1.6r channels (left) and Nav1.7r channels (right) showing the rate of recovery from inactivation at −80 mV. The standard repriming voltage protocol is shown below. The cells were prepulsed to −20 mV for 20 ms to inactivate all of the current, then brought back to the recovery potential (Vrec) for increasing recovery durations prior to the test pulse to 0 mV. The maximum pulse rate was 0.5 Hz. The times indicated for each trace shown in A correspond to the recovery duration for that trace. B, the time course for recovery from inactivation of peak Nav1.6r (left) and Nav1.7r (right) currents in A are shown (note different time scales on x-axes). The continuous curve is a single exponential function fitted to the data, with a time constant τ of 6.6 ms for the Nav1.6r currents and 72 ms for the Nav1.7r currents. C, the time constants for recovery from inactivation of Nav1.6r (left) and Nav1.7r (right) currents are shown plotted as a function of voltage (note different time scales on y-axes). Time constants were estimated from single exponential fits to time courses measured at recovery potentials ranging from −140 to −60 mV with the protocol shown in A for currents recorded from neurons transfected with Nav1.6r channels (n = 15) or Nav1.7r channels (n = 11).
Figure 3 compares the repriming time constants of Nav1.6r and Nav1.7r currents to those of native TTX-sensitive currents measured in large cutaneous afferent DRG neurons and small DRG neurons. The Nav1.6r repriming kinetics are similar to those of TTX-sensitive currents in large DRG neurons and the Nav1.7r repriming kinetics are similar to those measured in small DRG neurons.
Figure 3.
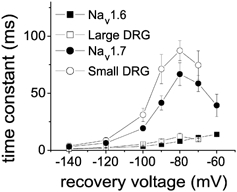
The recovery time constants for Nav1.6r (▪) and Nav1.7r channels (•) expressed in Nav1.8-null DRG neurons are compared to those for TTX-sensitive currents in large DRG neurons (▪; from Everill et al. 2001) and TTX-sensitive currents in small DRG neurons (○; from Cummins et al. 1998).
We used the same inactivating prepulse (20 ms at −20 mV) to examine the repriming kinetics of both Nav1.6 and Nav1.7. This is the same protocol that was used to examine the repriming kinetics of the native TTX-sensitive sodium currents in this and previous (Black et al. 1999; Everill et al. 2001) studies. More than 99.8 % of Nav1.6 and Nav1.7 current is inactivated at −20 mV under steady-state conditions (Fig. 1D) and, because the time constant for fast inactivation is ˜2–2.5 ms at −20 mV (Fig. 1F), more than 97 % of Nav1.6 and Nav1.7 current is inactivated after 20 ms at −20 mV. The 20 ms inactivating prepulse at −20 mV should therefore be sufficient to induce fast inactivation without inducing significant slow inactivation. However, to test for the possibility that stronger inactivating prepulses might affect the repriming kinetics, we also examined the repriming kinetics of Nav1.6 currents following a 20 ms inactivating prepulse to +20 mV and a 50 ms inactivating prepulse to −20 mV (Fig. 4A). The repriming kinetics of Nav1.6 currents were similar with all three inactivating prepulses (Fig. 4B). Similarly, the repriming kinetics of Nav1.7 currents were not affected by these different inactivating prepulses (data not shown). These data support the conclusion that Nav1.6 and Nav1.7 currents exhibit substantially different time courses for recovery from fast inactivation.
Figure 4. Recovery time constants for Nav1.6r channels are not altered by changes in the inactivating prepulse.
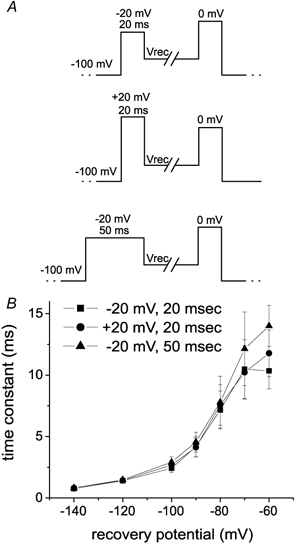
A, the three different inactivating prepulses used to examine the repriming kinetics of Nav1.6r channels are shown. B, the repriming time constants measured with the different protocols for Nav1.6r channels are shown. The first protocol used a 20 ms prepulse to −20 mV (▪), the second protocol used a 20 ms prepulse to +20 mV (•) and the third protocol used a 50 ms prespulse to −20 mV (▴) to inactivate the Nav1.6r channels. The repriming kinetics were always fast for Nav1.6r channels and were not altered by the use of different inactivating prepulses.
Development of closed-state inactivation
We have previously demonstrated that Nav1.7 channels exhibit fivefold slower closed-state inactivation than Nav1.4 channels in HEK293 cells, and we suggested that differences in closed-state inactivation might contribute to the firing properties of neurons (Cummins et al. 1998). The closed-state inactivation kinetics of Nav1.6 channels have not previously been characterized. Therefore we compared the development of inactivation kinetics of Nav1.6r and Nav1.7r channels in Nav1.8-null neurons. At −70 mV, the development of inactivation was rapid (τ˜20 ms) for Nav1.6r channels (Fig. 5A and B, left), but slow (τ˜150 ms) for Nav1.7r channels (Fig. 5A and B, right). The time constant for development of inactivation was estimated at voltages ranging from −90 to −40 mV and was relatively small throughout this voltage range for Nav1.6r channels expressed in Nav1.8-null DRG neurons (Fig. 5C, left) but relatively large for Nav1.7r channels (Fig. 5C, right).
Figure 5. Development of closed-state inactivation is more rapid for Nav1.6r than for Nav1.7r channels expressed in Nav1.8-null DRG neurons.
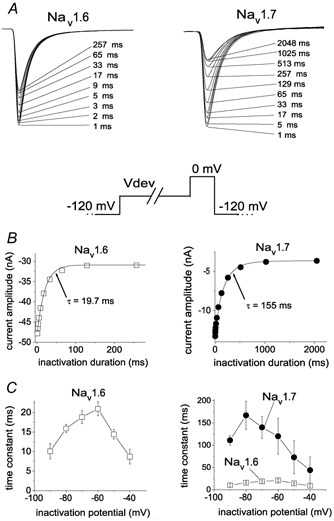
A, family of current traces showing the rate of development of inactivation for Nav1.6r channels (left) and Nav1.7r channels (right) at −70 mV. The standard development of inactivation voltage protocol is shown below. From a holding potential of −120 mV, the cells were prepulsed to −70 mV (Vdev) for increasing durations, then stepped to 0 mV to determine the fraction of current inactivated during the prepulse. The duration of the inactivation prepulse for each data trace is indicated. B, time course for development of inactivation for the peak currents for Nav1.6r and Nav1.7r in A are shown. The continuous curve is a single exponential function fitted to the data, with a time constant of 19.7 ms for Nav1.6r channels (left) and 155 ms for Nav1.7r channels (right). C, the time constants for development of inactivation are shown plotted as a function of voltage. Time constants were estimated from single exponential fits to time courses measured at recovery potentials ranging from −90 to −40 mV with the protocol shown in A for currents recorded from neurons transfected with Nav1.6r channels (▪, left and right; n = 14) and neurons transfected with Nav1.7r channels (•, right; n = 11). As can be seen in the right panel, the development of inactivation time constants for Nav1.6r currents was much faster than for Nav1.7 currents.
Characterization of subthreshold ramp currents
Previously we demonstrated that Nav1.7 channels expressed in HEK293 cells generate larger currents in response to slow ramp depolarizations than Nav1.4 (skeletal muscle) sodium channels. Like Nav1.6r channels, Nav1.4 channels exhibit fast recovery from inactivation and fast development of closed-state inactivation. Therefore we also measured the currents produced in Nav1.8-null neurons in response to slow ramp depolarizations (−100 mV to +30 mV over 500 ms). These ramp depolarizations elicited smaller inward currents in Nav1.8-null DRG neurons transfected with Nav1.6r channels (Fig. 6A) than with Nav1.7r channels (Fig. 6B). Because TTX was included in the bath, endogenous TTX-sensitive currents were blocked. The relative ramp current amplitude (normalized to peak current amplitude) elicited with this protocol averaged 1.4 ± 0.2 % (n = 37) for Nav1.6r channels and 2.4 ± 0.4 % (n = 9) for Nav1.7r channels (Fig. 6C) and this difference was significant (P < 0.05).
Figure 6. Ramp currents generated by Nav1.6r and Nav1.7r channels.
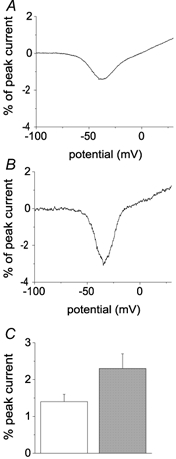
A, representative current elicited in a Nav1.8-null DRG neuron expressing Nav1.6r channels by a 500 ms ramp depolarization from −100 to +30 mV is shown. The peak sodium current amplitude elicited in this cell with step depolarizations was 65.1 nA. B, representative current elicited in a Nav1.8-null DRG neuron expressing Nav1.7r channels by a 500 ms ramp depolarization from −100 to +30 mV is shown. The peak sodium current amplitude elicited in this cell with step depolarizations was 75.4 nA. C, The relative ramp current amplitude was significantly larger in cells expressing Nav1.7r channels (grey bar) than in cells expressing Nav1.6r channels (open bar).
DISCUSSION
We have characterized the kinetic and voltage-dependent properties of the currents conducted by neuronal Nav1.6 and Nav1.7 sodium channels expressed in Nav1.8-null DRG neurons. These channels produced large fast-activating and inactivating currents following biolistic transfection of cultured Nav1.8-null DRG neurons. The Nav1.6 currents were distinct from the currents generated by other TTX-sensitive neuronal sodium channels that we have studied in mammalian cells. Our data provide insights into how sodium currents and firing properties in neurons can be regulated by altering the underlying sodium channel α-subunits that are expressed.
Comparison of Nav1.6 to other voltage-gated sodium channels
Voltage-dependent sodium currents underlie the rapid upstroke of the action potential in neurons. At least eight different voltage-gated sodium channel α-subunits have been detected in neurons and many neuronal cell types have been shown to express more than one α-subunit. While the roles of these different α-subunits in electrogenesis are not yet fully understood, it is clear that different α-subunits can have distinct voltage-dependent and kinetic properties. TTX-R neuronal isoforms have very distinctive properties and exhibit major differences in their activation and inactivation kinetics and voltage dependence (Akopian et al. 1996; Cummins et al. 1999; Dib-Hajj et al. 2002). The differences between the TTX-sensitive channels have been more difficult to elucidate. We show here that midpoints of the voltage dependence of activation and inactivation are 7–9 mV more positive for Nav1.6 than for Nav1.7 channels in DRG neurons. The comparable shifts in the voltage dependence of activation and steady-state inactivation are consistent with the hypothesis that activation and fast inactivation are coupled (Chahine et al. 1994).
We have also found that the different sodium channel isoforms exhibit substantial differences in recovery from inactivation. The time constant for recovery from inactivation for Nav1.6r channels at −80 mV is ˜10-fold faster than that for Nav1.7r channels and 2-fold faster than that for Nav1.3r channels expressed in DRG neurons (Cummins et al. 2001). Nav1.6 is the major sodium channel at nodes of Ranvier (Caldwell et al. 2000), and myelinated axons are known to follow high-frequency stimulation (Kocsis et al. 1983). Since repriming kinetics may help determine how fast a neuron can repetitively fire, this suggests that membranes expressing Nav1.6 channels should be able to sustain higher firing rates than membranes expressing Nav1.7 channels. Although Nav1.7 and Nav1.6 channels exhibit substantially different repriming kinetics, and our data suggest that this reflects recovery from the fast-inactivated state for both isoforms, it is not clear which channel structures account for this difference. Although the amino acid sequences of these two isoforms are well conserved (Goldin et al. 2000), the sequences differ in many places, especially in the loops that join the transmembrane segments.
Development of closed-state inactivation is also much faster for Nav1.6r channels than for either Nav1.7r or Nav1.3r channels. At −70 mV, Nav1.6r channels inactivated with a time constant of ˜20 ms, compared to ˜150 ms for Nav1.7r channels (see Fig. 5C) and ˜120 ms for Nav1.3 channels (Cummins et al. 2001). These data indicate that the rate of transitions between the closed-inactivated and closed states is significantly slower for Nav1.7 than Nav1.6 channels. This suggests that cells expressing Nav1.3 and Nav1.7 channels may generate more robust responses to slowly depolarizing inputs than cells expressing Nav1.6 channels, because Nav1.3 and Nav1.7 channels are less likely to undergo closed-state inactivation during slow depolarizations. Previously we proposed (Cummins et al. 1998) that slow closed-state inactivation of sodium currents can contribute to the development of large ramp currents. Indeed, the ramp currents generated by Nav1.6 channels (˜1.4 % of peak current amplitude) were smaller than Nav1.7 ramp currents (˜2.4 % of peak current amplitude) and Nav1.3 ramp currents (4–7 % of peak current amplitude; Cummins et al. 2001). The voltage ramps that we employed, ˜0.26 mV ms−1, were designed to mimic slow depolarizations that could be caused by natural stimuli such as generator potentials and postsynaptic potentials. Our data indicate that Nav1.3 and Nav1.7, which generated larger ramp currents, might be better than Nav1.6 at boosting slow depolarizing inputs to DRG sensory neurons. The attenuated response of Nav1.6 to small, slow depolarizations might be advantageous at nodes of Ranvier (where Nav1.6 is the major channel; Caldwell et al. 2000), since nodes act as repeaters, generating action potentials in response to the large, rapid depolarizations produced by firing of upstream nodes. Kocsis et al. (1983) showed that, while myelinated axons follow high-frequency stimulation, they do not fire in response to sustained or slow depolarizations. However, studies on the sodium currents in cerebellar Purkinje neurons from Nav1.6 knockout mice indicate that Nav1.6 underlies a significant proportion of the subthreshold ramp currents in these neurons (Raman et al. 1997) and the relative ramp current amplitude produced by Nav1.6r channels in DRG neurons was quite variable, ranging from 0.2 to 4.5 % of the peak current amplitude. These observations suggest that cellular factors may be able to modulate the Nav1.6 ramp current amplitude in neurons. Strupp et al. (1992) reported that the reducing agent glutathione could influence sodium channel inactivation in rat axonal membrane patches, indicating that redox processes, possibly altered by changes in the metabolic state of axons, might modulate the gating of axonal sodium channels and also influence the firing properties of axons.
A ramp current, while relatively small, was detectable in all cells expressing Nav1.6r that we studied. Because the density of sodium channels at the node is high (> 103μm−2; Ritchie & Rogart, 1977; Caffrey et al. 1992) it is possible that Nav1.6 channels provide the persistent current that has been shown (Stys et al. 1993) to drive damaging reverse Na+-Ca2+ exchange in myelinated axons after energy failure is induced by anoxia (Stys et al. 1992). Consistent with this hypothesis, the expression of Nav1.6 channels is downregulated in demyelinated CNS axons (Craner et al. 2003), and it is known that sensitivity to anoxic injury is decreased in these axons following demyelination (Imaizumi et al. 1998). The relatively small magnitude of the ramp currents produced by Nav1.6 may in fact be adaptive at nodes since the presence of other isoforms such as Nav1.7, which produce larger ramp currents, might increase the sensitivity of nodes to energy deprivation.
Interestingly, Craner et al. showed that a significant switch from Nav1.6 to Nav1.2 at nodes of Ranvier within the optic nerve of rats with experimental allergic encephalomyelitis (EAE), a model of multiple sclerosis. Although the properties of Nav1.2 have not been characterized in neurons, O'Leary (1998) characterized Nav1.2 channels expressed in HEK293 cells. Nav1.2 channels displayed repriming and closed-state inactivation kinetics in HEK293 cells that are ˜3-fold slower than that of the Nav1.6r currents reported here. Axons with predominantly Nav1.2 channels at their nodes might be less reliable in terms of following sustained high-frequency stimulation and more likely to fire in response to sustained or slow depolarizations. Thus a switch in isoform expression at nodes could also contribute to the abnormal firing associated with axonal pathologies.
Comparison to native DRG sodium currents
The TTX-sensitive sodium currents in large DRG neurons exhibit predominantly fast repriming kinetics (Everill et al. 2001) similar to those of Nav1.6r currents. The TTX-sensitive currents in small DRG neurons, however, exhibit predominantly slow repriming kinetics similar to those of Nav1.7r currents (Cummins et al. 1998), suggesting that the observed difference is due to a differential isoform expression pattern. Black et al. (1996) found that while the majority of large DRG neurons (which give rise to myelinated axons; Harper & Lawson, 1985) express moderate to high mRNA levels for Nav1.1 and Nav1.6, the majority of small DRG neurons express a low level of message for these isoforms. On the other hand, studies with Nav1.7-specific antibodies show more intense staining of small DRG neurons than large DRG neurons (Gould et al. 2000; Djouhri et al. 2003). Djouhri et al. (2003) reported that Nav1.7 immunoreactivity is negatively correlated with somal cell size and conduction velocity. However, in contrast to Nav1.9 sodium channel protein, which is predominantly found in small nociceptive DRG neurons (Fang et al. 2002), Nav1.7 immunoreactivity is found in both nociceptive and low-threshold mechanoreceptive neurons (Djouhri et al. 2003). Although these results in combination with our data suggest that at least the somal currents in small DRG neurons might be predominantly generated by Nav1.7 and those in large DRG neurons might be predominantly generated by Nav1.6, the lack of isoform-specific blockers prevents direct testing of these hypotheses. Furthermore, the repriming characteristics of Nav1.1 sodium channels have not been characterized in mammalian cells and therefore it is difficult to predict the contribution of this isoform to the firing properties of DRG neurons.
Conclusions
Our data show that Nav1.6 sodium channels generate currents in mammalian cells that are distinct from other TTX-sensitive sodium channels in several ways. The substantially faster repriming of Nav1.6 may contribute to the capability of large DRG neurons to fire at high frequencies, while the rapid development of closed-state inactivation might attenuate the response to slow, small depolarizations at nodes of Ranvier which act as repeaters, generating impulses in response to the large rapid depolarizations produced by firing at upstream nodes, and might protect nodes from damaging persistent sodium influx under conditions of energy deprivation. The different kinetics of TTX-sensitive neuronal sodium channel isoforms such as Nav1.3 (Cummins et al. 2001), Nav1.6 (this study) and Nav1.7 (this study and Cummins et al. 1998) suggest that selective expression of specific sodium channel isoforms can help tune the excitability and repetitive firing properties of neurons.
Acknowledgments
We thank L. Tyrell and B. Toftness for excellent technical support and Dr A. L. Goldin for generously providing the Nav1.6-pBS-SK− plasmid. This work was supported in part by grants from the National Multiple Sclerosis Society, and the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs (S.G.W.). We also thank the Eastern Paralyzed Veterans Association, the Paralyzed Veterans of America and the Nancy Davis Foundation for support.
REFERENCES
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel Nav1. 8 has a specialized function in pain pathways. Nature Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Black JA, Cummins TR, Plumpton C, Chen YH, Hormuzdiar W, Clare JJ, Waxman SG. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol. 1999;82:2776–2785. doi: 10.1152/jn.1999.82.5.2776. [DOI] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG. Spinal sensory neurons express multiple sodium channel α-subunit mRNAs. Brain Res Mol Brain Res. 1996;43:117–132. doi: 10.1016/s0169-328x(96)00163-5. [DOI] [PubMed] [Google Scholar]
- Black JA, Waxman SG. Sodium channel expression: a dynamic process in neurons and non-neuronal cells. Dev Neurosci. 1996;18:139–52. doi: 10.1159/000111403. [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Nav1. 6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97:5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD. Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res. 1992;592:283–297. doi: 10.1016/0006-8993(92)91687-a. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Lo AC, Black JA, Waxman SG. Abnormal sodium channel distribution in optic nerve axons in a model of inflammatory demyelination. Brain. 2003;126:1552–1561. doi: 10.1093/brain/awg153. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog R, Dib-Hajj SD, Waxman SG. Nav1. 3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences following expression in a mammalian cell line and in spinal sensory neurons. J Neurosci. 2001;21:5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in small primary sensory neurons. J Neurosci. 1999;19:1–6. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. RC43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Waxman SG, Donnelly DF. Characterization and developmental changes of Na+ currents of petrosal neurons with projections to the carotid body. J Neurophysiol. 2002;88:2993–3002. doi: 10.1152/jn.00350.2002. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation underlies tetrodotoxin-sensitive ramp currents in HEK293 cells expressing hNE sodium channels and in dorsal root ganglion neurons. J Neurosci. 1998;18:9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG. Down-regulation of TTX-resistant sodium currents and upregulation of a rapidly repriming TTX-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Black JA, Cummins TR, Waxman SG. NaN/Nav1. 9: a sodium channel with unique properties. Trends Neurosci. 2002;25:253–259. doi: 10.1016/s0166-2236(02)02150-1. [DOI] [PubMed] [Google Scholar]
- Djouhri L, Newton R, Levinson SR, Berry CM, Carruthers B, Lawson SN. Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav1). 7 (PN1) Na+ channel α-subunit protein. J Physiol. 2003;546:565–576. doi: 10.1113/jphysiol.2002.026559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott AA, Elliott JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol. 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everill B, Cummins TR, Waxman SG, Kocsis JD. Sodium currents of large (Aβ-type) adult cutaneous afferent dorsal root ganglion neurons display rapid recovery from inactivation before and after axotomy. Neuroscience. 2001;106:161–169. doi: 10.1016/s0306-4522(01)00258-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Djouhri L, Black JA, Dib-Hajj SD, Waxman SG, Lawson SN. The presence and role of the tetrodotoxin-resistant sodium channel Na(v)1. 9 (NaN) in nociceptive primary afferent neurons. J Neurosci. 2002;22:7425–7433. doi: 10.1523/JNEUROSCI.22-17-07425.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG. Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res. 1997;45:71–82. doi: 10.1016/s0169-328x(96)00241-0. [DOI] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Gould HJ, 3rd, Gould TN, England JD, Paul D, Liu ZP, Levinson SR. A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. 2000;854:19–29. doi: 10.1016/s0006-8993(99)02216-7. [DOI] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J Physiol. 1985;359:31–46. doi: 10.1113/jphysiol.1985.sp015573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi T, Kocsis JD, Waxman SG. Resistance to anoxic injury in the dorsal columns of adult rat spinal cord following demyelination. Brain Res. 1998;779:292–296. doi: 10.1016/s0006-8993(97)01171-2. [DOI] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis JD, Ruiz JA, Waxman SG. Maturation of mammalian myelinated fibers: changes in action-potential characteristics following 4-aminopyridine application. J Neurophysiol. 1983;50:449–463. doi: 10.1152/jn.1983.50.2.449. [DOI] [PubMed] [Google Scholar]
- Krzemien DM, Schaller KL, Levinson SR, Caldwell JH. Immunolocalization of sodium channel isoform NaCh6 in the nervous system. J Comp Neurol. 2000;420:70–83. [PubMed] [Google Scholar]
- O'Leary ME. Characterization of the isoform-specific differences in the gating of neuronal and muscle sodium channels. Can J Physiol Pharmacol. 1998;76:1041–1050. doi: 10.1139/cjpp-76-10-11-1041. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Ritchie JM, Rogart RB. Density of sodium channels in mammalian myelinated nerve fibers and nature of the axonal membrane under the myelin sheath. Proc Natl Acad Sci U S A. 1977;74:211–215. doi: 10.1073/pnas.74.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivilotti L, Okuse K, Akopian AN, Moss S, Wood JN. A single serine residue confers tetrodotoxin insensitivity on the rat sensory-neuron-specific sodium channel SNS. FEBS Lett. 1997;409:49–52. doi: 10.1016/s0014-5793(97)00479-1. [DOI] [PubMed] [Google Scholar]
- Smith RD, Goldin AL. Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci. 1998;18:811–820. doi: 10.1523/JNEUROSCI.18-03-00811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18:6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strupp M, Quasthoff S, Mitrovic N, Grafe P. Glutathione accelerates sodium channel inactivation in excised rat axonal membrane patches. Pflugers Arch. 1992;1421:283–285. doi: 10.1007/BF00374840. [DOI] [PubMed] [Google Scholar]
- Stys PK, Sontheimer H, Ransom BR, Waxman SG. Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci U S A. 1993;90:6976–6980. doi: 10.1073/pnas.90.15.6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+-Ca2+ exchanger. J Neurosci. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Moss BL, He Z-J, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, Mandel G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA. 1997;94:1527–1532. doi: 10.1073/pnas.94.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell PJ, Lawson SN. Electrophysiological properties of subpopulations of rat dorsal root ganglion neurons in vitro. Neuroscience. 1990;36:811–22. doi: 10.1016/0306-4522(90)90024-x. [DOI] [PubMed] [Google Scholar]
- Wellmann H, Kaltschmidt B, Kaltschmidt C. Optimized protocol for biolistic transfection of brain slices and dissociated cultured neurons with a hand-held gene gun. J Neurosci Methods. 1999;92:55–64. doi: 10.1016/s0165-0270(99)00094-1. [DOI] [PubMed] [Google Scholar]