

Abstract
Selenium (Se) and vitamin E are antioxidant micronutrients. Se functions through selenoproteins and vitamin E reacts with oxidizing molecules in membranes. The relationship of these micronutrients with the Nrf2-antioxidant response element (ARE) pathway was investigated using ARE-reporter mice and Nrf2−/− mice. Weanling males were fed Se-deficient (0 Se), vitamin E-deficient (0 E), or control diet for 16 or 22 weeks. The ARE-reporter was elevated 450 fold in 0 Se liver but was not elevated in 0 E liver. Antioxidant enzymes induced by Nrf2-ARE—glutathione S-transferase (GST), NAD(P)H quinone oxidoreductase (NQOR), and heme oxygenase-1 (HO-1)—were elevated in 0 Se livers but not in 0 E livers. Deletion of Nrf2 had varying effects on the inductions, with GST induction being abolished by it but induction of NQOR and HO-1 still occurring. Thus, Se deficiency, but not vitamin E deficiency, induces a number of enzymes that protect against oxidative stress and modify xenobiotic metabolism through Nrf2-ARE and other stress-response pathways. We conclude that Se deficiency causes cytosolic oxidative stress but that vitamin E deficiency does not. This suggests that the oxidant defense mechanisms in which these antioxidant nutrients function are autonomous from one another.
Keywords: selenium deficiency, vitamin E deficiency, mouse liver, oxidative stress, Nrf2-ARE pathway, cytosolic oxidant defense network, phase II enzymes
Introduction
Selenium and vitamin E are essential micronutrients that have antioxidant effects in vivo [1]. Selenium functions through selenoproteins that are redox enzymes and vitamin E functions by reacting directly with oxidizing intermediates. The antioxidant mechanisms supported by selenium and vitamin E appear to be constituents of a broad network of antioxidant mechanisms that overlap with one another functionally to varying degrees. Overall, the network of antioxidant mechanisms appears to be organized to protect against oxidative injury while still allowing signaling by oxidant molecules [2].
Many reports have linked selenium nutritional status with antioxidant mechanisms not containing the element. In 1976 we reported that heme oxygenase (HO) activity was elevated in the liver of the selenium-deficient rat [3]. More recently, our group showed that induction of HO-1, an oxidant defense enzyme, was the cause of the increased hepatic HO activity in selenium deficiency [4]. We also reported elevations of hepatic glutathione S-transferase (GST) and glutamate-cysteine ligase activities in selenium-deficient liver [5, 6]. A Swedish group reported that NAD(P)H quinone oxidoreductase (NQOR) activity was elevated in selenium-deficient rat liver [7]. German investigators reported alterations in a number of mouse liver enzyme activities in selenium deficiency [8]. We speculated that the activities of some of the phase II enzymes were increased to compensate for loss of selenoproteins that protect against oxidative injury [9].
The genes of the phase II enzymes induced in selenium deficiency, as well as the selenoproteins thioredoxin reductase-1 (TrxR1) and glutathione peroxidase-2 (Gpx2), contain the antioxidant response element (ARE) in their promoter regions [10, 11]. The ARE confers on these enzymes the property of being induced by activation of the redox-and electrophile-sensitive transcription factor NF-E2-related factor 2 (Nrf2). The present study was designed to evaluate the involvement of the Nrf2-ARE pathway in the induction of antioxidant enzymes by selenium deficiency. The effect of vitamin E deficiency on the Nrf2-ARE system was also examined.
Materials and Methods
Chemicals
NADPH was purchased from U.S. Biochemical Corp. (Cleveland, OH). Other reagents were purchased from Sigma Chemical Co. (St. Louis, MO) except as specified.
Animal Husbandry
Jeffrey A. Johnson, Ph.D. of the University of Wisconsin kindly provided the ARE-reporter mice. As described [12], the reporter construct contained a 51 basepair segment of rat NQOR1 promoter with the core ARE inserted into a TATA-Inr minimal promoter attached to the human placental alkaline phosphatase (hPAP) reporter gene. Male mice expressing the transgene were mated with female C57BL/6 mice. Male pups of those matings were genotyped [12] and those carrying the transgene were used for studies.
Nrf2+/− mice [13] were obtained from J.Y. Chan, M.D., Ph.D. of the University of California at Irvine. Separate Nrf2−/− and Nrf2+/+ breeding colonies were established.
The experimental diets were Torula yeast-based and identical except for their selenium and vitamin E contents. Supplements to the diets were 0.25 mg selenium/kg diet as sodium selenite and 50 I.U. vitamin E/kg diet as dl-alpha-tocopheryl acetate. Selenium-deficient, vitamin E-deficient, and replete (control) diets were prepared and pelleted to our specifications [14] by Harlan-Teklad (Madison, WI).
At weaning, male mice were fed the experimental diets. Mice were housed in an AAALAC-approved facility with a 10 h light:14 h dark light cycle. They had free access to food and water.
Mice were anesthetized with isoflurane prior to exsanguination from the inferior vena cava. Blood was treated with disodium EDTA (1 mg/ml) to prevent coagulation and plasma was separated by centrifugation for 2 min at 16,000 g. The liver was removed and cut into two pieces. One piece was quick-frozen in liquid N2. The other was used fresh. The frozen liver and the plasma were stored at −80°C until assayed.
Protocols
Male mice were fed control, selenium-deficient, or vitamin E-deficient diet beginning at weaning. Transgenic reporter mice were studied 16 weeks after initiation of the experimental diets. Groups of the Nrf2−/− and Nrf2+/+ mice were studied at 16 and at 22 weeks after initiation of the experimental diets. The Vanderbilt University Institutional Animal Care and Use Committee approved the experimental protocols.
Biochemical Assays
The fresh liver was homogenized by 3 passes of a motor-driven Teflon pestle in a glass vessel on ice. The homogenate, 20% in 0.1 M potassium phosphate, pH 7.5 containing 0.25 M sucrose, was centrifuged for 10 min at 18,000 g and the supernatant was used for HO activity and protein concentration measurements [4].
hPAP activity was measured using a chemiluminescence assay [12]. Briefly, 5% tissue homogenates were made from frozen liver in buffer containing 50 mM Tris-Cl, pH 7.5, 5 mM MgCl2, 0.1 M NaCl, and 4% CHAPS. The homogenates, diluted to 1% in buffer without CHAPS, were mixed with 0.2 M diethanolamine (1:3 v:v), and heated at 65°C for 15 min. Samples were then incubated with chemiluminescent substrate (Tropix Ready-To-Use CSPD Chemiluminescent Substrate with Emerald-II (Applied Biosystems, Foster City, CA)) for 20 min at room temperature in the dark. The chemiluminscent signal was detected using an Lmax Luminometer (Molecular Devices Corp., Sunnyvale, CA).
Other enzyme activities were measured in 105,000 g supernatant (cytosol) prepared from the 18,000 g supernatant used for HO activity determination. Cytosol was diluted in 0.1 M potassium phosphate prior to measurement of glutathione peroxidase, GST, and NQOR activities. Glutathione peroxidase activity was measured as previously described using 0.25 mM H2O2 as substrate [15]. GST activity was measured at 340 nm using 1 mM 1-chloro-2,4-dinitrobenzene and 1 mM GSH in 0.1 M potassium phosphate, pH 6.5, as substrates [16]. NQOR activity was measured as described [17]. Plasma ALT was measured according to instructions provided with the kit from Pointe Scientific, Inc. (Canton, MI).
F2 isoprostanes were measured after Folch extraction of liver tissue [18], base hydrolysis, and derivatization[19]. Determination of the derivatized isoprostanes was accomplished using gas chromatography/negative ion chemical ionization-mass spectrometry with [2H4]prostaglandin F2α as the internal standard [20].
Alpha-tocopherol was determined by HPLC with UV detection at 292 nm as described [21]. HPLC separation of alpha-tocopherol from heptane-extracted material was performed using an Åkta purifier system (Amersham Pharmacia Biosystems, Uppsala, Sweden) with an Ultra C18 column (5 μm, 250 mm × 4.6 mm, Restek Corp., Bellefonte, PA) at a flow rate of 0.75 ml/min. The solvent system was 30% methanol: 66.5% ethanol: 3.5% isopropanol (v:v:v). A standard curve was constructed daily with dl-alpha-tocopherol (Supelco, Bellefonte, PA). The lower limit of detection was 10 nmol of alpha-tocopherol/g tissue.
Protein concentration was determined using bicinchoninic acid reagent (Pierce Chemical Co., Rockford, IL).
Western analysis
Western detection of HO-1 and HO-2 was performed as described elsewhere [4]. Samples for western analysis (65 μg) were separated on 10% SDS-PAGE gels and blotted onto nitrocellulose membranes. After blocking with 5% non-fat milk powder in PBS containing 0.1% Tween-20 (PBS-T) for 1 h at room temperature, membranes were probed with polyclonal antibodies raised in rabbits against recombinant HO-1 and HO-2, respectively, (1:2000 in PBS-T, overnight at 4°C) [22]. After three washes with PBS-T (5 min each, room temperature), membranes were incubated with goat anti-rabbit IgG-horseradish peroxidase conjugate (1:5000, 1 h, room temperature) and washed 4 times with PBS-T (5 min each, room temperature). Immunoreactive proteins were visualized using the Western Lightning kit (Perkin-Elmer, Boston, USA) and exposure to X-ray film.
Statistics
Statistical comparison of groups was performed using the One Way-Analysis of Variance package in Prism, version 4.0b (GraphPad Software Inc., San Diego, CA). Bonferroni‘s Multiple Comparison Test was used following ANOVA to determine statistical differences between groups. Statistical calculations were performed on a Macintosh G5.
Results
Activation of the ARE by antioxidant micronutrient deficiency
In order to determine whether the hepatic Nrf2-ARE pathway becomes activated with deficiency of antioxidant micronutrients, we studied mice containing a transgene consisting of the coding sequence of human placental alkaline phosphatase (hPAP) with an ARE in its promoter. Thus, an increase in hPAP activity would indicate activation of ARE, presumably by Nrf2.
Male reporter mice were fed selenium-deficient, vitamin E-deficient, or control diet for 16 weeks beginning at weaning. This experiment was carried out at the same time and using the same diets as the experiment described in the next section. Figure 1 shows that selenium deficiency caused a 450-fold increase in the hepatic ARE-reporter enzyme activity. Vitamin E deficiency had no effect on reporter enzyme activity. These results indicate that the hepatic Nrf2-ARE system is highly activated by selenium deficiency but not by vitamin E deficiency of the severity produced in this experiment.
Figure 1.
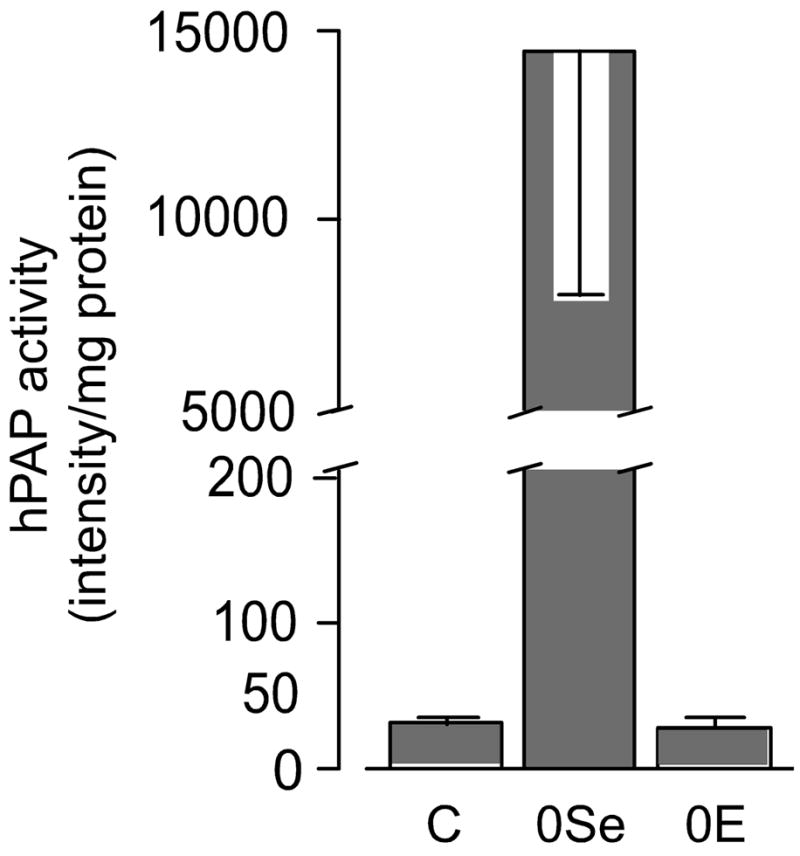
Activation of hepatic ARE in male mice fed diets deficient in selenium or vitamin E. The diets were fed for 16 weeks beginning at weaning. The ARE reporter enzyme was hPAP. The selenium-deficient value (0Se) was different (p<0.05) from the other values but control (C) and vitamin E-deficient (0E) values were not different from each other.
Production of selenium and vitamin E deficiencies in Nrf2−/− mice
Groups of Nrf2+/+ and Nrf2−/− weanling male mice were fed selenium-deficient, vitamin E-deficient, or control diet for 16 or for 22 weeks and then studied. The results obtained at 22 weeks were similar to those obtained at 16 weeks but were slightly more pronounced, in agreement with the longer exposure to deficient diet. The results presented, except as indicated, are from the mice fed the diets for 22 weeks. Table 1 shows that the desired nutritional deficiencies were achieved.
Table 1.
Liver Selenium and Vitamin E Status of Micea
| experimental diets | ||||
|---|---|---|---|---|
| micronutrient biomarker | mouse genotype | control | selenium-deficient | vitamin E-deficient |
| Gpx activityb
(U/mg protein) |
Nrf2+/+ | 370 ± 32 (5) | n.d. (5) | 330 ± 50 (5) |
| Nrf2−/− | 310 ± 21 (5) | n.d. (4) | 270 ± 39 (5) | |
| vitamin Ec
(nmol/g liver) |
Nrf2+/+ | 66 ± 25 (4) | 82 ± 20 (4) | n.d. (4) |
| Nrf2−/− | 91 ± 28 (4) | 73 ± 43 (4) | n.d. (4) | |
Values are means ± S.D. (n). n.d. indicates not detectable. Values of 0 U/mg protein and 10 nmol/g liver were used for detection limits of Gpx activity and vitamin E concentration, respectively, in statistical calculations.
Both selenium-deficient values were different (p<0.05) from all other values. No other values were different from each other.
Vitamin E determinations were made on livers from the 16-week experiment because there was insufficient liver available from the 22-week experiment. Both vitamin E-deficient values were different (p<0.05) from all other values. No other values were different from each other.
Figure 2 presents the growth curves of the mice. Neither nutritional deficiency affected growth of Nrf2+/+ mice. However, selenium deficiency led to failure of weight gain by Nrf2−/− mice at 16 and 22 weeks (Figure 2B). Despite the failure to gain weight, selenium-deficient Nrf2−/− mice did not exhibit debility or mortality. Thus, selenium deficiency, but not vitamin E deficiency, affects weight gain of Nrf2−/− mice.
Figure 2.
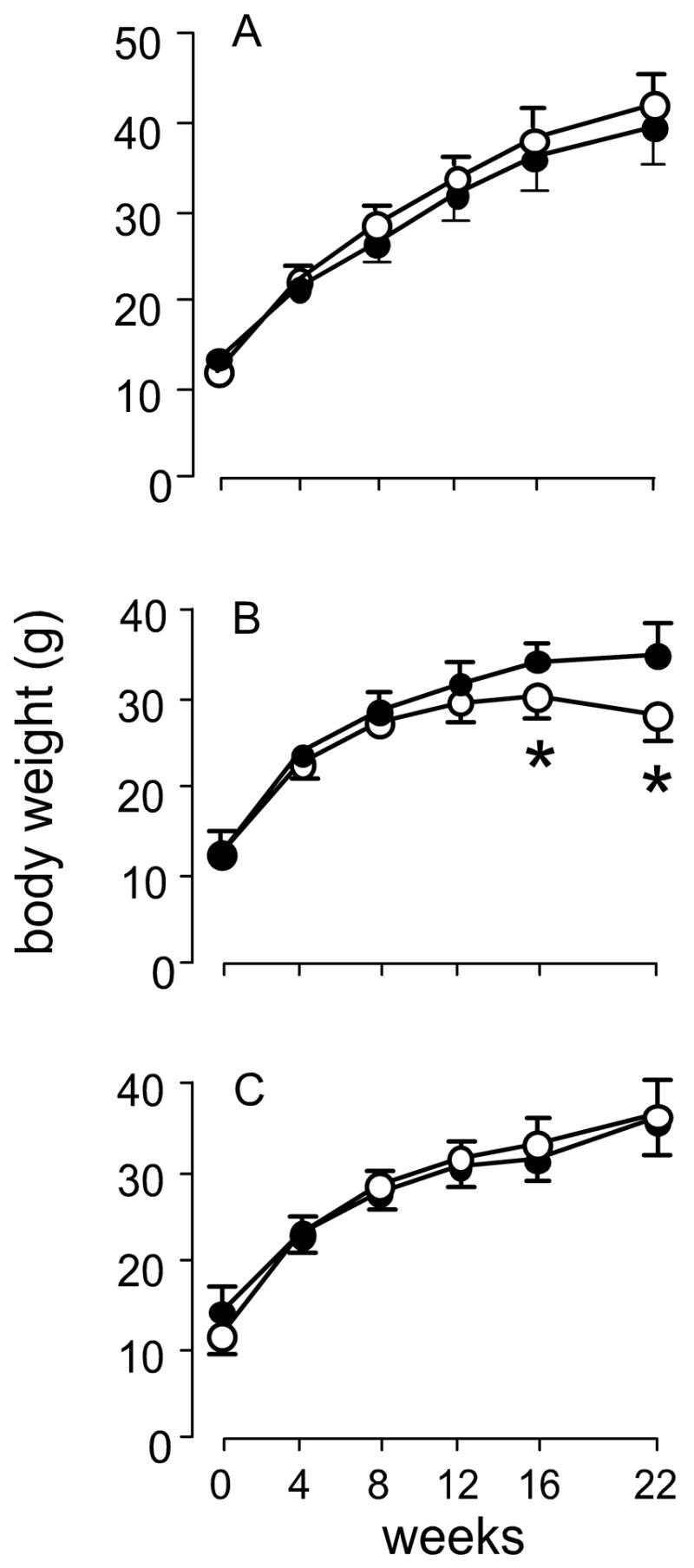
Growth curves of selenium-deficient and vitamin E-deficient Nrf2+/+ (solid circles) and Nrf2−/− (open circles) mice. Mice were fed control diet (panel A), selenium-deficient diet (panel B), or vitamin E-deficient diet (panel C) from the time of weaning. Weights are those of the same mice at each time point and are means + S.D., n=5. The asterisks indicate significant differences between mouse groups within a panel. The diet fed did not affect the weights of Nrf2+/+ mice.
ALT, a marker enzyme of liver injury, was measured in plasma. Table 2 shows that there were no statistically significant differences between diet groups. The marker of lipid peroxidation, F2 isoprostane concentration, was determined in livers. This marker was not affected by the micronutrient deficiencies produced in this experiment (Table 2). Thus, none of the experimental groups exhibited evidence of liver injury or lipid peroxidation.
Table 2.
Biochemical Markers of Liver Injury in Mice
| experimental diets | ||||
|---|---|---|---|---|
| mouse genotype | control | selenium-deficient | vitamin E-deficient | |
| ALTa
(IU/L plasma) |
Nrf2+/+ | 11 ± 4 | 11 ± 2 | 9 ± 1 |
| Nrf2−/− | 13 ± 5 | 14 ± 3 | 10 ± 3 | |
| F2 isoprostanesa
(ng/g liver) |
Nrf2+/+ | 7.9 ± 0.7 | 11 ± 3.8 | 11 ± 1.7 |
| Nrf2−/− | 7.9 ± 2.1 | 8.8 ± 0.4 (4) | 12 ± 4.0 | |
Values are means ± S.D., n=5 (except where indicated). There were no significant differences in ALT or F2 isoprostanes between diet groups.
Effects of Nrf2 deletion on induction of hepatic antioxidant enzymes by nutritional deficiency
GST activity was increased 180% by selenium deficiency in Nrf2+/+ mice but was not affected by vitamin E deficiency (Figure 3A). Deletion of Nrf2 abolished the induction by selenium deficiency and depressed the activity in all diet groups. Thus, the effect of selenium deficiency on GST activity appears to be mediated entirely by Nrf2.
Figure 3.
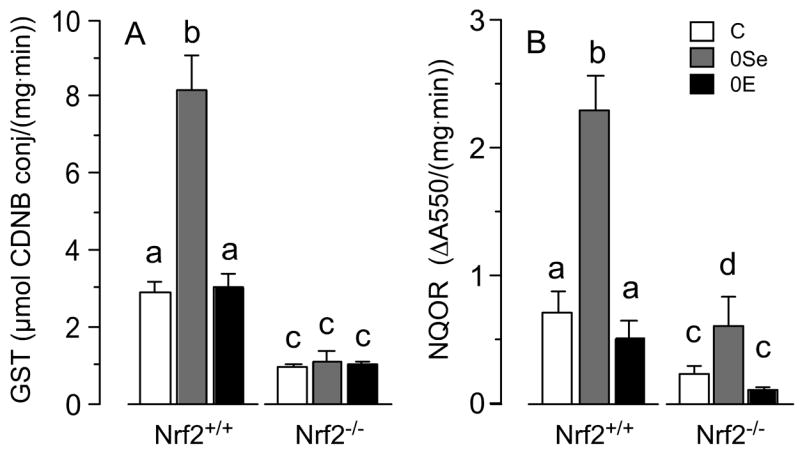
Hepatic glutathione S-transferase (GST) (panel A) and NAD(P)H quinone oxidoreductase (NQOR) (panel B) activities of Nrf2+/+ and Nrf2−/− mice fed the experimental diets for 22 weeks. Values are means + S.D., n=5. In each panel values were compared by diet within genotypes and by genotype within diets. Values not sharing letters were significantly different (p<0.05).
NQOR activity was increased 230% by selenium deficiency and was not significantly affected by vitamin E deficiency (Figure 3B). Deletion of Nrf2 depressed NQOR activity in each diet group but induction by selenium deficiency (170%) persisted. These results confirm that Nrf2 supports NQOR activity generally but suggest that it is not the factor, or at least, not the only factor, that causes induction of NQOR activity in selenium deficiency.
HO activity was increased 770% by selenium deficiency in Nrf2+/+ mouse liver (Figure 4A), confirming earlier observations [4]. It was not significantly affected by vitamin E deficiency. HO-2, the constitutive enzyme, and HO-1, the inducible enzyme, account for the HO activity. Using HO-2 as a reference for HO-1 on western blot (Figure 4B), a sharp increase in HO-1 protein is observed in selenium deficiency. This is compatible with previously published results showing that the increased HO activity in selenium-deficient liver was caused by induction of HO-1 [4].
Figure 4.

Hepatic heme oxygenase (HO) of Nrf2+/+ and Nrf2−/− mice fed the experimental diets for 22 weeks. Panel A presents HO activities as means + S.D., n=5. They were compared by diet within genotypes and by genotype within diets. Values not sharing letters were different (p<0.05). Panel B presents western blots of 18,000 g supernatants.
Deletion of Nrf2 had no effect on HO activity in mice fed control or vitamin E-deficient diet (Figure 4A). However, it attenuated the induction caused by selenium deficiency. Western analysis demonstrated induction of HO-1 in selenium-deficient Nrf2−/− mice (Figure 4B). Using the constitutive enzyme HO-2 as a reference, however, the induction in the absence of Nrf2 was smaller than when Nrf2 was present. These results confirm that HO-1 is responsible for the increase in HO activity observed in selenium deficiency. Moreover, they indicate that the induction of HO-1 in selenium deficiency is caused in part by Nrf2 and in part by other transcription factors.
Discussion
Results presented here demonstrate that selenium deficiency activates the Nrf2-ARE pathway in the livers of male mice (Figure 1). This pathway has many points of regulation [11, 23, 24] and examination of them is beyond the scope of this study. The major activity of the Nrf2-ARE pathway, however, is transduction of cytosolic oxidative stress into induction of ARE-responsive protective enzymes (Figure 5). Thus, the 450-fold induction of the ARE reporter enzyme indicates that selenium deficiency greatly raises the level of oxidative stress in the liver.
Figure 5.
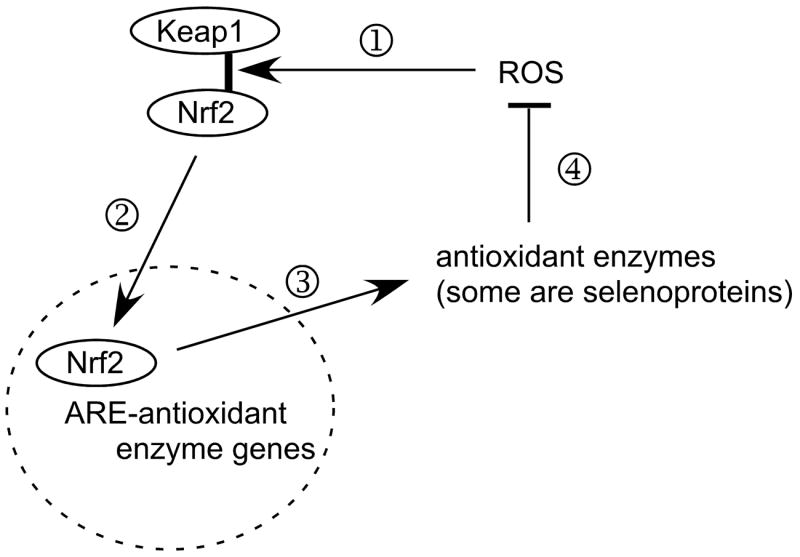
Greatly simplified scheme of postulated function of the Nrf2-antioxidant response element (ARE) system in combating oxidative stress. Under basal conditions, endogenous ROS (reactive oxygen species) cause release (1) of Nrf2 from Keap1 and (2) Nrf2 enters the nucleus (broken-line circle) and binds to AREs in the promoter regions of genes of some antioxidant enzymes. As a result of the Nrf2-ARE interaction, (3) antioxidant enzymes are produced, (4) reducing the concentrations and effects of ROS and lessening the effect of ROS on Keap1 binding of Nrf2.
Selenium deficiency decreases the antioxidant selenoproteins, allowing the ROS to increase and activate the Nrf2-ARE system. Antioxidant enzymes that are not selenoproteins are induced until the ROS are brought into balance at a higher concentration that maintains the activation of the Nrf2-ARE system.
A number of selenoproteins exhibit antioxidant enzyme activities, e.g., the glutathione peroxidases and the thioredoxin reductases. The activities of these enzymes are sharply decreased in selenium-deficient liver [25]. Two of them, TrxR1 and Gpx2, have AREs in their promoter regions [10, 11]. This implies that these two cytosolic enzymes respond to Nrf2 activation and that diminution of their activities would increase the level of oxidative stress. We demonstrated that chemical inhibition of TrxR caused induction of HO-1 [26], which is consistent with the loss of TrxR1 activity increasing oxidative stress [27, 28]. In contrast, deletion of Gpx1, the most abundant selenoprotein in liver, did not cause induction of HO-1 [26], implying that its loss did not affect endogenous cytosolic oxidative stress. However, Gpx1 has been shown to protect against the severe oxidative stress caused by administration of high doses of paraquat to selenium-replete mice [29]. Thus, it appears that some selenoproteins (TrxR1, Gpx2, and perhaps others) protect against endogenous oxidative stress while Gpx1 has an antioxidant role that is reserved for very severe oxidative stresses.
The increased oxidative stress that is present in selenium-deficient liver would be predicted to activate protective pathways such as heat shock, NFκB, and activator protein-1 systems in addition to the Nrf2-ARE pathway. While these additional pathways were not examined directly in these experiments, the inductions of NQOR activity and HO-1 that occurred in the livers of selenium-deficient Nrf2−/− mice (Figures 3B and 4) provide evidence that some of those pathways become activated in selenium deficiency. This increases the potential complexity of the response of protective enzymes to selenium deficiency.
The enzyme activities that protect the liver against oxidative stress and xenobiotics are drastically altered in selenium deficiency. As a consequence, the selenium-deficient liver remains compensated and intact under steady-state conditions without evidence of lipid peroxidation or cell injury (Table 2). When stressed, however, it responds in a different manner than does the selenium-replete liver. An example is the response of the rat liver to diquat, a redox cycling compound. A modest dose of diquat in a selenium-deficient rat causes massive liver necrosis and lipid peroxidation leading to death within hours [30]. The same dose does not injure a selenium-replete rat. This strongly suggests that one of the antioxidant selenoproteins is needed to protect against the type of oxidative stress caused by diquat and that the induced activities cannot adequately compensate for it. In contrast, selenium deficiency actually protects against liver injury in the rat by acetaminophen and aflatoxin [31]. Both these compounds are detoxified by glutathione conjugation of reactive metabolites, and glutathione synthesis and conjugation are enhanced in selenium-deficient liver (fig. 3A, [5]). Other examples of selenium deficiency altering responses to stress have been reported [8, 9]. Outside the U.S. there are human populations with suboptimal selenium nutritional status. Assessment of their susceptibility to injury by oxidative stress and xenobiotics is needed to evaluate their need for selenium supplementation.
Vitamin E deficiency did not activate the Nrf2-ARE pathway. Neither did it have a significant effect on the antioxidant enzymes that were measured (Figures 3 and 4). Because of its lipid nature, vitamin E is restricted to membranes and other lipid domains. The results of this study are consistent with a relative separation between the cytosolic and membrane antioxidant systems. It is likely, however, that cytosolic constituents, such as vitamin C, interact with vitamin E in membranes to maintain vitamin E activity, as has been suggested by in vitro [32] and in vivo [33, 34] experiments. The activity of such a vitamin E-based antioxidant system would appear to be confined to the lipid phase, however.
This study was designed to examine relationships between antioxidant mechanisms and not to establish their antioxidant properties. However, some may interpret the results we obtained in vitamin E-deficient mice as supporting the view that vitamin E does not function as an antioxidant. We would not agree with that interpretation because more severe vitamin E deficiency than was imposed in this study has been shown to cause lipid peroxidation in vivo [33, 35–37]. It was not our intention to produce such a severe deficiency that tissue damage would occur. Had lipid peroxidation been provoked, its electrophilic products might have caused activation of the stress response pathways, complicating interpretation of the results.
In conclusion, nutritional deficiency of selenium activates the Nrf2-ARE pathway and other stress response pathways in the liver. This indicates that selenium deficiency increases endogenous cytosolic oxidative stress. Moreover, activation of the stress response pathways by selenium deficiency explains the induction of numerous enzymes that protect against injury by oxidative stress and xenobiotics. Deficiency of the membrane antioxidant vitamin E does not activate the Nrf2-ARE pathway, suggesting that coordination between cytosolic and membrane antioxidant systems does not occur or is limited.
Acknowledgments
The authors are grateful to Dr. Jeffrey A. Johnson of the University of Wisconsin for the gift of the ARE-reporter mice and to Dr. Jefferson Y. Chan of the University of California at Irvine for the gift of the Nrf2 knockout mice. This study was supported by NIH grants R37 ES02497 and P30 ES00267.
Supported by: NIH grants R37 ES02497 and P01 ES00267
List of Abbreviations
- ALT
alanine amino transferase
- ARE
antioxidant response element
- Gpx
glutathione peroxidase
- GST
glutathione S-transferase
- HO
heme oxygenase
- hPAP
human placental alkaline phosphatase
- NQOR
NAD(P)H quinone oxidoreductase
- Nrf2
NF-E2-related factor 2
- PBS-T
phosphate buffered saline-Tween
- TrxR
thioredoxin reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Institute of Medicine. Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. Washington, D.C: National Academy Press; 2000. pp. 284–324. [PubMed] [Google Scholar]
- 2.Desaint S, Luriau S, Aude JC, Rousselet G, Toledano MB. Mammalian antioxidant defenses are not inducible by H2O2. J Biol Chem. 2004;279:31157–31163. doi: 10.1074/jbc.M401888200. [DOI] [PubMed] [Google Scholar]
- 3.Correia MA, Burk RF. Hepatic heme metabolism in selenium-deficient rats: effect of phenobarbital. Arch Biochem Biophys. 1976;177:642–644. doi: 10.1016/0003-9861(76)90476-8. [DOI] [PubMed] [Google Scholar]
- 4.Mostert V, Hill KE, Ferris CD, Burk RF. Selective induction of liver parenchymal cell heme oxygenase-1 in selenium-deficient rats. Biol Chem. 2003;384:681–687. doi: 10.1515/BC.2003.076. [DOI] [PubMed] [Google Scholar]
- 5.Hill KE, Burk RF. Effect of selenium deficiency and vitamin E deficiency on glutathione metabolism in isolated rat hepatocytes. J Biol Chem. 1982;257:10668–10672. [PubMed] [Google Scholar]
- 6.Lawrence RA, Parkhill LK, Burk RF. Hepatic cytosolic non selenium-dependent glutathione peroxidase activity: Its nature and the effect of selenium deficiency. J Nutr. 1978;108:981–987. doi: 10.1093/jn/108.6.981. [DOI] [PubMed] [Google Scholar]
- 7.Olsson U, Lundgren B, Segura-Aguilar J, Eriksson AM, Andersson K, DePierre JW. Effects of selenium deficiency on xenobiotic-metabolizing and other enzymes in rat liver. Int J Vit Nutr Res. 1993;63:31–37. [PubMed] [Google Scholar]
- 8.Reiter R, Wendel A. Selenium and drug metabolism--I. Multiple modulations of mouse liver enzymes. Biochem Pharmacol. 1983;32:3063–3067. doi: 10.1016/0006-2952(83)90250-2. [DOI] [PubMed] [Google Scholar]
- 9.Burk RF. Biological activity of selenium. Annu Rev Nutr. 1983;3:53–70. doi: 10.1146/annurev.nu.03.070183.000413. [DOI] [PubMed] [Google Scholar]
- 10.Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohe R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol. 2005;25:4914–4923. doi: 10.1128/MCB.25.12.4914-4923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm638. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- 13.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF. Neurological dysfunction occurs in mice with targeted deletion of selenoprotein P gene. J Nutr. 2004;134:157–161. doi: 10.1093/jn/134.1.157. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem Biophys Res Commun. 1976;71:952–958. doi: 10.1016/0006-291x(76)90747-6. [DOI] [PubMed] [Google Scholar]
- 16.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 17.Floreani M, Napoli E, Quintieri L, Palatini P. Oral administration of trans-resveratrol to guinea pigs increases cardiac DT-diaphorase and catalase activities, and protects isolated atria from menadione toxicity. Life Sci. 2003;72:2741–2750. doi: 10.1016/s0024-3205(03)00179-6. [DOI] [PubMed] [Google Scholar]
- 18.Patton GM, Robins SJ. Extraction and analysis of phospholipid molecular species. Meth Enzymol. 1990;187:195–215. doi: 10.1016/0076-6879(90)87025-x. [DOI] [PubMed] [Google Scholar]
- 19.Morrow JD, Awad JA, Kato T, Takahashi K, Badr KF, Roberts LJ, Burk RF. Formation of novel non-cyclooxygenase derived prostanoids (F2-isoprostanes) in carbon tetrachloride hepatotoxicity, an animal model of lipid peroxidation. J Clin Invest. 1992;90:2502–2507. doi: 10.1172/JCI116143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrow JD, Harris TM, Roberts LJ. Noncyclooxygenase oxidative formation of a series of novel prostaglandins: analytical ramifications for measurement of eicosanoids. Anal Biochem. 1990;184:1–10. doi: 10.1016/0003-2697(90)90002-q. [DOI] [PubMed] [Google Scholar]
- 21.Lang JK, Gohil K, Packer L. Simultaneous determination of tocopherols, ubiquinols, and ubiquinones in blood, plasma, tissue homogenates, and subcellular fractions. Anal Biochem. 1986;157:106–116. doi: 10.1016/0003-2697(86)90203-4. [DOI] [PubMed] [Google Scholar]
- 22.Baranano DE, Wolosker H, Bae BI, Barrow RK, Snyder SH, Ferris CD. A mammalian iron ATPase induced by iron. J Biol Chem. 2000;275:15166–15173. doi: 10.1074/jbc.275.20.15166. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Yu SW, Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem. 2006;281:27251–27263. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- 24.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Hill KE, McCollum GW, Boeglin ME, Burk RF. Thioredoxin reductase activity is decreased by selenium deficiency. Biochem Biophys Res Commun. 1997;234:293–295. doi: 10.1006/bbrc.1997.6618. [DOI] [PubMed] [Google Scholar]
- 26.Mostert V, Hill KE, Burk RF. Loss of activity of the selenoenzyme thioredoxin reductase causes induction of hepatic heme oxygenase-1. FEBS Lett. 2003;541:85–88. doi: 10.1016/s0014-5793(03)00309-0. [DOI] [PubMed] [Google Scholar]
- 27.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Bio. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 28.Trigona WL, Mullarky IK, Cao Y, Sordillo LM. Thioredoxin reductase regulates the induction of haem oxygenase-1 expression in aortic endothelial cells. Biochem J. 2006;394:207–216. doi: 10.1042/BJ20050712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng WH, Quimby FW, Lei XG. Impacts of glutathione peroxidase-1 knockout on the protection by injected selenium against the pro-oxidant-induced liver aponecrosis and signaling in selenium-deficient mice. Free Radic Biol Med. 2003;34:918–927. doi: 10.1016/s0891-5849(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 30.Burk RF, Lawrence RA, Lane JM. Liver necrosis and lipid peroxidation in the rat due to paraquat and diquat administration. J Clin Invest. 1980;65:1024–1031. doi: 10.1172/JCI109754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burk RF, Lane JM. Modification of chemical toxicity by selenium deficiency. Fund Appl Toxicol. 1983;3:218–221. doi: 10.1016/s0272-0590(83)80129-8. [DOI] [PubMed] [Google Scholar]
- 32.Packer JE, Slater TF, Willson RL. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature. 1979;278:737–738. doi: 10.1038/278737a0. [DOI] [PubMed] [Google Scholar]
- 33.de Cabo R, Burgess JR, Navas P. Adaptations to oxidative stress induced by vitamin E deficiency in rat liver. J Bioenerg Biomembr. 2006;38:309–317. doi: 10.1007/s10863-006-9050-1. [DOI] [PubMed] [Google Scholar]
- 34.Hill KE, Montine TJ, Motley AK, Li X, May JM, Burk RF. Combined deficiency of vitamins E and C causes paralysis and death in guinea pigs. Am J Clin Nutr. 2003;77:1484–1488. doi: 10.1093/ajcn/77.6.1484. [DOI] [PubMed] [Google Scholar]
- 35.Awad JA, Morrow JD, Hill KE, Roberts LJ, Burk RF. Detection and localization of lipid peroxidation in selenium- and vitamin E-deficient rats using F2-isoprostanes. J Nutr. 1994;124:810–816. doi: 10.1093/jn/124.6.810. [DOI] [PubMed] [Google Scholar]
- 36.Burk RF, Lane JM. Ethane production and liver necrosis in rats after administration of drugs and other chemicals. Toxicol Appl Pharmacol. 1979;50:467–478. doi: 10.1016/0041-008x(79)90400-9. [DOI] [PubMed] [Google Scholar]
- 37.Hafeman DG, Hoekstra WG. Lipid peroxidation in vivo during vitamin E and selenium deficiency in the rat as monitored by ethane evolution. J Nutr. 1977;107:666–672. doi: 10.1093/jn/107.4.666. [DOI] [PubMed] [Google Scholar]