

Abstract
Stimulation of regulated secretory cells promotes protein release via the fusion of cytoplasmic storage vesicles with the plasma membrane. In Tetrahymena thermophila, brief exposure to secretagogue results in synchronous fusion of the entire set of docked dense-core granules with the plasma membrane. We show that stimulation is followed by rapid new dense-core granule synthesis involving gene induction. Two genes encoding granule matrix proteins, GRL1 and GRL4, are shown to undergo induction following stimulation, resulting in ≈10-fold message accumulation within 1 h. The mechanism of induction involves transcriptional regulation, and the upstream region of GRL1 functions in vivo as an inducible promoter in a heterologous reporter construct using the gene encoding green fluorescent protein. Taking advantage of the characterized exocytosis (exo−) mutants available in this system, we asked whether the signals for regranulation were generated directly by the initial stimulation, or whether downstream events were required for transcription activation. Three mutants, with defects at three distinct stages in the regulated secretory pathway, failed to show induction of GRL1 and GRL4 after exposure to secretagogue. These results argue that regranulation depends upon signals generated by the final steps in exocytosis.
Keywords: inducible promoter, GRL, dense core granule, regranulation, green fluorescent protein
Endocrine, exocrine, and neuronal cells, as well as numerous unicellular eukaryotes, can secrete proteins rapidly from dense-core granules (DCGs) in response to extracellular stimulation, a phenomenon called regulated exocytosis (1). Upon cell stimulation, fusion of the vesicle and plasma membranes results in release of the vesicle contents (2). Stimulation also promotes synthesis of new DCGs to replace the secreted set (see references in ref. 3). Since the extent of secretion can be graded to the degree of stimulation, the signals that direct regranulation must be similarly graded. This is suggested by the fact that brief stimulation of pancreatic β cells augments insulin synthesis primarily via translation initiation, whereas prolonged stimulation increases insulin synthesis through transcription (reviewed in ref. 4). In addition, unidentified signals during regranulation can potentially modulate the composition of the new granules (5, 6). Though the mechanistic details are unknown, these phenomena suggest that regulation of exocytotic recovery can serve as an important element of an adaptable secretory response (7).
What is the relationship between the secretagogue-generated signals that promote membrane fusion and postexocytotic events? In principle, identical signals could trigger both pathways. For example, localized elevation of cytosolic calcium can trigger membrane fusion (8) as well as activate transcription factors (9, 10). Furthermore, the targets of transcriptional activation can be specified by the subcellular localization of calcium (11). Alternatively, signals directing postexocytotic recovery may be generated in whole or in part by events related directly to membrane fusion. For example, cytoskeletal rearrangement can regulate granule fusion (12, 13) as well as gene expression (14, 15). These two models make distinct predictions regarding whether postexocytotic events might be induced in the absence of membrane fusion. In the first model, but not the second, stimulation of cells that are blocked in fusion could still promote post-exocytotic events. Such uncoupling has been observed in pancreatic β cells under conditions in which glucose-stimulated insulin release is inhibited by chelation of extracellular calcium (16) or somatostatin treatment (17). Under these conditions, glucose addition can still stimulate insulin synthesis.
We have examined coupling in a system where exocytotic inhibition is achieved via genetic lesion, using mutants in the unicellular ciliate Tetrahymena thermophila. Tetrahymena possess DCGs with features of neuroendocrine granules (18), and ≈99% of these granules fuse within seconds upon stimulation (19). Ciliates are genetically tractable, and numerous exocytosis mutants have been characterized in both Tetrahymena (20–22) and in Paramecium (23). Efficient transformation also makes Tetrahymena amenable to molecular genetic approaches (24–26).
In this paper, we examine the stimulation of DCG synthesis following exocytosis in Tetrahymena. The expression levels of two granule protein genes serve as markers for DCG biosynthesis. GRL1 encodes an abundant DCG acidic calcium-binding protein that plays an essential role in organization of the DCG matrix, as shown by gene disruption (27). GRL4 encodes a second abundant protein in the granule matrix. Both genes are transcriptionally activated during a period of DCG regeneration. We looked for uncoupling between the stimulatory signals leading to exocytosis and transcriptional activation in three mutants with defects at distinct stages in the regulated secretory pathway.
MATERIALS AND METHODS
Cells and Cell Culture.
T. thermophila strains are designated by their micronuclear diploid genotype, followed by their macronuclear-determined phenotype (28). CU428.1, MPRR/MPRR (6-methylpurine-sensitive, VII) and B2086, mprS/mprS (6-methylpurine-sensitive, II) were provided by Peter Bruns (Cornell University). Homozygous mutant strains were generated from CU428.1 by Tim Soelter and Eric Cole, (St. Olaf College, MN) by nitrosoguanidine mutagenesis and uniparental cytogamy (29). MN173 and MN175, both MPRR/MPRR (6-methylpurine-resistant), were subsequently identified in a screen for exocytosis deficiencies. SB281 (18, 30) was a gift of Eduardo Orias (University of California, Santa Barbara). Gene nomenclature and cell culture have been described (27). Transformed strains were grown in 1% proteose peptone, 0.2% dextrose, 0.1% yeast extract, 0.003% ferric EDTA, and 120 μg/ml paromomycin sulfate.
Exocytosis Stimulation and Recovery.
Exocytosis was triggered using Alcian blue 8GX (Sigma) (19). Growing cultures (400 ml at 1.5–2 × 105 cells/ml) were pelleted in 50 ml conical tubes (≈810 × g, 45 s) washed once and starved in DMC (0.1 mM Na2HPO4/0.1 mM NaH2PO4/0.65 mM CaCl2/0.1 mM MgCl2/0.2 mM sodium citrate, pH 7.1) (21) for 16 h at room temperature with shaking. The resulting starved cultures were approximately twice the density of the starting culture. Aliquots (50 ml) of starved cells were stimulated as follows. Cells were pelleted as above and resuspended at 10% initial volume. Alcian blue (2%) was added to 0.05% and mixed by tube inversion. The mixture was diluted immediately with 9 volumes of 0.25% proteose peptone, 0.5 mM CaCl2. Cells were washed once in DMC and resuspended for recovery in DMC at room temperature with shaking. Most importantly, cells remained active throughout the stimulation and recovery period.
Cloning of GRL4 Partial cDNA.
Secreted protein was purified from a dibucaine-stimulated culture as described (27), fractionated by SDS/PAGE (31), and transferred to polyvinylidene difluoride (Bio-Rad). A Coomassie blue-staining band of MWa ≈ 20 kDa, named Grl4p, was excised and yielded the amino-terminal sequence FDEQRLAEVISKLQTIQAAIQASYIED (Protein Chemistry Laboratory, Washington University).
Based on known codon usage in T. thermophila (32) (M. Gorovsky, University of Rochester, personal communication), degenerate primers were used to amplify by PCR a portion of the GRL4 coding sequence from a T. thermophila λgt10 cDNA library provided by Tohru Takemasa (University of Tsukuba, Japan). The forward primer 5′-GAA (C/T)A (A/G)AGA (C/T)T (A/G/C/T)GC (C/T)GAAGT-3′ corresponded to polypeptide residues 3–9; the reverse primer 5′-TCTTC (A/G)AT (A/G)TA (A/G)GA (A/G)GCTT-3′ corresponded to residues 22–27. The 50 μl reactions contained ≈108 pfu in 5 μl SM buffer, 2.5 U Taq polymerase (Boehringer Mannheim), 10 mM Tris⋅HCl (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, 250 μM each dNTP, and 1 μM each primer; 30 cycles of 94° for 30 s, 55° for 30 s, and 72° for 30 s were performed. The expected product was cloned into pCRII (Invitrogen) and its identity confirmed by sequencing (Applied Biosystems).
Isolation of Total RNA and Northern and Slot-Blot Analysis.
Total RNA was isolated as described (27). RNA samples were normalized for poly(A)+ content following the methods of Fornance and coworkers (33, 34) as modified by Farrell (35). Northern blot analyses were performed as described by Farrell (35). For slot-blot analysis, total RNA was denatured with 3 volumes of 6.15 M formaldehyde and 10× SSPE and heated at 67°C for 15 min. Samples were transferred to nylon membranes using a vacuum manifold and cross-linked using 150 mJ UV light (Stratalinker, Bio-Rad). Blots were hybridized overnight in 5× SSPE, 50% formamide, 0.1% SDS, 5× Denhardt’s solution, and 100 μg/ml denatured salmon sperm DNA at a probe concentration of ≈5 × 106 cpm/ml. Specific gene probes were generated using gel-purified DNA fragments from the following sources: for histone H4, an EcoRI–HindIII fragment from plasmid pGB 508.8 (36) (gift of M. Gorovsky); for CyP, two PstI fragments from plasmid pBC11 (37) (gift of K. Karrer, Marquette University); for GRL1, an EcoRI fragment of GRL1 cDNA (27); for green fluorescent protein (GFP), GFP cDNA amplified using the PCR conditions described below. Probes were labeled by random priming (38) with [32P]dTTP and randomly generated hexamers (Pharmacia). A GRL4 cDNA probe was labeled by incorporation of [32P]dTTP by PCR, as described (39), using conditions as above.
GFP Constructs.
The replicative plasmid pH4T2-3, based on the endogenous rDNA chromosome (24), was modified for expression of GFP. First, PCR was used to create a copy of the histone H4-I promoter (H4) and β-tubulin (BTU) transcription terminator flanking a unique EcoRI site. This H4/BTU cassette (generously provided by N. D. Chilcoat, University of Chicago) was derived from plasmid p4T2–1 (gift of J. Gaertig, University of Georgia, Athens) (24). A NotI fragment containing the H4/BTU cassette was inserted into pH4T2–3 to create pNRC.EC. A GFP variant, mut3b (Ser-65 → Gly, Ser-72 → Ala), was chosen to take advantage of its higher fluorescence intensity and faster folding relative to the wild-type Aequorea victoria GFP (40) (kindly provided by W. Buikema, University of Chicago). GFP mut3b cDNA was first amplified by PCR using pfu polymerase (Stratagene) following the supplier’s conditions. For correct expression of GFP in Tetrahymena, two primer-incorporated changes were introduced into the GFP cDNA. The original TAA stop codon was changed to the unique Tetrahymena stop codon, TGA, and the nucleotide at position −1 (relative to the start of translation) was changed to match the consensus found at the translational start sites of most Tetrahymena genes (41). This modified GFP cDNA (GFP1) was cloned into the EcoRI site of pBluescript (Stratagene), and its sequence was confirmed. The EcoRI fragment containing GFP1 was inserted into pNRC.EC to create the construct pH4.GFP1 (shown in Fig. 6A). Two additional GFP1 constructs were derived from this plasmid: pΔH4.GFP1 was made by removal of the EcoRV fragment of pH4.GFP1 containing the H4 promoter, and pGRL1.GFP1 was made by the insertion of a 1.2-kb BsmI–HindII fragment containing the GRL1 upstream sequence (27) into the EcoRV site of pΔH4.GFP1.
Figure 6.

A 1.2-kb genomic fragment upstream of GRL1 functions as an inducible promoter element in vivo. (A) GFP1 reporter constructs. For a putative promoter, pH4.GFP1 utilizes the 300-bp histone H4-I promoter; pGRL1.GFP1 utilizes the 1200-bp genomic fragment upstream of GRL1; pΔH4.GFP1 utilizes a 300-bp fragment of the plasmid backbone (denoted by ∗). (B) Transformants bearing each of the three constructs were starved, stimulated, and followed during recovery. Total RNA was prepared at several time points, as in Fig. 4. Northern blot analyses of samples normalized for poly(A)+ RNA were probed with 32P-labeled GFP1 cDNA. To the left of each blot is a diagram of the GFP1 locus designating each of the three reporter constructs. The size of the GFP1 mRNA is ≈900 bp.
Transformation of Tetrahymena.
Replicative transformants were obtained as described (24). Mating pairs of B2086 and CU428.1 were electroporated using the ECM 600 (BTX, San Diego) with 25 μg of purified plasmid DNA (Plasmid Maxi Kit, Qiagen, Chatsworth, CA). Paromomycin was added at 120 μg/ml 6 h following electroporation. After 5–7 days, cells were inoculated in drug-containing medium, grown to a high density, and then maintained as tube stocks.
Immunofluorescence.
Cells were stained using the monoclonal antibody 4D11 (gift of Marlo Nelson and Joseph Frankel, University of Iowa), which recognizes an 80-kDa protein present in the DCG matrix (22). Confocal images were collected with a Zeiss LSM 4 laser scanning microscope, courtesy of Susan Lindquist (University of Chicago). GFP autofluorescence was visualized using an epifluorescence microscope fitted with a standard fluorescein filter.
RESULTS
Exocytosis and Replacement of Secretory Granules in Tetrahymena.
Tetrahymena DCGs, called mucocysts, are positioned at regularly spaced sites along the plasma membrane (42) and form a periodic array when visualized by indirect immunofluorescence (22) (Fig. 1, prestimulation). The immunofluorescent staining of DCGs provides a convenient method to monitor granule exocytosis and replacement. Immediately following stimulation, the granule staining pattern disappeared (Fig. 1, 0 min poststimulation), indicating rapid and extensive exocytosis of docked DCGs. By 60 min poststimulation, cells contained a small number of granules (Fig. 1, 60 min post-). The number of granules increased for ≈240 min, when cells appeared to be completely regranulated (Fig. 1, 240 min post-). These experiments were done with starved cells, which show optimal stimulation (21). Such starved cells are viable, though translationally repressed (43). That a new set of DCGs could be synthesized within this brief time suggested that regranulation was activated, either directly or indirectly, by secretagogue treatment.
Figure 1.
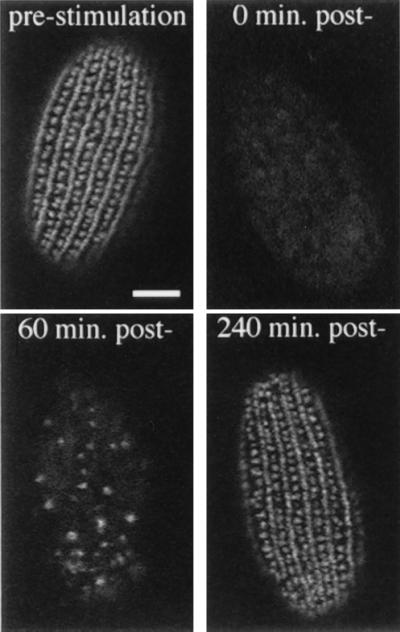
Stimulated exocytosis and recovery of wild-type cells. Immunofluorescent visualization of DCGs in wild-type cells prior to stimulation (16 h of starvation) and at three time points following stimulation of exocytosis. Before stimulation, granules are arranged along the cell surface in rows corresponding with cytoskeletal features called meridians. The difference in granule staining in alternating rows is due to the fact that primary meridians also contain interspersed basal bodies. Immediately following the stimulus (0 min) there is virtually no granule staining, reflecting the rapid and synchronous exocytotic response. At 60 min poststimulation, cells are already seen to be regranulating, as indicated by an increase in granule staining. By 240 min poststimulation, newly formed granules have docked and the cells are largely regranulated. (Bar = 10 μm.)
Expression of DCG-Related mRNAs During Regranulation.
Two genes were monitored as indicators of the DCG biosynthetic pathway. GRL1 and GRL4 encode DCG matrix proteins that are secreted during exocytosis. As in Fig. 1, cells were stimulated with secretagogue and allowed to recover. Total RNA was prepared from cultures before and after stimulation, and transcript levels were measured.
GRL1 transcript was readily detected in growing cells, becoming slightly less abundant in cells starved for 16 h (Fig. 2A). Following stimulation, the transcript rapidly accumulated to peak within 60 min, and then returned to prestimulation levels by ≈360 min poststimulation (data not shown). The GRL4 gene showed a similar pattern of expression (Fig. 2A). The GRL1 transcript peak at 60 min was ≈8- to 10-fold relative to prestimulation levels (Fig. 2B), whereas that for GRL4 was ≈12-fold.
Figure 2.

Expression of mRNAs for GRL1, GRL4-CyP, and H4 genes in wild-type cells. Total RNA was isolated at various times of starvation (0 h = growing culture) and following exocytotic stimulation. Equal amounts of poly(A)+ RNA were loaded for each blot. (A) Northern blot analysis showing changes in abundance for four genes. Sizes of transcripts are as follows: GRL1, 1.4 kb; GRL4, 1.4 kb; CyP, 1.3 kb; H4, 1.0 kb and 870 bp, respectively. (B) Quantitation of mRNAs using slot blot hybridization to total cellular RNA. Typical results are shown normalized to the 0 h starvation time point. GRL1 mRNA increases 8- to 10-fold within 60 min of triggering exocytosis (n = 3) compared with prestimulation levels. CyP mRNA first accumulates during starvation; it then decreases following stimulation and shows a second increase toward the end of recovery.
The increased mRNA levels seen for GRL1 and GRL4 were specific and not seen in genes unrelated to DCG function. Both CyP, a starvation-induced cysteine protease (44, 45), and histone H4 showed patterns of transcript regulation different from those of the DCG proteins. CyP mRNA was undetectable in growing cells, but it rapidly accumulated when cells were starved (Fig. 2A). Transcript abundance rapidly decreased following secretagogue treatment and then showed a second peak of accumulation during the later recovery time points. Transcript for histone H4 decreased with the onset of starvation and remained relatively stable for the duration of the experiment. These results demonstrate that at least two DCG matrix proteins show increased transcript abundance following exocytosis, consistent with the specific activation of a DCG biosynthetic pathway.
exo− Mutants Do Not Exhibit GRL1 mRNA Accumulation Following Stimulation.
To ask whether stimulation and gene induction could be uncoupled, we monitored mRNA accumulation in exo− mutants. Mutants with defects at three distinct stages of the regulated exocytosis pathway were chosen to minimize the possibility of being misled by nonspecific defects in signal generation. SB281 fails to accumulate either mature DCGs or recognizable precursors (30) (Fig. 3A). MN173 synthesizes DCGs which, however, do not dock at the plasma membrane (27). The immunofluorescence pattern reflects the accumulation of cytoplasmic granules (Fig. 3B). MN175 shows a normal pattern of docked DCGs, but these do not undergo exocytosis in response to stimulation with either Alcian blue or another secretagogue, dibucaine (data not shown) (Fig. 3C).
Figure 3.

Granule immunolocalization in three exo− mutants. Cells were fixed at 16 h of starvation (pre-) and immediately following stimulation (post-). In all cases, the pattern of granule immunoreactivity is unchanged following stimulation. (A) SB281 does not synthesize granules and shows no immunoreactivity. (B) MN173 accumulates mature granules in the cytoplasm, and exhibits an unorganized granule staining pattern. (C) MN175 is blocked in exocytosis per se and shows a granule staining pattern identical to wild type.
Using these mutants, we asked whether exposure to secretagogue in the absence of exocytosis could induce transcript accumulation of GRL1 or GRL4. None of these strains showed any quantitative (Fig. 4 A and B) accumulation of GRL1 or GRL4 (data not shown) transcript following stimulation. The expression patterns of CyP and histone H4 were unchanged, suggesting that the changes in the mutants were specific for the DCG-associated proteins (data not shown). These results indicate that secretagogue exposure is not sufficient to generate specific gene activation.
Figure 4.

None of the three exo− mutants shows induction of GRL1 mRNA following stimulation. Total RNA was isolated from wild-type and exo− mutants (SB281, MN173, and MN175) at 0 and 16 h of starvation and 60 and 240 min following stimulation. (A) Slot-blot hybridization of poly(A)+ normalized total RNA samples using a 32P-labeled GRL1 cDNA probe. Wild type shows the characteristic increase in GRL1 mRNA abundance at 60 min poststimulation. In the exo− mutants, there is no increase in GRL1 mRNA abundance following stimulation. (B) Quantitation of slot blots hybridized with the GRL1 specific probe. The patterns of mRNA abundance for both CyP and histone H4 are similar to wild type (data not shown).
A Genomic Region Upstream of GRL1 Functions as an Inducible Promoter Element in Vivo.
We asked whether the upstream genomic region of GRL1 could direct inducible expression of a linked ORF. A modified variant of GFP, called GFP1, was used as a heterologous marker. The GFP1 coding region was inserted 3′ to the 1200-bp genomic region that is immediately upstream of GRL1, which could therefore serve as a potential promoter element. Two other constructs were made in which GFP1 was linked downstream to either (i) the 300-bp upstream element from the T. thermophila histone H4-I gene [which is a functional promoter (46)], or (ii) a 300-bp region of the plasmid backbone. These constructs, inserted in a replicative vector, are shown in Fig. 6A. Wild-type Tetrahymena were transformed with each of these constructs, and the GFP1 expression patterns were observed.
GFP autofluorescence was evident in growing cultures of cells harboring the GFP1 construct driven from the H4 promoter (Fig. 5). GFP expression has not previously been reported in Tetrahymena, and our results indicate it will be a useful marker in this organism. GFP autofluorescence diminished when cells were transferred to starvation buffer in preparation for the stimulation regimen (data not shown). This was expected, because endogenous histone H4 expression is sharply reduced under similar conditions (47). It was, however, still possible to detect and measure promoter activity by the accumulation of the GFP1 transcript.
Figure 5.

GFP autofluorescence in Tetrahymena. Cells harboring an expression construct without (A) or with (B) the GFP1 cDNA under the control of the histone H4-I promoter were fixed during exponential growth. Diffuse GFP autofluorescence was readily observed under these conditions. We frequently observed GFP autofluorescence in what appears to be the macronucleus. (Bar = 10 μm.)
Northern blots probed with GFP1 cDNA revealed an ≈900-bp GFP1 transcript in transformants harboring the GFP1 construct driven from the histone H4 promoter (Fig. 6B). This transcript was most abundant in growing cultures and was sharply reduced in starved cells or in cells during regranulation, similar to the expression pattern of the endogenous histone H4 gene (Fig. 2A). Transformants harboring the GFP1 construct downstream of the 300-bp plasmid backbone sequence showed no detectable GFP1 mRNA at any time.
Analysis of strains transformed with the GFP1 construct linked to the putative GRL1 promoter showed a pattern of GFP1 transcript accumulation similar to that of the endogenous GRL1 gene, though the absolute level of transcript was lower (Fig. 6B). A faint GFP1 band was present in growing cells and disappeared during starvation. At 1 h poststimulation, a strong GFP1 transcript appeared and persisted up to 4 h poststimulation. These results indicate that the 1.2-kb genomic sequence upstream of GRL1 can function in vivo to confer qualitatively similar patterns of transcript accumulation as is seen for the endogenous gene, and therefore constitutes an exocytosis-inducible promoter.
DISCUSSION
We observed rapid replacement of secreted DCGs in starved cultures of Tetrahymena. Bulk protein synthesis in such cells is <1% of that in growing cultures (43). The speed and extent of regranulation suggested that a period of accelerated DCG synthesis might involve the coordinated induction of granule-related genes, as has been seen in other systems (48). Indeed, the transcripts encoding two granule matrix proteins, Grl1p and Grl4p, increased ≈10-fold in abundance within 1 h after stimulation, and decreased to prestimulation levels following regranulation. The results imply that genes encoding all of the granule contents, which together form a well-ordered matrix (27), are likely to be similarly induced. By screening for mRNAs induced after complete exocytosis, we have identified a large set that show expression patterns similar to GRL1 and GRL4 (A.H., unpublished data). From a mechanistic perspective, the phenomena of synchronized exocytosis and rapid regranulation in these cells allows for examination of the coupling of these events.
Transcriptional activation appears to be a common regulatory feature of Tetrahymena (49). The increased abundance of GRL1 mRNA is accounted for at least in part by this mechanism, as shown by the ability of a GRL1 promoter region to direct GFP1 transcription following exocytotic stimulation. Under these conditions GFP1 message levels were low and protein autofluorescence was undetectable. In contrast, GFP autofluorescence was readily visible in growing cells expressing GFP1 driven by the histone H4 promoter, in which much higher levels of GFP1 mRNA were present.
A major question addressed in this paper is whether cells blocked in exocytosis would undergo transcriptional induction following exposure to secretagogue. In three exo− mutants, stimulation uncoupled from exocytosis was not sufficient to induce gene expression. These mutants were generated by nitrosoguanidine mutagenesis, and the precise lesions are unknown. However, they are unlikely to involve plasma membrane signal transduction itself. SB281 makes no visible granules and maps as a single recessive mutation (20). MN173 accumulates cytoplasmic granules, and these undergo efficient docking shortly after mutants are conjugated with wild-type cells, when a cytoplasmic bridge is established between the pair (A.P.T., unpublished data). This finding suggests that the defect lies in a cytoplasmic docking factor. MN175 shows accumulation of docked granules that do not respond to stimulation, one of nine Tetrahymena mutants with this phenotype (A.P.T., unpublished data). Unlike the other eight, MN175 granules do not undergo exocytosis even when cells are stimulated with dibucaine, which is believed to raise directly the level of cytosolic calcium by membrane disruption. Because elevation of intracellular calcium, in ciliates as in other regulated secretory cells, can trigger exocytosis (50), this argues that the MN175 defect lies downstream to signal transduction.
A simple hypothesis to explain our results is that transcription induction in Tetrahymena depends upon signals generated, directly or indirectly, by the exocytotic event. This may be different from mammalian cells, for which results discussed earlier have suggested uncoupling of exocytosis and related gene induction. Relative to that of cells within a tissue, a unicell’s environment is inherently unstable. The exocytotic stimuli for unicellular organisms may be directly generated by environmental variations such as temperature and osmotic strength, as well as by interactions with other organisms. Because such environmental stimuli can affect many pathways simultaneously, a unicell’s transcriptional response during a recovery period may be more economically tuned to the consequences of the stimulus, rather than to the stimulus itself.
Acknowledgments
Tim Soelter and Eric Cole (St. Olaf College, Northfield, MN) helped to isolate MN173 and MN175. We gratefully acknowledge Doane Chilcoat, Adam Lindstedt, Martin Gorovsky, and Don Steiner for discussion and manuscript review. This work was supported by grants to A.P.T. from the National Institutes of Health (GM 50946) and the American Cancer Society (IRG-41-34). A.H. was supported by predoctoral training Grant GM 071836 from the National Institutes of Health.
ABBREVIATIONS
- DCG
dense-core granule
- GFP
green fluorescent protein
Footnotes
A commentary on this article begins on page 10490.
References
- 1.Burgoyne R D, Morgan A. Biochem J. 1993;293:305–316. doi: 10.1042/bj2930305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almers W. Annu Rev Physiol. 1990;52:607–624. doi: 10.1146/annurev.ph.52.030190.003135. [DOI] [PubMed] [Google Scholar]
- 3.Tang K, Wu H, Mahata S K, Taupenot L, Rozansky D J, Parmer R J, O’Connor D T. J Biol Chem. 1996;271:28382–28390. doi: 10.1074/jbc.271.45.28382. [DOI] [PubMed] [Google Scholar]
- 4.Docherty K, Clark A R. FASEB J. 1994;8:20–27. doi: 10.1096/fasebj.8.1.8299887. [DOI] [PubMed] [Google Scholar]
- 5.Fischer-Colbrie R, Iacangelo A, Eiden L E. Proc Natl Acad Sci USA. 1988;85:3240–3244. doi: 10.1073/pnas.85.9.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laslop A, Mahata S K, Wolkersdorfer M, Mahata M, Srivastava M, Seidah N G, Fischer-Colbrie R, Winkler H. J Neurochem. 1994;62:2448–2456. doi: 10.1046/j.1471-4159.1994.62062448.x. [DOI] [PubMed] [Google Scholar]
- 7.Desnos C, Laran M P, Langley K, Aunis D, Henry J P. J Biol Chem. 1995;270:16030–16038. doi: 10.1074/jbc.270.27.16030. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee A, Kowalchyk J A, DasGupta B R, Martin T F J. J Biol Chem. 1996;271:20227–20230. doi: 10.1074/jbc.271.34.20227. [DOI] [PubMed] [Google Scholar]
- 9.Bading H, Ginty D D, Greenburg M E. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 10.Eckert B, Schwaninger M, Knepel W. Endocrinology. 1996;137:225–233. doi: 10.1210/endo.137.1.8536617. [DOI] [PubMed] [Google Scholar]
- 11.Hardingham G E, Chawla S, Johnson C M, Bading H. Nature (London) 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 12.Muallem S, Kwiatkowska K, Xu X, Yin H L. J Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrin D, Langley O K, Aunis D. Nature (London) 1987;326:498–501. doi: 10.1038/326498a0. [DOI] [PubMed] [Google Scholar]
- 14.Hesketh J W, Pryme I F. Biochem J. 1991;277:1–10. doi: 10.1042/bj2770001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu L, Gaertig J, Stargell L, Gorovsky M A. Mol Cell Biol. 1995;15:5173–5179. doi: 10.1128/mcb.15.9.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guest P C, Hutton J C. In: Biosynthesis of Insulin Secretory Granule Proteins. Flatt P R, editor. London: Portland Press; 1992. pp. 59–82. [Google Scholar]
- 17.Olsson S-E, Andersson A, Petersson B, Hellerstrom C. Diabete Metab. 1976;2:199–202. [PubMed] [Google Scholar]
- 18.Turkewitz A P, Madeddu L, Kelly R B. EMBO J. 1991;10:1979–1987. doi: 10.1002/j.1460-2075.1991.tb07727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiedtke A. Naturwissenschaften. 1976;63:93–94. doi: 10.1007/BF00622415. [DOI] [PubMed] [Google Scholar]
- 20.Gutierrez J C, Orias E. Dev Genet. 1992;13:160–166. doi: 10.1002/dvg.1020130210. [DOI] [PubMed] [Google Scholar]
- 21.Orias E, Flacks M, Satir B H. J Cell Sci. 1983;64:49–67. doi: 10.1242/jcs.64.1.49. [DOI] [PubMed] [Google Scholar]
- 22.Turkewitz A P, Kelly R B. Dev Genet. 1992;13:151–159. doi: 10.1002/dvg.1020130209. [DOI] [PubMed] [Google Scholar]
- 23.Bonnemain H, Gulik-Krzywicki T, Grandchamp C, Cohen J. Genetics. 1992;130:461–470. doi: 10.1093/genetics/130.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaertig J, Gu L, Hai B, Gorovsky M A. Nucleic Acids Res. 1994;22:5391–5398. doi: 10.1093/nar/22.24.5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cassidy-Hanley D, Bowen J, Lee J H, Cole E, A, L, VerPlank L A, Gaertig J, Gorovsky M A, Bruns P J. Genetics. 1997;146:135–147. doi: 10.1093/genetics/146.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hai B, Gorovsky M A. Proc Natl Acad Sci USA. 1997;94:1310–1315. doi: 10.1073/pnas.94.4.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chilcoat N D, Melia S M, Haddad A, Turkewitz A P. J Cell Biol. 1996;135:1775–1787. doi: 10.1083/jcb.135.6.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruns P J. In: Genetic Organization of Tetrahymena. Gall J G, editor. New York: Academic; 1986. pp. 27–44. [Google Scholar]
- 29.Cole E S, Bruns P J. Genetics. 1993;132:1017–1031. doi: 10.1093/genetics/132.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maihle N J, Satir B H. J Cell Sci. 1985;78:49–65. doi: 10.1242/jcs.78.1.49. [DOI] [PubMed] [Google Scholar]
- 31.Laemmli U K. Nature (London) 1970;227:680–682. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 32.Martindale D W. J Protozool. 1989;36:29–34. doi: 10.1111/j.1550-7408.1989.tb02679.x. [DOI] [PubMed] [Google Scholar]
- 33.Hollander M C, Fornance A J. BioTechniques. 1990;9:174–179. [PubMed] [Google Scholar]
- 34.Fornance A J, Mitchel J. Nucleic Acids Res. 1986;14:5793–5811. doi: 10.1093/nar/14.14.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farrell R E J. RNA Methodologies: A Laboratory Guide for Isolation and Characterization. New York: Academic; 1993. [Google Scholar]
- 36.Bannon G A, Bowen J K, Yao M-C, Gorovsky M A. Nucleic Acids Res. 1984;12:1961–1975. doi: 10.1093/nar/12.4.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karrer K M, Stein G S. J Protozool. 1990;37:409–414. doi: 10.1111/j.1550-7408.1990.tb01165.x. [DOI] [PubMed] [Google Scholar]
- 38.Sambrook J, Fritsch E J, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 39.Schowalter D B, Sommer S S. Anal Biochem. 1989;177:90–94. doi: 10.1016/0003-2697(89)90019-5. [DOI] [PubMed] [Google Scholar]
- 40.Cormack B P, Valdivia R, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 41.Brunk C F, Sadler L A. Nucleic Acids Res. 1990;18:323–329. doi: 10.1093/nar/18.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allen R D. J Protozool. 1967;14:553–565. doi: 10.1111/j.1550-7408.1967.tb02042.x. [DOI] [PubMed] [Google Scholar]
- 43.Calzone F J, Stathopoulos V A, Grass D, Gorovsky M A, Angerer R C. J Biol Chem. 1983;258:6899–6905. [PubMed] [Google Scholar]
- 44.Rogers M B, Karrer K M. Dev Biol. 1989;131:261–268. doi: 10.1016/s0012-1606(89)80057-0. [DOI] [PubMed] [Google Scholar]
- 45.Karrer K M, Peiffer S L, DiTomas M E. Proc Natl Acad Sci USA. 1993;90:3063–3067. doi: 10.1073/pnas.90.7.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kahn R W, Andersen B H, Brunk C F. Proc Natl Acad Sci USA. 1993;90:9295–9299. doi: 10.1073/pnas.90.20.9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bannon G A, Calzone F J, Bowen J K, Allis C D, Gorovsky M A. Nucleic Acids Res. 1983;11:3903–3917. doi: 10.1093/nar/11.12.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holthuis J C M, Martens G J M. J Neurochem. 1996;66:2248–2256. doi: 10.1046/j.1471-4159.1996.66062248.x. [DOI] [PubMed] [Google Scholar]
- 49.Stargell L A, Karrer K M, Gorovsky M A. Nucleic Acids Res. 1990;18:6637–6639. doi: 10.1093/nar/18.22.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kerboeuf D, Cohen J. Biol Cell. 1996;86:39–43. doi: 10.1111/j.1768-322x.1996.tb00953.x. [DOI] [PubMed] [Google Scholar]