

Abstract
N1-ethyl-N11-[(cyclopropyl)methyl]-4,8,-diazaundecane (CPENSpm) is a polyamine analogue that represents a new class of antitumor agents that demonstrate phenotype-specific cytotoxic activity. However, the precise mechanism of its selective cytotoxic activity is not known. CPENSpm treatment results in the superinduction of the polyamine catabolic enzyme spermidine/spermine N1-acetyltransferase (SSAT) in sensitive cell types and has been demonstrated to induce programmed cell death (PCD). The catalysis of polyamines by the SSAT/polyamine oxidase (PAO) pathway produces H2O2 as one product, suggesting that PCD produced by CPENSpm may be, in part, due to oxidative stress as a result of H2O2 production. In the sensitive human nonsmall cell line H157, the coaddition of catalase significantly reduces high molecular weight (HMW) DNA (≥50 kb) and nuclear fragmentation. Important to note, specific inhibition of PAO by N,N′-bis(2,3-butadienyl)-1,4-butane-diamine results in a significant reduction of the formation of HMW DNA and nuclear fragmentation. In contrast, the coaddition of catalase or PAO inhibitor has no effect on reducing HMW DNA fragmentation induced by N1-ethyl-N11-[(cycloheptyl)methyl]-4,8,-diazaundecane, which does not induce SSAT and does not deplete intracellular polyamines. These results strongly suggest that H2O2 production by PAO has a role in CPENSpm cytotoxicity in sensitive cells via PCD and demonstrate a potential basis for differential sensitivity to this promising new class of antineoplastic agents. Furthermore, the data suggest a general mechanism by which, under certain stimuli, cells can commit suicide through catabolism of the ubiquitous intracellular polyamines.
Keywords: spermidine, spermine N1-acetyltransferase, polyamine oxidase
Programmed cell death (PCD) is a fundamental biological regulatory mechanism involving selective cell deletion. It is an active and irreversible process in which cells activate the intrinsic death program for their own demise. PCD is absolutely required for the natural development and homeostasis of tissues in complex multicellular organisms (1–4). Morphological characteristics of PCD include cell shrinkage, nuclear condensation and fragmentation, plasma and nuclear membrane budding, and apoptotic bodies (3). PCD is biochemically characterized by activation of nucleases that cleave chromosomal DNA into high molecular weight (HMW) and/or low molecular weight oligonucleosomal DNA fragments (5). PCD can be induced by normal physiological processes and by multiple nonphysiological stimuli, including oxidative stress and chemotherapeutic agents (4–7).
The polyamines spermidine and spermine and their diamine precursor putrescine are intracellular cationic molecules that are essential for cell proliferation and differentiation (8, 9). The intracellular concentration of these ubiquitous molecules is highly regulated by the polyamine metabolic pathway, which influences the synthesis, degradation, uptake, and excretion of the cations (9). High ornithine decarboxylase (ODC) activity, the first rate-limiting step of polyamine biosynthesis, and increased levels of intracellular polyamines are known to occur in rapidly proliferating cells or cells undergoing differentiation and transformation. Depletion of intracellular polyamines by direct inhibition of polyamine biosynthesis is generally associated with a decrease in proliferation and has been the primary focus in past antiproliferative studies (10).
However, a more recent strategy has been to design polyamine analogues that exploit the self-regulating nature of polyamine metabolism. Porter, Bergeon, and colleagues (11, 12) have led the field in the design and testing of the symmetric bis(ethyl)polyamines that were designed specifically to down-regulate polyamine biosynthesis by feedback mechanisms rather than by direct enzyme inhibition. We and others have described an additional action of these compounds that, in a cell type-specific manner, leads to a superinduction of spermidine/spermine N1-acetyltransferase (SSAT), the first rate-limiting step in the catabolism of spermine and spermidine (13). The cell type-specific superinduction of SSAT has been associated with, but not causally linked to, the cytotoxic response to several polyamine analogues that have demonstrated significant antitumor activity against important solid tumors. We have, therefore, focused our attention on the design and testing of polyamine analogues that maintain tumor type-specific SSAT induction and cytotoxicity. Current evidence suggests that the regulation of the intracellular polyamine levels plays a pivotal role not only in cell proliferation and differentiation but also in PCD. The deregulation of the intracellular polyamine levels and abnormal polyamine metabolic enzyme activity have been reported in cells undergoing PCD (14–20).
We have demonstrated that the cell type-specific cytotoxicity induced by N1-ethyl-N11-[(cyclopropyl)methyl]-4,8,-diazaundecane (CPENSpm) in the human nonsmall cell lung carcinoma line NCI H157 occurs via a PCD pathway (21). Similar results were observed in breast (22) and prostate cancer cell lines (23). However, the mechanism of analogue-induced PCD is not known. CPENSpm treatment leads to a superinduction of SSAT (24), and the resultant N1-acetylspermine and N1-acetylspermidine are substrates for the constitutive, FAD-requiring, intracellular polyamine oxidase (PAO). This enzyme is present in most cell types and cleaves N1-acetylated polyamines to produce spermidine or putrescine, H2O2, and 3-acetamidopropanal (25).
The goal of the present study was to determine if the production H2O2 resultant from analogue induced SSAT is associated with the observed CPENSpm-induced DNA and nuclear fragmentation. The results presented here demonstrate that the inhibition of PAO, thus H2O2 production, significantly reduced the CPENSpm-induced HMW DNA and nuclear fragmentation and delayed the onset of PCD in the NCI H157 cells. To verify that SSAT induction plays a role in the observed induction of PCD, we compared the effects of CPENSpm to those of N1-ethyl-N11-((cycloheptyl)methyl)-4,8,-diazaundecane (CHENSpm), a related analogue that does not superinduce SSAT. The data also underscore the possibility that intracellular polyamine catabolism may function in a general cellular suicide mechanism in response to various stimuli.
MATERIALS AND METHODS
Chemicals and Cell Culture.
CPENSpm and CHENSpm were synthesized as reported (26). The stock solutions of analogues were prepared at a concentration of 1 mM in 0.1 N HCl. N,N′-bis(2,3-butadienyl)-1,4-butane-diamine (MDL 72, 527) was a generous gift from Eugene Gerner (University of Arizona, Tucson, AZ) and Hirak S. Basu (University of Wisconsin, Madison, WI). Catalase from bovine liver, Cu/Zn-superoxide dismutase (Cu/Zn-SOD) from bovine erythrocytes, N-acetyl-l-cysteine, butylated hydroxytoluene, butylated hydroxyanisole, ZnCl2, aminoguanidine, and aurintricarboxylic acid were purchased from Sigma. Proteinase K and RNase A were purchased from GIBCO/BRL. 5-(and -6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), mixed isomers, was purchased from Molecular Probes. The nonsmall cell lung carcinoma line NCI H157 cell was maintained as reported (21). Intracellular polyamine pools and SSAT and ODC activities were measured using cellular extracts as reported (21).
Field-Inversion Gel Electrophoresis and Quantitation of HMW DNA Fragmentation.
DNA from treated and control cells was resolved using field-inversion gel electrophoresis, as described (21). DNA fragmentation was assayed quantitatively using a modification of the method described by Hoyt et al. (27). For qualitative analysis, DNA was stained with ethidium bromide and photographed using the Eagle Eye system (Stratagene). For quantitative analysis, DNA was transferred to a ZetaProbe nylon membrane (Bio-Rad) and hybridized to a 32P-labeled human AluI DNA sequence (from Barry Nelkin, Johns Hopkins University School of Medicine, Baltimore) using published methods (28). Phosphor image analysis was performed on a Molecular Dynamics PhosphorImager using imagequant software (Sunnyvale, CA). Five hundred thousand cells, which were shown to be within the linear range of the quantitative detection without saturation (data not shown), were analyzed for the HMW DNA fragmentation. Percentage of HMW DNA fragmentation = DNA migrated/total DNA (DNA remaining in the well + DNA migrated) × 100.
Assessment of Morphology.
Exponentially growing NCI H157 cells were treated with 10 μM CPENSpm with or without cotreatment of 500 units/ml catalase or 250 μM MDL 72,527 for various times. Adherent cells were harvested with trypsin and combined with cells floating in the medium. Cells were then washed with 1X PBS, fixed with 300 μl PBS, 20 μl 10% Nonidet P-40 in PBS, 36 μl 37% formaldehyde, and 2 μl of 1 mg/ml Hoechst 33342 (Calbiochem–Behring), visualized under UV excitation, and photographed with a Nikon Labophot microscope.
Flow Cytometric Detection of Peroxides by CM-H2DCFDA.
Control cells and cells treated for 1 h with 10 μM CPENSpm with or without cotreatment of 500 units/ml catalase or 250 μM MDL 72,527 were analyzed for increase in changes in fluorescence indicating a change in H2O2 production. Adherent cells were harvested with trypsin and combined with cells floating in the medium. Cells were then washed with 1X PBS, and 1 × 106 cells were treated with 10 μM CM-H2DCFDA for 20 min at 37°C. One hundred thousand cells were then analyzed on a Becton Dickinson FACScan as reported (29).
Flow Cytometry.
Flow cytometric analysis of CPENSpm- and CHENSpm-treated cells was performed using the propidium iodide (Sigma) staining method as described (30). Stained nuclei were analyzed on a Becton Dickinson FACScan with an argon ion laser at an excitation wavelength of 488 nm.
RESULTS
The Formation of HMW DNA Fragments Induced by CPENSpm Treatment in NCI H157 Cells Occurs Concurrently with SSAT Superinduction.
CPENSpm was found to induce formation of HMW DNA fragments at a concentration as low as 8 μM in 24 h, and fragments could be detected as early as 18 h with 10 μM CPENSpm treatment (data not shown). Based on these results, the treatment of 10 μM CPENSpm for 24 h was chosen for further experimentation. Treatment with 10 μM CPENSpm produced 37 ± 5% fragmented DNA after 24 h (Table 1). The formation of apoptotic nuclei (condensed or fragmented) was observed with 10 μM CPENSpm treatment at 24 h (Fig. 1B). During this period, cells became detached from the flask and aggregated. During the same treatment time, CPENSpm-induced SSAT activity from 146 pmol/mg/min to >28,000 pmol/mg/min (Table 1) and reduced ODC activity (>95%) from 8980 ± 70 pmol/mg/h to 290 ± 3 pmol/mg/h. The increased SSAT activity and reduced ODC activity were accompanied by a significant decrease in intracellular polyamine pools and accumulation of the analogue (Table 1). These results are in contrast to those observed with CHENSpm, which does not induce SSAT to nearly the same extent as CPENSpm and does not deplete intracellular polyamine to the same levels (Table 1). However, 10 μM CHENSpm does produce significant HMW DNA fragmentation (Fig. 2 A and B).
Figure 1.
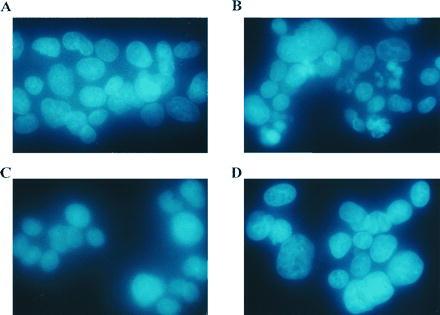
The effects of catalase and MDL 72,527 on CPENSpm-induced apoptotic nuclei. Cells were (A) untreated, (B) treated with 10 μM CPENSpm, (C) treated with 10 μM CPENSpm plus 500 units/ml catalase, or (D) treated with 10 μM CPENSpm plus 250 μM MDL 72,527. All treatment times were 24 h. Cells were fixed with Hoechst dye, visualized under UV excitation, and photographed with a Nikon Labophot microscope.
Figure 2.
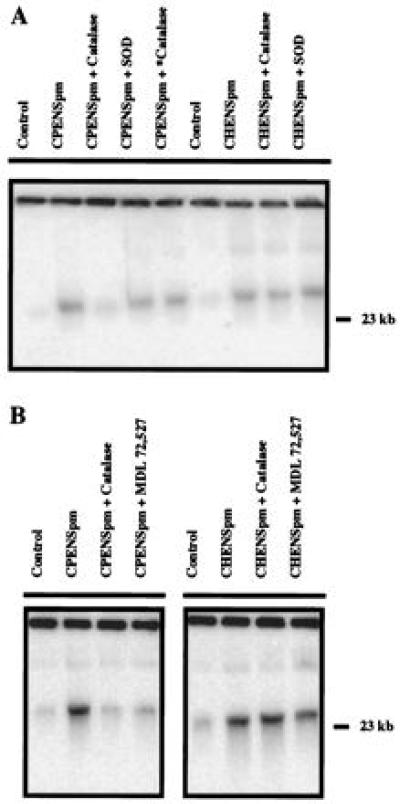
HMW DNA fragmentation induced by polyamine analogues. (A) The effects of catalase, heat-inactivated catalase (∗), and Cu/Zn-SOD on HMW (≥50 kb) DNA fragmentation induced by 10 μM CPENSpm and CHENSpm in NCI H157 cells. Cells were exposed to 10 μM CPENSpm for 24 h with or without 500 units/ml catalase, 500 units/ml heat-inactivated catalase, or 1000 units/ml Cu/Zn-SOD and analyzed by field-inversion gel electrophoresis to assess HMW (≥50 kb) DNA fragmentation. Quantitation of HMW DNA fragmentation was determined by hybridizing transferred DNA to a 32P-labeled human AluI DNA sequence as described in Materials and Methods. (B) The effects of catalase and MDL 72,527 on HMW (≥50 kb) DNA fragmentation induced by 10 μM CPENSpm or CHENSpm in NCI H157 cells. Cells were exposed to 10 μM CPENSpm or CHENSpm for 24 h with or without 500 units/ml catalase or 250 μM MDL 72,527.
Prevention of the Formation of HMW DNA Fragmentation by Antioxidants.
To determine whether CPENSpm-induced DNA fragmentation was mediated by the production of H2O2, we examined the effect of catalase on CPENSpm-induced DNA fragmentation. Catalase inhibited the formation of HMW DNA fragments by 77% compared with the treatment of CPENSpm alone (Fig. 2 A and B; Table 1). The prevention of the formation of HMW DNA fragments was seen at ≥250 units/ml of catalase. Heat-inactivated catalase and Cu/Zn-SOD did not effect the formation of HMW DNA fragments (Fig. 2A). However, the protective effect of catalase was observed to diminish with time. Few CPENSpm-induced apoptotic nuclei were observed with cotreatment of catalase at 24 h (Fig. 1C), but an increasing number of fragmented nuclei were observed by 48 h (data not shown). N-acetyl-l-cysteine (10 μM) also was found to reduce CPENSpm-induced formation of HMW DNA fragmentation by ≈42% during 24-h exposure. To a lesser extent, butylated hydroxytoluene (400 μM) and butylated hydroxyanisole (400 μM) reduced CPENSpm-induced formation of HMW DNA fragmentation (data not shown). Catalase was most effective in preventing the CPENSpm-induced formation of HMW DNA fragmentation among the antioxidants examined. In contrast, neither catalase nor Cu/Zn-SOD had a significant effect on CHENSpm-induced DNA fragmentation (Fig. 2 A and B).
Source of Reactive Oxygen Species.
The above results demonstrated that NCI H157 cells treated with CPENSpm were under oxidative stress. Therefore, the possibility that the breakdown of the natural polyamines was the source of the reactive oxygen species that induces HMW DNA fragmentation was examined. Both the copper requiring serum amine oxidase and the FAD-dependent intracellular PAO produce H2O2 as a by-product of polyamine catabolism. Therefore, to verify which enzyme was responsible for producing H2O2, the effects of inhibitors of these enzymes on the CPENSpm-induced formation of HMW DNA fragmentation was determined. Increasing concentrations of aminoguanidine (0.1–2 mM), an inhibitor of serum amine oxidase, were used alone and in combination with 10 μM CPENSpm. Aminoguanidine produced no damage on its own and was unable to inhibit HMW DNA fragmentation produced by 24-h CPENSpm treatment (data not shown). MDL 72,527, a specific inhibitor of PAO, was similarly tested alone and in combination with CPENSpm. Based on the result of a dose-response analysis, 250 μM MDL 72,527 was chosen for further testing. At 250 μM, MDL 72,527 was found to significantly reduce the generation of HMW DNA fragmentation in CPENSpm-treated cells by 67% (Fig. 2C; Table 1). Similar to results with catalase, there was little CPENSpm-induced nuclear fragmentation with cotreatment of MDL 72,527 at 24 h (Fig. 1D), but the nuclear fragmentation was again observable at later time points (48 h). As stated above, CHENSpm is not a potent inducer of SSAT and has a relatively minor effect on intracellular polyamine concentrations (Table 1). Therefore, it is highly significant that neither aminoguanidine nor the specific PAO inhibitor had a significant effect on CHENSpm-induced DNA fragmentation (Fig. 2 A and B), suggesting that CHENSpm-induced damaged is not mediated through the same oxidative stress pathway. These results strongly suggest that PAO activity is a source of reactive oxygen species in CPENSpm-treated NCI H157 cells.
The results with catalase and MDL 72,527 indicate that the generated reactive oxygen species is H2O2. In an attempt to substantiate this hypothesis, CM-H2DCFDA, an oxidation-sensitive fluorescent probe, was used. CM-H2DCFDA is oxidized to the fluorescent compound dichlorodihydrofluorscein, which is retained in the cell. The induction of SSAT by CPENSpm and similar compounds occurs very rapidly (13). Therefore, a 1-h exposure to CPENSpm was chosen to examine the early effects on H2O2 production. During the 1-h treatment, there is a large increase in enzyme activity, and high concentrations of the natural polyamines are available as substrates. CPENSpm treatment alone produced a significant increase in detected fluorescence (Fig. 3). However, cotreatment with either catalase or the PAO inhibitor resulted in no increased fluorescence over untreated cells. It should be noted that Fig. 3 represents one of four experiments that demonstrated identical trends. However, the baseline fluorescence of controls varied between experiments. This shift in background fluorescence was possibly caused by the necessity of trypsinization of the monolayer cells.
Figure 3.

The effects of catalase and MDL 72,527 on CPENSpm-induced fluorescence detected by CM-H2DCFDA. Cells were (A) treated with 10 μM CPENSpm, (B) treated with 10 μM CPENSpm plus 500 units/ml catalase, or (C) treated with 10 μM CPENSpm plus 250 μM MDL 72,527. After a 1-h treatment, cells were harvested and treated with 10 μM CM-H2DCFDA for 20 min, and 1 × 105 cells were analyzed by flow cytometry.
CHENSpm-, but not CPENSpm-Induced PCD, is Associated with G2/M Arrest.
Cell cycle analysis of H157 cells after treatment with CPENSpm and CHENSpm demonstrated distinctly different profiles. The cell cycle profile of cells treated with 10 μM CPENSpm for 24 h was essentially unchanged compared with control although cells treated with 10 μM CPENSpm were clearly undergoing PCD by this time (Fig. 4). By contrast, cells treated with 10 μM CHENSpm for 24 h demonstrated a profound G2/M block (≈61% cells in G2/M), which was observable as early as 16 h (Fig. 4).
Figure 4.
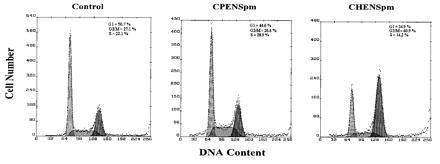
Flow cytometric analysis of NCI H157 nonsmall lung carcinoma cells. Cells were untreated, treated with 10 μM CPENSpm, and treated with 10 μM CHENSpm for 24 h. Cell number and DNA content are represented by the ordinate and abscissa, respectively. The G1, S, and G2/M fractions were shaded and quantitated by using the multicycle software package.
DISCUSSION
The unsymmetrically substituted polyamine analogue CPENSpm has been shown to exhibit a cell type-specific cytotoxic activity against human nonsmall cell lung carcinoma lines in culture (24). Although the treatment of analogue-sensitive cells is accompanied by a superinduction of SSAT activity, reduction of ODC activity, depletion of intracellular polyamines, and accumulation of analogue, the precise mechanisms underlying the cytotoxic response have not been elucidated. It recently has been demonstrated that CPENSpm-treated NCI H157 cells undergo PCD (21). The results of the current study demonstrate that the PCD is associated with HMW DNA and nuclear fragmentation in CPENSpm-treated NCI H157 cells and suggest a mechanism, demonstrating that the observed damage is at least partially a result of oxidative stress from the production of H2O2 by the SSAT/PAO pathway.
Treatment with exogenous catalase significantly reduced CPENSpm-induced HMW DNA fragmentation, demonstrating that H2O2 contributes to the DNA fragmentation produced in CPENSpm-treated cells. It should be noted that exogenous catalase is thought to act on endogenously produced H2O2 in at least two ways. First, H2O2 is a long-lived, readily diffused, reactive oxygen species, and once outside the cell it can be detoxified by the exogenous enzyme (30). Alternatively, a cell type-specific accumulation of exogenous catalase through an apparent receptor-mediated, energy-dependent system has been observed (31). It is currently not known which mechanism, or combination of mechanisms, is operative in the NCI H157 cells. Also consistent with the hypothesis that H2O2 is contributing to the induction of PCD is the trend observed in the CM-H2DCFDA experiments. These experiments indicated that there was an increase in H2O2 production leading to an increase in fluorescence in response to CPENSpm treatment. However, treatment with exogenous catalase has no significant effect on PCD produced by CHENSpm. The extracellular serum amine oxidase was excluded as a possible source of H2O2 through the lack of effect of the serum amine oxidase inhibitor, aminoguanidine. By contrast, the specific inhibitor of the intracellular PAO, MDL 72,527, significantly decreased the amount of CPENSpm-induced HMW DNA fragmentation. The effect of MDL 72,527 was not related to competition for uptake of CPENSpm by MDL 72,527; the treatment with the PAO inhibitor had no effect on the ability of cells to accumulate CPENSpm. The results with the fluorescent probe in the cotreatment experiments suggest that both catalase and MDL 72,527 reduced the amount of H2O2 produced by CPENSpm treatment, completely consistent with the results of the HMW DNA fragmentation experiments. However, similar to results with catalase, the PAO inhibitor had no effect on CHENSpm-induced PCD as measured by HMW DNA fragmentation, suggesting that cells undergoing PCD induced by CHENSpm are not under oxidative stress similar to those cells treated with CPENSpm. Basu et al. (32) have demonstrated that small changes in the structure of polyamine analogues can have significant effects on the interaction with DNA. Their data suggest the potential of different direct effects on DNA by CPENSpm and CHENSpm. It is also interesting to note that, in the 24-h exposure experiments, CHENSpm appeared to accumulate to a greater extent in those cells treated with the PAO inhibitor compared with cells treated only with CHENSpm. These results suggest the possibility that CHENSpm may be a substrate of PAO. However, no breakdown products attributable to CHENSpm were observed in the HPLC analysis of these samples.
That CPENSpm and CHENSpm initially kill cells by different mechanisms is underscored by the completely different cell cycle profiles observed after treatment with the individual agents. The cytotoxic activity of CPENSpm does not appear to have a profound effect on the cell cycle, killing cells without an apparent block at any stage of the cell cycle progression. In contrast, CHENSpm treatment produces a significant G2/M block concurrent with its cytotoxic activity. This effect by CHENSpm is somewhat unusual in that interference with polyamines metabolism and polyamine depletion generally produce a G1/S block if they have any measurable effect on the cell cycle at all (33, 34).
It should be noted that protection against CPENSpm toxicity provided by treatment with exogenous catalase, other antioxidants, or the PAO inhibitor in these experiments was neither complete nor permanent. Therefore, it must be stressed that these results indicate that oxidative stress is only one component of CPENSpm-induced PCD. Inhibition of only one component in a multi-component system may be expected to only influence the kinetics of the PCD process. These data are consistent with the possibility that CPENSpm and CHENSpm may share a common reactive oxygen species-independent mechanism that cannot be altered by antioxidants.
It is unlikely that polyamine depletion alone is responsible for the observed apoptosis because, in NCI H157 cells, depletion of natural polyamines by 2-difluoromethylornithine does not result in cell death (36), and CHENSpm only has a minor effect on polyamine levels. These results are consistent with the results of Albanese et al., which demonstrated in ODC overproducing cells that the accumulation of N1,N12-bis(ethyl)spermine, not polyamine depletion, corresponded best with the cytotoxic effect of the analogue (37).
Extra- and intracellular polyamines have been shown to be involved in the cell death process. Pierce, Parchment, and colleagues first postulated that H2O2 from extracellular serum amine oxidase-dependent catabolism of polyamines was a mediator of PCD in the murine embryo, limb buds, and blastocysts (38–41). Other recent studies have implicated the intracellular polyamines to be involved in the cell death process. Induction of ODC (both mRNA and activity), depletion of intracellular polyamines, and induction of SSAT activity were shown in dexamethasone-induced PCD in rat thymocytes (14). An imbalance of polyamine metabolism was proposed to be a trigger of PCD in heat shock treatment- and γ-irradiation-induced apoptosis, in which induction of ODC mRNA and activity was observed without subsequent increase intracellular spermidine and spermine levels (20). Packham and Cleveland (18) suggested the possibility that the production of H2O2 by the SSAT/PAO pathway might mediate PCD in interleukin 3-dependent murine myeloid cells. In their study, PCD was induced by enforced expression of ODC concurrent with overexpression of c-myc, and inhibition of ODC reduced this apoptotic process. They proposed that excessive intracellular polyamines produced by overexpression of ODC might be catalyzed by the SSAT/PAO pathway, thus producing H2O2, which mediates PCD. However, in a more recent study, Packham et al. (42) have demonstrated that, in a myeloid system, cytokine withdrawal or c-myc-enforced death can occur without increases in reactive oxygen species.
In summary, CPENSpm treatment of NCI H157 cells superinduces SSAT activity concurrently with the production of HMW DNA fragmentation. The early generation of HMW DNA fragments can be inhibited by various antioxidant compounds, suggesting that the molecular insult is a reactive oxygen species. The prevention of DNA fragmentation by catalase, but not by Cu/Zn-SOD, suggests that the reactive oxygen species is H2O2. Most importantly, the specific inhibition of PAO by MDL 72,527 significantly reduces the amount of HMW DNA fragmentation, suggesting that the source of H2O2 is from the two-step polyamine catabolic pathway. These results provide the first evidence that the superinduction of SSAT may have a direct role in DNA damage and cell death in specific cell types. The mechanism of CPENSpm-induced cytotoxicity described here has important implications for the development of new antitumor agents. Specifically, it provides a unique pathway that may be exploited because the superinduction of SSAT is a relatively rare, tumor-specific response to certain agents (43, 44). Furthermore, the possibility that the production of H2O2 by polyamine catabolism may be a general mechanism for cellular suicide must be considered. Additional studies will be necessary to determine how widespread this phenomenon is and what other mechanisms are responsible for the apparent H2O2-independent DNA damage observed.
Table 1.
Comparison of effects of 10 μM CPENSpm and CHENSpm with or without catalase or MDL 72,527 on HMW DNA fragmentation in NCI H157 cells
| Treatment* | Polyamines, nmol/mg protein†
|
SSAT activity, pmol/mg protein/min§ | HMW DNA fragmentation, %¶ | Relative inhibition of DNA fragmentation, % | |||
|---|---|---|---|---|---|---|---|
| Put | Spd | Spm | Analogue | ||||
| None | 3.1 | 12.9 | 24.0 | 146 ± 3 | 1 ± 1 | — | |
| CPENSpm | ND‡ | ND | 1.2 | 53.8 | 28,561 ± 286 | 37 ± 5 | — |
| CHENSpm | 8.5 | 9.3 | 19.7 | 35.5 | 468 ± 2 | 43 ± 7 | |
| CPENSpm/catalase | 3.4 | 0.5 | 2.1 | 45.0 | 29,718 ± 892 | 8 ± 3 | 77 |
| CHENSpm/catalase | 18.5 | 7.0 | 10.9 | 36.3 | 532 ± 2 | 39 ± 9 | 9 |
| CPENSpm/MDL 72,527 | ND | ND | 2.4 | 47.7 | 24,073 ± 722 | 12 ± 8 | 67 |
| CHENSpm/MDL 72,527 | 5.5 | 6.7 | 18.2 | 43.0 | 580 ± 3 | 38 ± 9 | 12 |
Treatment of NCI H157 cells, where indicated, was performed with 10 μM CPENSpm for 24 h in the presence or absence of 500 units/ml catalase or 250 μM MDL 72,527.
Values represent the means of duplicate determinations.
ND, <0.05 nmol/mg protein.
Values for SSAT enzyme activities represent the means of triplicate determinations ± SD.
Values represent the means of triplicate determination SD, quantitation based on phosphoimage analysis as described in Materials and Methods.
Acknowledgments
We thank Drs. John T. Isaac and Christophe Lengauer for their advice concerning the morphologic studies. This research was supported in part by National Institutes of Health Grants ES07141, CA57545, CA63552, and CA51085.
ABBREVIATIONS
- CHENSpm
N1-ethyl-N11-[(cycloheptyl)methyl]-4,8,-diazaundecane
- CM-H2DCFDA
5-(and -6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (mixed isomers)
- CPENSpm
N1-ethyl-N11-[(cyclopropyl)methyl]-4,8,-diazaundecane
- Cu/Zn-SOD
Cu/Zn-superoxide dismutase
- HMW
high molecular weight
- MDL 72
527, N,N′-butadienyl)-1,4-butane-diamine
- ODC
ornithine decarboxylase
- PAO
polyamine oxidase
- PCD
programmed cell death
- SSAT
spermidine/spermine N1-acetyltransferase
References
- 1.Kerr J F R, Wyllie A H, Currie A R. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wyllie A H, Kerr J F R, Currie A R. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 3.Majno G, Joris I. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 4.Ellis R E, Yuan J, Horvitz H R. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 5.Stellar H. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 6.Vaux D L, Strasser A. Proc Natl Acad Sci USA. 1996;93:2239–2244. doi: 10.1073/pnas.93.6.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corcoran G B, Fix L, Jones D P, Moslen M T, Nicotera P, Oberhammer F A, Buttyan R. Toxicol Appl Pharmacol. 1994;128:169–181. doi: 10.1006/taap.1994.1195. [DOI] [PubMed] [Google Scholar]
- 8.Pegg A E. Cancer Res. 1988;48:759–774. [PubMed] [Google Scholar]
- 9.Marton L J, Pegg A E. Annu Rev Pharmacol. 1995;35:55–91. doi: 10.1146/annurev.pa.35.040195.000415. [DOI] [PubMed] [Google Scholar]
- 10.Janne J, Alhonen L, Leinonen P. Ann Med. 1991;23:241–259. doi: 10.3109/07853899109148056. [DOI] [PubMed] [Google Scholar]
- 11.Porter C W, Bernacki R J, Miller J, Bergeron R J. Cancer Res. 1993;53:581–586. [PubMed] [Google Scholar]
- 12.Bergeron R J, Neims A H, McManis J, Hawthorne J S, Vinson J R T, Bortell R, Ingeno M J. J Med Chem. 1988;31:1183–1190. doi: 10.1021/jm00401a019. [DOI] [PubMed] [Google Scholar]
- 13.Casero R A, Pegg A E. FASEB J. 1993;7:653–661. [PubMed] [Google Scholar]
- 14.Desiderio M A, Grassilli E, Bellesia E, Salomoni P, Franceschi C. Cell Growth Diff. 1995;6:505–513. [PubMed] [Google Scholar]
- 15.Poulin R, Pelletier G P, Pegg A E. Biochem J. 1995;311:723–727. doi: 10.1042/bj3110723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dypbuket J M, Ankarcrona M, Burkitt M, Sjoholm A, Strom K, Orrenius S, Nicotera P. J Biol Chem. 1994;269:30553–30560. [PubMed] [Google Scholar]
- 17.Tobias K E, Kahana C. Cell Growth Diff. 1995;6:1279–1285. [PubMed] [Google Scholar]
- 18.Packham G, Cleveland J L. Mol Cell Biol. 1994;14:5741–5747. doi: 10.1128/mcb.14.9.5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Min A, Hasuma T, Yano Y, Matsui-Yusas I, Otani S. J Cell Physiol. 1995;165:615–623. doi: 10.1002/jcp.1041650320. [DOI] [PubMed] [Google Scholar]
- 20.Grassilli E, Desiderio M A, Bellesia E, Salomoni P, Benatti F, Franceschi C. Biochem Biophys Res Commun. 1995;216:708–714. doi: 10.1006/bbrc.1995.2679. [DOI] [PubMed] [Google Scholar]
- 21.McCloskey D E, Yang J, Woster P M, Davidson N E, Casero R A. Clin Cancer Res. 1996;2:441–446. [PubMed] [Google Scholar]
- 22.McCloskey D E, Casero R A, Woster P M, Davidson N E. Cancer Res. 1995;55:3233–3236. [PubMed] [Google Scholar]
- 23.McCloskey D E, Prestigiacomo L J, Woster P M, Casero R A, Davidson N E. Proc Am Assoc Can Res. 1996;37:400. (abstr.). [Google Scholar]
- 24.Casero R A, Mank A R, Saab N H, Wu R, Dyer W, Woster P M. Cancer Chemother Pharmacol. 1995;36:69–74. doi: 10.1007/BF00685735. [DOI] [PubMed] [Google Scholar]
- 25.Seiler N. Prog Brain Res. 1995;106:333–344. doi: 10.1016/s0079-6123(08)61229-7. [DOI] [PubMed] [Google Scholar]
- 26.Woster P M. In: Polyamines: Regulation and Molecular Interaction. Casero R A, editor; Casero R A, editor. Austin, TX: Landes; 1995. pp. 171–186. [Google Scholar]
- 27.Hoyt D G, Mannix R J, Rusnak J M, Pitt B R, Lazo J S. Am J Physiol. 1995;269:L171–L177. doi: 10.1152/ajplung.1995.269.2.L171. [DOI] [PubMed] [Google Scholar]
- 28.Southern E M. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 29.Hockenbery D M, Oltvai Z N, Yin X-M, Milliman C L, Korsmeyer S J. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 30.Dietch A D, Law H, White R D. J Histochem Cytochem. 1982;30:967–972. doi: 10.1177/30.9.6182188. [DOI] [PubMed] [Google Scholar]
- 31.Al-Mehdi A, Shuman H, Fisher A B. Lab Invest. 1994;70:579–587. [PubMed] [Google Scholar]
- 32.Sundaresan M, Yu Z-X, Ferrans V J, Irani K, Finkel T. Science. 1996;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 33.Basu H S, Schwietert H C A, Feuerstein B G, Marton L J. Biochem J. 1990;269:329–334. doi: 10.1042/bj2690329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pohjanpelto P, Nordling S, Knuutila S. Cytometry. 1994;16:331–338. doi: 10.1002/cyto.990160407. [DOI] [PubMed] [Google Scholar]
- 35.Bergeron C J, Basu H S, Marton L J, Deen D F, Pellarin M, Feuerstein B G. Cancer Chemother Pharmacol. 1995;36:411–417. doi: 10.1007/BF00686190. [DOI] [PubMed] [Google Scholar]
- 36.Casero R A, Go B, Theiss H W, Smith J, Baylin S B, Luk G D. Cancer Res. 1987;47:3964–3967. [PubMed] [Google Scholar]
- 37.Albanese L, Bergeron R J, Pegg A E. Biochem J. 1993;291:131–137. doi: 10.1042/bj2910131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierce G B, Lewellyn A L, Parchment R E. Proc Natl Acad Sci USA. 1989;86:3654–3658. doi: 10.1073/pnas.86.10.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parchment R E, Lewellyn A, Swartzendruber D, Pierce G B. Proc Natl Acad Sci USA. 1990;87:4340–4344. doi: 10.1073/pnas.87.11.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parchment R E, Pierce G B. Cancer Res. 1989;49:6680–6686. [PubMed] [Google Scholar]
- 41.Parchment R E. Int J Dev Biol. 1993;37:75–83. [PubMed] [Google Scholar]
- 42.Packhamn G, Ashmun R A, Cleveland J L. J Immunol. 1996;156:2792–2800. [PubMed] [Google Scholar]
- 43.Casero R A, Mank A R, Xiao L, Smith J, Bergeron R J, Celano P. Cancer Res. 1992;52:5359–5363. [PubMed] [Google Scholar]
- 44.Shappell N W, Miller J T, Bergeron R J, Porter C W. Anticancer Res. 1992;12:1083–1090. [PubMed] [Google Scholar]