

Abstract
The past decade has seen an expansion of research and knowledge on pharmacotherapy for the treatment of alcohol dependence. The Food and Drug Administration (FDA)–approved medications naltrexone and acamprosate have shown mixed results in clinical trials. Oral naltrexone and naltrexone depot formulations have generally demonstrated efficacy at treating alcohol dependence, but their treatment effect size is small, and more research is needed to compare the effects of different doses on drinking outcome. Acamprosate has demonstrated efficacy for treating alcohol dependence in European trials, but with a small effect size. In U.S. trials, acamprosate has not proved to be efficacious. Research continues to explore which types of alcohol-dependent individual would benefit the most from treatment with naltrexone or acamprosate. The combination of the two medications demonstrated efficacy for treating alcohol dependence in one European study but not in a multi-site U.S. study. Another FDA-approved medication, disulfiram, is an aversive agent that does not diminish craving for alcohol. Disulfiram is most effective when given to those who are highly compliant or who are receiving their medication under supervision. Of the non-approved medications, topiramate is among the most promising, with a medium effect size in clinical trials. Another promising medication, baclofen, has shown efficacy in small trials. Serotonergic agents such as selective serotonin reuptake inhibitors and the serotonin-3 receptor antagonist, ondansetron, appear to be efficacious only among certain genetic subtypes of alcoholic. As neuroscientific research progresses, other promising medications, as well as medication combinations, for treating alcohol dependence continue to be explored.
INTRODUCTION
Alcohol dependence is a common disorder. Globally and in the U.S., alcohol dependence ranks 5th and 3rd, respectively, on the list of preventable causes of morbidity and mortality [1]. In 2000, the U.S. had 20,687 alcohol-related deaths, excluding accidents and homicides, with an overall estimated cost to the nation of about $185 billion [1].
Alcohol dependence is a chronic relapsing medical disorder [2]. Notwithstanding its psychological and social ramifications, once established, alcohol dependence is essentially a brain disorder that bears many of the characteristics of other medical relapsing disorders such as diabetes and hypertension. Indeed, without a pharmacological adjunct to psychosocial therapy, the clinical outcome is poor, with up to 70% of patients resuming drinking within one year [3,4].
Alcohol dependence is a treatable disorder when efficacious medicines are added to enhance the effects of psychosocial treatment. The development of these medicines has been facilitated by advances in the neurosciences that have implicated several target neurotransmitter systems, such as those within the cortico-mesolimbic dopamine (CMDA) pathway, which mediate alcohol’s reinforcing effects associated with its abuse liability. Additionally, it is now known that some alcoholics may possess a biological predisposition to the disease. These biologically vulnerable alcoholics can be expected to benefit from specific adjunctive medication targeted toward correcting or ameliorating the underlying abnormalities. Further, we are now better at controlling the “dose” of psychosocial treatments through a manual-guided treatment approach, thereby enabling the optimization of how particular medicines can be combined with adjunctive psychosocial treatment.
Recently, the treatment of alcohol dependence has been advanced by development of new models as well as broader therapeutic objectives. An important model is that with appropriate pharmacotherapy it is possible to initiate treatment for alcohol dependence while the individual is still drinking heavily and at the point of maximum crisis and help-seeking behavior [5]. To broaden access to treatment, effective but brief and standardized behavioral treatment has been developed to accompany medication delivery; thus, these medicines can now be provided more readily in the general practice setting [6,7]. Finally, it is now better recognized that although abstinence remains the ultimate goal in treating alcohol-dependent individuals, reducing the frequency of heavy drinking has the major impact of decreasing alcohol-related consequences and improving quality of life [5].
In this review, I focus on the development of those medications for which there is clinical information and that have been designed to reduce the desire to drink, to promote abstinence, or both. Basically, of the numerous neurotransmitter systems that have been identified for the development of new medicines, the most promising compounds appear to be those that modulate the function of opioids, glutamate with or without gamma-aminobutyric acid (GABA), and serotonin (5-HT). Other putative therapeutic medications including direct modulators of dopamine function and enzyme inhibitors also shall be discussed. Each subsection of this article provides an overview of the basic science, clinical studies, and future directions for the development of specific promising medications from these neurobiological systems. Emphasis is made in places where the development of a particular medicine has advanced the development of a new treatment model or broadened therapeutic objectives. I conclude the article with remarks pertaining to current barriers to treatment and how they might be overcome.
OPIOIDS: MU RECEPTOR ANTAGONIST — NALTREXONE
Basic science and human laboratory studies
The endogenous opioid system, particularly through its interactions with the CMDA system, is involved in the expression of alcohol’s reinforcing effects [8-14] (Fig. 1). Obviously, opioid receptors also have interactions with other neurotransmitters, including those in the glutamate [15], GABA [16], 5-HT [17], cannabinoid [18] and perhaps glycine [19] systems, that contribute to its effects on ethanol intake.
Fig. 1.
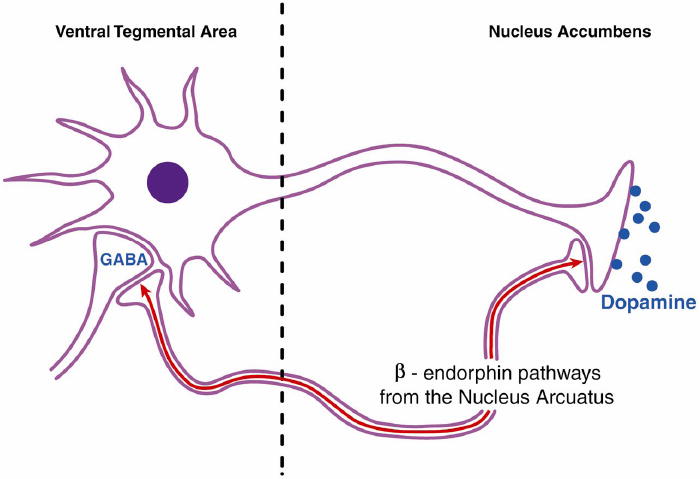
Schematic representation of opioid interactions with the cortico-mesolimbic dopamine reward pathway. Functional activity of beta-endorphin pathways primarily originating from the nucleus arcuatus can lead to increased dopamine release in the nucleus accumbens via two mechanisms. First, beta-endorphins can disinhibit the tonic inhibition of gamma-aminobutyric acid (GABA) neurons on dopamine cells in the ventral tegmental area [10-12]. Second, beta-endorphins can stimulate dopamine cells in the nucleus accumbens directly. Both mechanisms may be important for alcohol reward. Alcohol stimulates beta-endorphin release in both the nucleus accumbens and ventral tegmental area [13]. Mu receptor antagonists such as naloxone and naltrexone block these central effects of beta-endorphins [9,13]. Embellished from Gianoulakis [13]. Reprinted from Figure 1 in Johnson and Ait-Daoud [14], with the kind permission of Springer Science and Business Media.
Even though naltrexone has some affinity for the kappa-opioid receptor [20], its principal pharmacological effect on alcohol consumption is through blockade of the mu-opioid receptor as mice that lack the mu-opioid receptor do not self-administer alcohol [21]. Further, alcohol intake increases beta-endorphin release in brain regions such as the nucleus accumbens [22-24], an effect that is blocked by naltrexone [25]. Mu receptor antagonists such as naltrexone and naloxone also suppress ethanol intake across a wide range of animal paradigms [26-36] cf. [37-39]. More recently, there also has been interest in elucidating the role of the hypothalamic-pituitary-adrenocortical axis in stress-induced ethanol consumption and sensitivity and how this might be influenced by naltrexone treatment [40].
Ethanol has complex neurobiological interactions that affect the production, secretion, and binding of opioids to their receptors [41], thereby hinting at a fundamental mechanistic process linking the two. This relationship does, however, remain imperfectly understood. For example, animals bred for high ethanol preference exhibit an exaggerated reactive rise in beta-endorphin level following ethanol intake [42]. Yet, naltrexone’s ability to suppress ethanol-associated increases in beta-endorphin level appears greater in animals bred for low rather than high preference for alcohol [25]. Indeed, from a group of animals in the beta-endorphin-deficient mutant mouse line — C57BL/6-Pomc1(tm1Low) — the highest ethanol consumption occurred in the heterozygotes (50% beta-endorphin deficient) and not the homozygotes (no beta-endorphin) or control group of sibling wild type mice from the same strain [43]. These findings do, however, suggest that molecular genetic differences that alter beta-endorphin expression, not simply its plasma levels, modulate the level of response to naltrexone. Nevertheless, there is growing evidence in humans that differences in the OPRM1 mu-opioid receptor gene are associated with differential therapeutic response to naltrexone — a theme that is explored in detail later in this review.
Human laboratory studies that have evaluated naltrexone’s effects on alcohol-induced positive subjective mood and craving have yielded mixed results. Although it has been shown that naltrexone can reduce alcohol-induced positive subjective mood, albeit with increased sedation [44], and increase the latency to consume alcohol among social drinkers [45], others have reported no effect [46]. It does, however, appear that a positive familial loading for alcoholism might predict the potential anti-drinking and anti-craving effects of naltrexone in human laboratory studies. For example, King et al. [47] showed that social drinkers with a familial loading for alcoholism were more likely than those without it to exhibit a decrease in the stimulant effects of alcohol following naltrexone treatment. Nevertheless, they also reported concomitant negative mood exemplified by increased tension, fatigue, and confusion and decreased vigor, as well as notable adverse events such as nausea and vomiting following naltrexone. More recently, Krishnan-Sarin et al. [48] have shown that individuals with a family history of alcoholism, compared with their family history-negative counterparts, consumed less alcohol in a laboratory paradigm. Obviously, these results would lead to the speculation that a genetic explanation for differential response to naltrexone’s effects on craving and alcohol consumption among alcohol-dependent individuals is being studied in the human laboratory. Nevertheless, even here, what has been demonstrated is that naltrexone increases the urge to drink among alcohol-dependent individuals who are aspartate (Asp) carriers of the OPRM1 gene but has no effect on their homozygote, i.e., asparagine-carrying, counterparts in a cue-reactivity laboratory paradigm [49]. Despite the dissimilarities between studies, including the subject’s motivation toward seeking treatment, experimental set, setting, expectations, and paradigm, these results do appear to be in contrast with the report that naltrexone preferentially protected against relapse in Asp-carrying alcohol-dependent individuals [50]. The implications of these findings are discussed in the clinical subsection below.
In sum, basic science studies support the finding that naltrexone can reduce ethanol drinking and related behaviors in animals. Naltrexone appears most effective in suppressing the expected ethanol-induced increase in beta-endorphin level among animals that exhibit an exaggerated beta-endorphin response. The molecular genetic construct for understanding preferential response to naltrexone is not well understood and is even contrary to expectations. Generally, human laboratory studies provide some support for naltrexone as a medication that can reduce craving for alcohol as well as its consumption; however, these effects appear to be more readily demonstrable among individuals with high familial loading for alcoholism. An initial molecular genetic exploration did not demonstrate that naltrexone’s anti-drinking effect is greatest among non-treatment-seeking, alcohol-dependent individuals who carry the Asp variant of the OPRM1 gene.
Clinical studies with oral naltrexone
In 1994, the Food and Drug Administration (FDA) approved naltrexone for the treatment of alcohol dependence based on data from two relatively small (total N = 167) studies [51,52]. In those studies, recently abstinent, alcohol-dependent individuals who received naltrexone (50 mg/day), compared with their counterparts who got placebo, were less likely to relapse during the treatment period of 12 weeks. Nevertheless, 5 months after treatment, the relapse rates for the naltrexone and placebo groups were similar. The anti-alcohol-craving effects that were ascribed to naltrexone were based on three findings. First, individuals with the highest level of baseline craving appeared to benefit the most from naltrexone [53]. Second, abstinent individuals who had received naltrexone had less of an impulse to initiate drinking [54]. Third, even among those who sampled alcohol, less pleasure was derived from the beverage [55]. These earlier studies were limited by the fact that only male veterans were tested in one of the studies [52], and either there was no biomarker used to corroborate the self-reported data [51] or when the liver enzyme gamma-glutamyl transferase (GGT) was used as a biomarker the results were not contributory [52] — presumably due to the relative insensitivity of this measure to capture transient drinking patterns.
Notably, in two large meta-analytic studies [56,57], naltrexone has been demonstrated to be efficacious at reducing the risk of relapse among recently abstinent, alcohol-dependent individuals. What emerged from this literature review was that naltrexone’s effect size was small, with a corresponding number needed to treat (i.e., the number of individuals who need to be treated to prevent relapse in a single individual) of 7. Another threat to demonstrating efficacy for naltrexone is not having quite high enough levels of medication compliance. Indeed, in a 3-month follow-up and systematic replication of their study, Volpicelli et al. [58] only found a significant effect of naltrexone treatment compared with placebo recipients if the pill taking rate exceeded 90%; even here, the difference in the percentage of drinking days between the naltrexone and placebo groups was small — 3% and 11%, respectively. Perhaps because of this small effect size, some studies have failed to demonstrate naltrexone’s efficacy in treating alcohol dependence. For instance, in the UK collaborative trial led by Chick et al., no overall difference was found between the naltrexone 50 mg/day and placebo groups on any of the endpoint measures; however, when individuals with less than 80% pill-taking compliance were excluded from the analysis, naltrexone was associated with a lower percentage of days drinking compared with placebo — 12% vs. 20%, respectively [59,60]. With naltrexone treatment, reduced pill-taking compliance is typically the result of adverse events such as nausea that can be reported as significant in up to 15% of trial participants [61]. Therefore, new technologies that aim to improve compliance by delivering naltrexone in depot form might possess a therapeutic advantage to the oral formulation. These technologies are discussed later in this section. Importantly, the recent publication of the results of the NIAAA-sponsored COMBINE study (N = 1383) has served to underscore that naltrexone (100 mg/day) plus medication management to enhance compliance compared with placebo reduced the risk of a heavy drinking day (hazard ratio = 0.72; 97.5% CI = 0.53–0.98; p = 0.02) [62]. Uniquely, this study used a higher naltrexone dose (i.e., 100 mg/day vs. 50 mg/day), and the high compliance rate of pill-taking — 85.4% — improved clinical outcome.
Recently, it has been proposed that individuals with the Asp variant of the OPRM1 gene exhibited preferentially higher relapse prevention rates when receiving naltrexone treatment [50]. As described previously, a similar response to naltrexone treatment on cue-elicited craving was not observed among non-treatment-seeking, alcohol-dependent individuals in a human laboratory study [49]. Further, a recent clinical trial did not find a preferential effect of naltrexone treatment on any of the variants of the OPRM1 gene [63]. Notably, the functional importance of variation in the OPRMI gene is still being elucidated. Although earlier studies in transfected cells suggested that the OPRM1-Asp40 variant had a 3-fold higher affinity for beta-endorphin than OPRM1-Asn40, which would suggest enhanced function [64], this has not been corroborated by others [65,66]. Recent in vitro transfection studies have, however, suggested that the G118 allele might be associated with lower OPRM1 protein expression than the A118 allele [67]. A further complication to estimating the general clinical significance of the effects of the Asp40 allele on pharmacotherapeutic response to naltrexone is that its frequency can vary considerably between populations — from as low as 0.047 in African Americans to 0.154 in European Americans, and as high as 0.485 among those of Asian descent [68,69]. More molecular genetic studies are, therefore, needed to elucidate fully the mechanistic effects of the Asp40 allele, and to establish whether or not naltrexone response varies by variation at the OPRM1 gene.
Certain clinical characteristics have, however, been associated with good clinical response to naltrexone, and these include a family history of alcohol dependence [53,70,71] or strong cravings or urges for alcohol [71].
In sum, the majority of the data confirm that naltrexone is an efficacious medication for treating alcohol dependence. The therapeutic treatment effect size is, however, small, and poor pill-taking compliance can be associated with poor clinical outcome. There remains a dearth of published studies on the effects of different doses of naltrexone on drinking outcome. Further research is needed to establish whether naltrexone’s therapeutic efficacy in treating alcohol dependence differs among individuals who have variants of the OPRM1 gene. Alcohol-dependent individuals with a positive family history for the disease and individuals with strong cravings for alcohol appear to benefit the most from naltrexone treatment.
Clinical studies with depot naltrexone
Three extended-release formulations of naltrexone for deep intramuscular injection have been developed — Vivitrol® (Alkermes, Inc., Cambridge, MA, USA), Naltrel® (Drug Abuse Sciences, Inc., Paris, France), and Depotrex® (Biotek, Inc., Woburn, MA, USA). The premise for developing these depot formulations of naltrexone is three-fold. First, a well formulated depot preparation can maintain relatively constant plasma levels by producing a slow but regular release of naltrexone. Individuals who take oral naltrexone and have notable adverse events such as nausea that can lead to study discontinuation probably experience this phenomenon due to the rapid rise in plasma levels following initial doses of oral naltrexone. Hence, a depot formulation might be expected to decrease these initial adverse events if it provided a more gradual rise in naltrexone plasma levels. Second, by providing a monthly depot preparation, compliance with receiving the medication is optimized and should be greater than reliance on remembering to take tablets. Third, because plasma levels should remain relatively constant throughout the month following the administration of a depot preparation, there should be relatively greater exposure to the therapeutic dose, thereby facilitating good clinical outcome. Information pertaining to the three depot preparations of naltrexone that are being tested is provided below.
Vivitrex® or Vivitrol®
Vivitrex®, or Vivitrol® as it is known now, is naltrexone formulated into poly-(lactide-co-glycolide) [72], small-diameter (<100 μm), injectable microspheres that contain other proprietary active moieties, which lead to its extended-release properties lasting for several weeks [73]. In 2004, Johnson et al. [74] published the initial safety, tolerability, and efficacy trial of Vivitrex® for treating alcohol dependence. The design of the study was a 16-week randomized, placebo-controlled, double-blind clinical trial. Of the 25 alcohol-dependent individuals who participated in the trial, five of them got placebo and the remainder (n = 20) got 400 mg of Vivitrex®. Results of that trial showed the safety of Vivitrex®, with the most common adverse events being non-specific abdominal pain, nausea, pain at the injection site, and headaches. None of the placebo recipients dropped out due to adverse events; in contrast, two of those who got Vivitrex® discontinued for that reason. Due to the unbalanced design and small subject numbers, any inferences regarding efficacy had to be viewed quite cautiously. Nevertheless, there was a trend for those on Vivitrex®, compared with placebo, to have a lower percentage of heavy drinking days — 11.7% vs. 25.3%. Later, in a large placebo-controlled, double-blind, randomized, multi-site, 24-week clinical trial, Garbutt et al. [75] showed that high-dose Vivitrex® (380 mg) recipients had a significantly lower percentage of heavy drinking days than those who got placebo (hazard ratio = 0.75; 95% CI = 0.60–0.94; p = 0.02). Recipients of low-dose Vivitrex® (190 mg) had outcomes similar to those who got placebo. The treatment response signal in the high-dose Vivitrex® recipients came from the male participants as the effect of both Vivitrex® doses was no different from that in women who took placebo (hazard ratio = 1.23; 95% CI = 0.85–1.78; p = 0.28). The lack of efficacy for Vivitrol® in women has been ascribed to greater subclinical affective symptoms, less of a family history of alcoholism (which is meant to be associated with good clinical outcomes to naltrexone), more responsiveness to placebo, and more clinical heterogeneity in the sample. In contrast with the premise for developing depot preparations, the dropout rate of 14.1% in the high-dose Vivitrex® group was similar to that reported in studies with oral naltrexone. The chosen objective biomarker to corroborate the self-reported data — GGT — did not show a difference between any of the Vivitrex® doses and the placebo group. The common reasons for study discontinuation were injection site reactions, headaches, and nausea. Serious adverse events were reported in two participants taking active medication that resulted in an interstitial pneumonia and an allergic-type eosinophilic pneumonia, both of which resolved after medical treatment. Thus, the evidence remains that Vivitrol® appears to be efficacious in preventing heavy drinking in men; however, it was approved by the FDA for treatment of both men and women based on the extant literature on naltrexone as a treatment for alcohol dependence. The expected advantage of Vivitrol® to increase compliance did not materialize quickly although this might become more manifest in generic treatment settings rather than a closely monitored clinical trial. The potential for hypersensitivity reactions to Vivitrol®, while small, does require post-marketing evaluation by the FDA.
Naltrel®
Naltrel® consists of naltrexone incorporated into microspheres of poly-(DL-lactide) polymer. These microspheres, stored in single-dose vials, are suspended in a diluent that contains carboxymethylcellulose, mannitol, polysorbate 80, and water for injection. The polylactide polymer is metabolized to water and carbon dioxide. Then, as the microspheres degrade, naltrexone is released. In 2004, Kranzler et al. [76] studied the safety and efficacy of Naltrel® in treating male and female alcohol-dependent individuals receiving monthly motivation enhancement-based therapy in a double-blind, placebo-controlled, 3-month randomized controlled trial (N = 157). The initial dose of Naltrel® (150 mg) was delivered as a deep intramuscular injection into each buttock, and subsequent monthly doses were just 150 mg. Placebo injections were provided at the same frequency and constitution but lacked the active compound. Adverse events reported significantly more frequently in the Naltrel® group than in the placebo group included injection site reactions, chest pain, and upper abdominal pain. Placebo recipients were, however, more likely to report irritability than those who got Naltrel®. While 6 (3.8%) of the placebo recipients dropped out, 13 (8.2%) of those who got Naltrel® discontinued treatment. Naltrel® was superior to placebo at increasing the mean number of cumulative abstinent days (52.8 days, 95% CI 48.5–57.2 days, vs. 45.6 days, 95% CI 41.1–50.0 days, respectively; p = 0.018) and having a longer median time to first drink (5 days, 95% CI 3–9 days, vs. 3 days, 95% CI 2–4 days, respectively; p = 0.003). The effects of gender on treatment outcome were not examined.
Somewhat in contrast, a single-site, 6-week trial of 16 alcohol-dependent individuals who received one intramuscular dose of Naltrel® (300 mg) [77] suggested low tolerability, with 198 adverse events being reported. Of these, 17 were considered to be severe and included fatigue, gastrointestinal pain, irritability, nausea, somnolence (two reports), headache (four reports from three subjects), injection site pain, injection site mass, lethargy, depression, increased level of GGT (an index of heavy drinking [78]), back pain, and flatulence. No serious adverse events were reported. Drinking outcomes showed an improving trend over the duration of the trial.
Nevertheless, further studies on the safety and efficacy of the Naltrel® formulation are warranted. Additional data are needed to determine whether, as with Vivitrol®, there is a differential response on drinking outcomes between men and women who get Naltrel®.
Depotrex®
Rather little public information is available on the Depotrex® depot formulation. Like the other depot formulations, Depotrex® appears to provide steady increases in plasma naltrexone levels [79] and is an effective mu-opioid receptor antagonist [80,81]. Pharmacokinetic data from 12 heroin-dependent individuals who received low and high doses of Depotrex® — 192 mg and 384 mg, respectively — showed that both doses maintained plasma naltrexone levels above 1 ng/ml for up to 4 weeks [82]. Average peak levels for the low and high doses of Depotrex® were 3.8 ng/ml and 8.9 ng/ml, respectively. Plasma beta-naltrexol, the major metabolite of naltrexone, was greater proportionately but could not be detected 5 weeks following Depotrex® administration. Both doses of Depotrex® antagonized the positive subjective effects of heroin. Reported adverse events were minimal and included mild discomfort at the injection site, with no irritation or erythema. The promising earlier study by Kranzler et al. [79] of Depotrex® (206 mg) in the treatment of alcohol dependence needs to be followed up.
In sum, depot formulations of naltrexone may offer some advantages such as increased compliance over the oral formulations. This advantage has, however, been difficult to demonstrate in randomized controlled trials but might become more apparent when these depot formulations are used in generic practices. Depot formulations do not appear to be more efficacious than the oral formulations, and with one of these — Vivitrol® — no therapeutic effect in women has been demonstrated. The adverse events profiles of depot formulations of naltrexone that have been reported in randomized controlled trials appear similar in frequency and intensity to those observed for the oral formulation. The different depot formulations do appear to be similar in characteristics and profile, and more clinical information about which one to select to treat a particular alcohol-dependent patient, if all are approved by the FDA, shall be needed.
GLUTAMATE
Metabotropic glutamate receptor-5 (mGluR5) modulator and N-methyl-D-aspartate (NMDA) antagonist — acamprosate
Acamprosate’s principal neurochemical effects have been attributed to antagonism of NMDA glutamate receptors [83,84], which restores the balance between excitatory and inhibitory neurotransmission that is dysregulated following chronic alcohol consumption [85]. Recently, however, it also has been proposed that acamprosate modulates glutamate neurotransmission at metabotropic-5 glutamate receptors (mGluR5) [86]. Evidence that acamprosate modulates a novel site of action at mGluR5 comes from the finding that it inhibits the binding and neurotoxic effects of ±-1-aminocyclopentane-trans-1,3-dicarboxylic acid [86]. Acamprosate has been shown to decrease: a) ethanol consumption in rodents [87-89], but this effect may not be specific in food-deprived C57BL/6J mice as both ethanol and water were reduced in a schedule-induced polydipsia task [90]; b) dopamine hyperexcitability in the nucleus accumbens during alcohol withdrawal [91,92]; c) general neuronal hyperexcitability [93,94]; d) glutamatergic neurotransmission in alcohol-dependent rats [91,95]; e) voltage-gated calcium channel activity, and f) the expression of brain c-fos, an immediate early gene associated with alcohol withdrawal [96,97]. Nevertheless, it is acamprosate’s ability to suppress alcohol-induced glutamate receptor sensitivity [98], as well as conditioned cue responses to ethanol in previously dependent animals even after prolonged abstinence [99-102], that has been linked with its therapeutic effect in humans — dampening negative affect and craving post-abstinence [14,103] (Fig. 2).
Fig. 2.
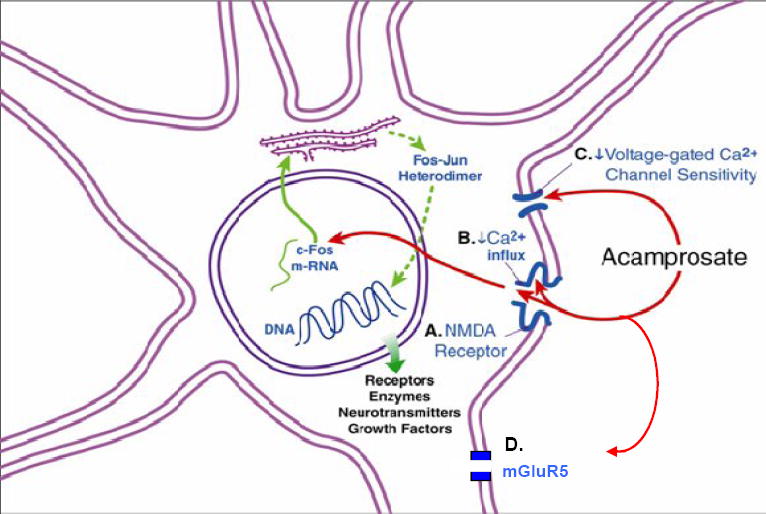
Schematic representation of acamprosate’s effects. Acamprosate has four principal effects: A) reducing post-synaptic excitatory amino acid neurotransmission at N-methyl-D-aspartate (NMDA); B) diminishing Ca2+ influx into the cell, which interferes with expression of the immediate early gene c-fos; C) decreasing the sensitivity of voltage-gated calcium channels, and D) modulating metabotropic-5 glutamate receptors (mGluR5). mGluR5 are post-synaptic and are coupled to their associated ion channels by a second messenger cascade system (not shown). Also shown in this representation is synthesis of c-fos and c-jun in the endoplasmic reticulum, which can bind with DNA to alter the transcription of late effector genes. Late effector genes regulate long-term changes in cellular activity such as the function of receptors, enzymes, growth factors, and the production of neurotransmitters. Embellished from Spanagel and Zieglgansberger [103]. Adapted from Figure 2 in Johnson and Ait-Daoud [14], with the kind permission of Springer Science and Business Media.
Interestingly, there has been a paucity of human laboratory studies that have examined the potential effects of acamprosate on alcohol-related behaviors associated with its abuse liability. Evidence from a human magnetic resonance imaging study does, however, support acamprosate’s ability to modulate glutamate neurotransmission as it decreases activity in brain regions rich in N-acetylaspartate and glutamate [95]. Human laboratory studies in both volunteers [104] and alcohol-dependent individuals [105] also have shown that acamprosate — i.e., calcium acetyl homotaurinate — is relatively safe, with the most important adverse events being diarrhea, nervousness, and fatigue, especially at a relatively high dose (3 g/day). Since acamprosate is excreted unchanged in the kidneys, there is no risk of hepatotoxicity, but it should be used with caution in those with renal impairment [104,105]. Acamprosate has no significant clinical interaction with alcohol. Recently, it was shown that acamprosate can reduce heart rate response but not the increase in cortisol or subjective craving following the presentation of alcohol cues — a finding that suggests utility for acamprosate in managing autonomic dysregulation in abstinent alcoholics exposed to a high risk for relapse situations [106].
Most of the clinical evidence for the efficacy of acamprosate in the treatment of alcohol dependence comes from a series of European studies. In 2004, Mann et al. [107] wrote a meta-analysis of 17 published studies that included 4087 alcohol-dependent individuals. In that report, continuous abstinence rates at 6 months were greater than for those who got placebo (acamprosate, 36.1%; placebo, 23.4%; relative benefit, 1.47; 95% CI = 1.29–1.69; p < 0.001). The overall pooled difference in success rates between acamprosate and placebo was 13.3% (95% CI = 7.8–18.7%), and the number needed to treat was 7.5. Similar results were obtained from another meta-analysis conducted at about the same time [56]. Generally, the effect size of acamprosate is small — 0.14 for increasing the percentage of non-heavy drinking days [108] and 0.23 for reducing the relapse to heavy drinking [109]. Early studies also had some methodological problems, including non-standardization of diagnostic criteria and the psychosocial adjunct to the medication, which were resolved in later trials.
Despite approval by the FDA on July 29, 2004, for the use of acamprosate in the treatment of alcohol dependence, largely based on the data from European studies, the results of U.S. studies have been disappointing. In the U.S. multi-site trial by Lipha Pharmaceuticals, Inc., there was no overall clinical evidence that acamprosate was superior to placebo among a heterogeneous cohort of alcohol-dependent individuals; however, post-hoc analysis suggested that a subgroup of alcoholics with a treatment goal of abstinence might derive benefit [110]. Further, in 2006, the multi-site COMBINE project also failed to find any therapeutic benefit of acamprosate compared with placebo on any drinking outcome measures [62]. Obviously, the findings of these U.S. studies have reduced the enthusiasm for using it by addiction specialists in the U.S. From a scientific perspective, these findings do beg the questions as to what type of alcohol-dependent individual benefits the most from acamprosate and why there is an important discrepancy between the results of U.S. and European studies.
From the European studies, acamprosate appears to benefit alcohol-dependent individuals with increased levels of anxiety, physiological dependence, negative family history, late age of onset, and female gender [111].
There are at least four possible explanations for the discrepancy between U.S. and European studies. First, the populations sampled differ, with European, compared with U.S., studies having alcohol-dependent individuals with more prolonged drinking histories and alcohol-related neurological and psychosocial impairments. Thus, it is tempting to speculate that European studies might have included individuals with greater neuroplasticity and, therefore, higher response to the ameliorating effects of anti-glutamatergic agents such as acamprosate. Second, U.S., compared with European, studies have tended to have higher levels of standardized psychosocial intervention as an adjunct to acamprosate, thereby masking the effect of the medication. Third, the therapeutic effect of acamprosate is small; hence, by chance, some trials can be expected to fail, especially those conducted in a multi-site rather than a single-site environment due to the greater heterogeneity and variability of the cohort and research settings. Fourth, it is possible that future research might uncover other important differences between U.S. and European cohorts to explain the discrepant findings such as potential differences in patient subtype, stage of the alcoholism disease, or bio-molecular constitution.
In sum, European studies have clearly demonstrated efficacy for acamprosate as a treatment for alcohol dependence. Acamprosate was FDA approved in the U.S. largely based on the results of the European studies. Acamprosate’s therapeutic effect is small, but it is well tolerated, with the most prominent adverse events being diarrhea, nervousness, and fatigue, especially at a relatively high dose (3 g/day). In contrast, U.S. studies have, to date, been unable to find efficacy for acamprosate among a heterogeneous group of alcohol-dependent individuals. The reason for this discrepancy between the results of U.S. and European studies has not been established. Perhaps, however, this discrepant finding might be due to differences in patient selection, subtype, stage of the alcoholism disease, or bio-molecular constitution that are yet to be determined. Future studies are needed to delineate more clearly what type of alcohol-dependent individual can benefit the most from acamprosate treatment.
Other N-methyl-D-aspartate (NMDA) receptor antagonists
Other NMDA receptor antagonists such as memantine and neramexane are being studied for the treatment of alcohol dependence. Both compounds have been shown in animal models to suppress ethanol-induced NMDA receptor up-regulation, thereby reducing ethanol sensitization and the propensity for subsequent drug use (for a review, see Nagy [112] and Kotlinska et al. [113]). In a human laboratory study, memantine reduced alcohol craving prior to but not after the experimental administration of alcohol. This would suggest that memantine might have the effect of reducing post-cessation craving for alcohol [114]. This finding is supported by a later report that memantine might have comparable effects to diazepam at ameliorating alcohol withdrawal symptoms [115]. Nevertheless, despite the early preliminary findings, a recent pilot clinical trial comparing memantine with placebo for the treatment of alcohol dependence reported that the greater therapeutic effect at reducing the percentage of heavy drinking days and increasing the percentage of days abstinent [116] occurred among the placebo group. Although this pilot study did not provide support for memantine as an efficacious treatment for alcohol dependence, further studies are needed to make a final determination of memantine’s therapeutic potential for this indication. No human study on the therapeutic effects of neramexane in treating alcohol dependence has been published.
Alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid and kainate glutamate receptor antagonist — topiramate
Topiramate, a sulfamate-substituted fructopyranose derivative, has six important mechanisms of action. Additional to its ability to antagonize alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors and kainate glutamate receptors [117-119], topiramate also facilitates inhibitory GABAA-mediated currents at non-benzodiazepine sites on the GABAA receptor [120,121], inhibits L-type calcium channels and limits calcium-dependent second messenger systems [122], reduces activity-dependent depolarization and excitability of voltage-dependent sodium channels [123], activates potassium conductance [124], and is a weak inhibitor of carbonic anhydrase isoenzymes, CA-II and CA-IV [125], which are found in both neuronal and peripheral tissues. In renal tubules, carbonic anhydrase isoenzyme inhibition reduces hydrogen ion secretion and increases secretion of Na+, K+, , and water, thereby enhancing the likelihood of acidosis and renal stone formation [125,126].
Johnson [127,128] has proposed a neuropharmacological model by which topiramate can decrease alcohol reinforcement and the propensity to drink (Fig. 3). Nevertheless, few studies on the effects of topiramate on ethanol consumption in animals have been published. An initial animal study had shown complex effects of topiramate on ethanol drinking in C57BL/6 mice. In that study, high-dose (50 mg/kg) but not low-dose (1, 5, and 10 mg/kg) topiramate suppressed ethanol intake 2 hours after it was injected into the animal. Topiramate also decreased saccharin preference, but its ability to suppress ethanol preference was associated with some increase in water intake [129]. Notably, in an elegant, recent animal study, Nguyen et al. [130] demonstrated that topiramate can suppress ethanol drinking in C57BL/6 mice; additionally, in contrast with the effects of naltrexone and tiagabine in the same animals, the mice treated with topiramate did not develop any tolerance to its anti-drinking effects. Further, topiramate also has been shown to suppress alcohol drinking moderately in both alcohol-preferring (P) and Wistar rats [131]. Additional to its ethanol-suppressing effects, there is evidence that topiramate can reduce alcohol withdrawal symptoms in a model of handling induced convulsions [132]. Hence, the preponderance of the animal literature does support topiramate as a promising medication for the treatment of alcohol dependence. Nevertheless, the effect of topiramate on ethanol drinking in animals appears to be less striking than that on drinking outcomes in humans, which are presented below. This challenges the notion that animal models can predict directly treatment response in humans, especially when a variety of models have not been used or been available to characterize or “fingerprint” response [133]. The results of additional animal experiments examining topiramate’s mechanistic effects on ethanol consumption or related behaviors in animals are, therefore, awaited eagerly.
Fig. 3.
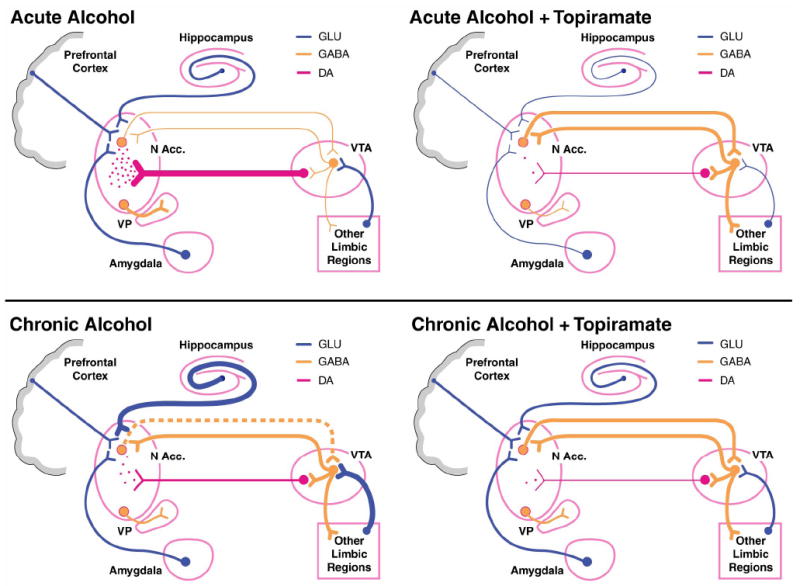
Schematic illustration of the hypothesized effects of acute and chronic alcohol, both with and without topiramate, on the cortico-mesolimbic dopamine (DA) reward circuit [127]. (Upper left) Acute alcohol suppresses the firing rate of ventral tegmental area (VTA) gamma-aminobutyric acid (GABA) neurons, which leads to less suppression of VTA DA neuronal activity. This disinhibition leads to VTA DA neuronal firing and DA release in the nucleus accumbens (N Acc.) [127]. (Lower left) With chronic drinking, VTA GABA neurons are hyperexcitable, mainly because of increased glutamatergic input, less GABA tone from the N Acc., and rebound firing of GABA neurons because of their long-term suppression from repeated alcohol ingestion. This leads to VTA DA hypofunction and decreased release (compared with the acute condition) of DA in the N Acc. [127]. (Upper right) During acute drinking, the GABAergic influence of topiramate probably predominates, particularly in the N Acc. This leads to greater inhibition of N Acc. DA neurons, and greater GABA tone from the N Acc. to the VTA suppresses VTA DA cell firing. Topiramate concomitantly inhibits the excitatory effects of glutamatergic neurons on DA neurons in the VTA and N Acc. These combined actions of topiramate should lead to profound suppression of DA neuronal activity and DA release in the N Acc. Hence, topiramate reduces the DA-mediated reinforcing effects of acute alcohol [127]. (Lower right) During chronic drinking, the predominant neuronal activity resides with the hyperexcitable state of VTA GABA neurons. Because of GABA-mediated inhibition and glutamatergic blockade of these neurons, topiramate “normalizes” VTA GABA neuronal activity. Although this would, at first, suggest that DA release in the N Acc. would be enhanced, this does not occur, and DA release in the N Acc. is most likely reduced because these N Acc. terminals are contemporaneously inhibited by GABA inhibition and blockade of glutamate (GLU). In the chronic drinker, the anti-glutamatergic and L-type calcium channel effects of topiramate to block sensitization might predominate. Hence, topiramate would make it easier for a chronic alcoholic to withdraw from alcohol because rebound DA release would not occur (if drinking were ceased abruptly), and topiramate would aid in relapse prevention because alcohol’s reinforcing effects would be decreased [127]. Line weights represent relative strengths of neuronal activity (heavy, medium, and light). The broken line represents decreased tone. VP, ventral pallidum. Reprinted from Figure 1 in Johnson [127], with the permission of Blackwell Publishing, Inc.
Recently, Johnson et al. [5,134] and Ma et al. [135] showed in a double-blind, randomized clinical trial that topiramate (up to 300 mg/day), compared with placebo, improved all drinking outcomes, decreased craving, and improved the quality of life of alcohol-dependent individuals who received 12 weeks of weekly brief behavioral compliance enhancement treatment [6]. The improvements in self-reported drinking outcomes were confirmed by plasma GGT, an objective biochemical measure of alcohol consumption [78]. The therapeutic effect size for the primary efficacy variable — percentage of heavy drinking days — was 0.63.
In a 6-week experimental study of 76 heavy drinkers who were not seeking treatment, Miranda et al. [136] showed that low- and high-dose topiramate — 200 mg/day and 300 mg/day, respectively — were significantly better than placebo at decreasing the percentage of heavy drinking days.
Further, in a subsequent 17-site (N = 371) U.S. trial, topiramate (up to 300 mg/day) was again superior to placebo at improving all self-reported drinking outcomes, GGT level, and some measures of quality of life among alcohol-dependent individuals who received 14 weeks of weekly brief behavioral compliance enhancement treatment. Topiramate’s therapeutic effect size for the reduction in percentage of heavy drinking days was 0.52, and the number needed to treat was 3.4 [137].
Taken together, these clinical studies provide strong evidence that topiramate is a promising medication for the treatment of alcohol dependence. Encouragingly, topiramate’s therapeutic effect size is in the moderate range, and the clinical effects appear to increase with greater length of time on the medication.
Generally, topiramate has a favorable adverse event profile, with most reported symptoms being classified as mild to moderate [138]. The most common adverse events are paresthesia, anorexia, difficulty with memory or concentration, and taste perversion. Slow titration to the ceiling dose (up to 300 mg/day) for 6 to 8 weeks is critical to minimizing adverse events and improving tolerability; however, about 10% of individuals taking topiramate may experience some cognitive difficulty irrespective of the dose titration schedule [139]. Topiramate use has been linked with acute but rare visual adverse events. As of January 2005, there had been 371 spontaneous reports of myopia, angle-closure glaucoma, or increased intraocular pressure, for a rate of 12.7 reports per 100,000 patient-years exposure. Usually, the syndrome of acute bilateral myopia associated with secondary angle-closure glaucoma presents as the acute onset of visual blurring, ocular pain, or both. Associated bilateral ophthalmologic findings can include myopia, shallowing of the anterior chamber, conjunctival hyperemia, and raised intraocular pressure. This syndrome resolves within a few days of discontinuing topiramate administration [138].
In sum, predicated upon a neuropharmacological conceptual model, there is now strong clinical support for topiramate as a promising medication for the treatment of alcohol dependence. Topiramate’s therapeutic effects appear to be robust, with a medium effect size, thereby potentially ushering in a new era of a reliably efficacious medicine for the treatment of alcohol dependence. Intriguingly, although the animal data do provide support for topiramate’s anti-drinking effects, more research is needed to characterize fully or “fingerprint” the pattern of response. Such preclinical studies should enable us to elucidate more clearly the basic mechanistic processes that underlie topiramate’s efficacy as a treatment for alcohol dependence.
SEROTONIN (5-HT)
For almost three decades, there has been intense interest in the effects of serotonergic agents in the treatment of alcohol dependence. Encouraged by increased knowledge about the various 5-HT receptor subtypes, researchers have examined the effects of various medications that bind to specific receptor sites. Here, I provide a synopsis of the preclinical and clinical studies that have been done on these 5-HT function-altering medications in the treatment of alcohol dependence.
Serotonin reuptake inhibitors
For decades, it has been known that pharmacological manipulations that deplete the brain of 5-HT decrease the preference for ethanol [140,141]. Using preference paradigms, pharmacological agents that inhibit 5-HT reuptake from the synapse reduce the voluntary consumption of ethanol solutions using the preference paradigm [142-147]. Knockout mice at the 5-HT transporter do, however, exhibit a general decrease in ethanol preference and consumption [148]. Thus, there is ample preclinical support for the notion that selective serotonin reuptake inhibitors (SSRIs) suppress ethanol consumption in animals.
Although these preclinical studies have shown that SSRIs can reduce ethanol consumption, the selectivity of this effect on reinforcement as opposed to general consummatory behaviors has been questioned [149-151].
The inhibition of 5-HT reuptake function has complicated the effects on food intake and fluid consumption [152]. SSRIs do suppress food intake [153,154] and fluid consumption [152] and decrease palatability [155]. Yet, motivational factors exert some control on the expression of these behaviors [156]. For instance, SSRIs enhance satiety [150] but selectively reduce preference for certain macronutrients (i.e., sweet items and carbohydrates) [ 157-159] cf. [160,161] that increase the palatability and rewarding effects of food [162-164]. Hence, SSRIs might decrease ethanol consumption via the suppression of non-specific general consummatory behaviors and specific anti-reinforcing effects.
Studies conducted using operant techniques have also supported a role for SSRIs in the suppression of ethanol consumption. Haraguchi et al. [165] showed that same-day pretreatments with fluoxetine dose-dependently reduced ethanol responding. Nevertheless, whereas the chronic administration of SSRIs to C57BL/6J male mice produced an initial suppression of lever pressing for ethanol, there was a later rebound to baseline levels of responding for ethanol and ethanol consumption [166]. These results are somewhat similar to those of Murphy et al. [167], who observed that fluoxetine administered to rats in a single daily infusion produced a significant reduction in ethanol-reinforced responding that started on the first day of treatment and increased on subsequent days of the 7-day treatment regimen. Responding for ethanol returned to pretreatment levels following cessation of fluoxetine treatment. Food intake, while somewhat suppressed initially, appeared to return to baseline levels on subsequent treatment days. Again, these results demonstrate that the suppression of ethanol intake by SSRIs follows a pattern of initial suppression of consummatory behavior followed by a reduction in reinforcement; thus, when the SSRIs are discontinued, there is an extinction-like pattern of a return to the baseline behavior.
Despite the promise of these preclinical results, there is, at present, little support for the proposal that SSRIs are an efficacious treatment for a heterogeneous group of alcohol-dependent individuals. Initial studies of small sample size reported that SSRIs can produce short-term (1–4 weeks) decreases in alcohol consumption among problem drinkers [168-172]. Nevertheless, these studies were limited by at least three factors. First, most of the studies were conducted in men, thereby limiting the generalizability of the results to the general population [168-170]. Second, the adjunctive psychosocial treatment, which can decrease the apparent efficacy of the putative therapeutic medication because this too can have an important effect on drinking outcomes, was not standardized. Third, the treatment periods were short; thus, it was not possible to determine whether these initial effects, which could be due to non-specific factors, would be sustained. Indeed, the problem with studies of short duration that focus on a chronic relapsing disorder such as alcohol dependence was highlighted in a later study by Gorelick and Paredes [173], who found that there also was an effect for fluoxetine, compared with placebo, to decrease alcohol consumption by about 15% in the first 4 weeks of the trial but not over the entire length of the trial. Also, Naranjo et al. [174] did not demonstrate that citalopram (40 mg/day) was superior to placebo in a 12-week treatment trial. Further, neither Kabel and Petty [175] nor Kanzler et al. [176] in two separate 12-week studies found fluoxetine (60 mg/day) to be superior to placebo for the treatment of alcohol dependence.
There has been renewed understanding about how the administration of functionally different serotonergic agents can lead to different drinking outcomes among various subtypes of alcoholic (for a review, see Johnson [177]). Adapted from Cloninger’s classification scheme [178], two methods for subtyping alcoholics have been used in these pharmacotherapy studies. Basically, a particular type of alcoholic (i.e., Type A-like or late onset) characterized by a later age of onset of problem drinking (typically over the age of 25 years), a preponderance of psychosocial morbidity, and low familial loading can experience improved drinking outcomes after SSRI treatment.
Although early human laboratory studies showed that Type B-like or early-onset alcoholics, characterized by an early age of problem drinking onset (i.e., before the age of 25 years), high familial loading for alcohol dependence, and a range of impulsive or antisocial traits, might be centrally deficient in the major metabolite of 5-HT, 5-hydroxyindoleaceteic acid [179-181], the implications of this finding were, perhaps, oversimplified. At a cursory glance, it would appear that an SSRI, by increasing 5-HT turnover, would compensate for this dysfunction; thus, these Type B-like or early-onset alcoholics would then be expected to experience improved drinking outcomes following SSRI treatment. Remarkably, the literature has demonstrated quite the opposite. For instance, Kranzler et al. [182] observed that fluoxetine treatment appeared to worsen the clinical benefit of the adjunctive cognitive behavioral treatment and there was no difference from placebo. Actually, Type A-like or late-onset alcoholics, with presumably more normative 5-HT function, have been observed to experience improved drinking outcomes from sertraline both during active treatment [183] and at 6-month follow-up [184]. Also, Chick et al. [185] have shown that early-onset or Type B-like alcoholics were more likely to relapse than their late-onset or Type A-like counterparts following fluvoxamine treatment.
Obviously, the relationship between serotonergic dysfunction and Type B-like or early-onset alcoholism is not the simple result of a deficiency state. Indeed, Johnson [177] has hypothesized that an explanation for this effect might be allelic variation at the 5-HT transporter, which leads to the differential expression of 5-HT function. Of course, other bio-molecular explanations are possible, and further research is needed to elucidate this important area of research.
While outside the scope of this review, it has been proposed that SSRIs might be of therapeutic benefit in the treatment of alcohol-dependent individuals with suicidal tendencies and severe comorbid depression [186]. Nevertheless, a recent study did not find that sertraline treatment was more beneficial than placebo to depressed alcohol-dependent individuals irrespective of the severity of depression [187]; nor has it been shown that the reduction in dysphoria in depressed alcoholics is associated with concomitant decreases in alcohol consumption [188,189]. Hence, the only conclusion that can be drawn at present is that except for a subtype of depressed alcoholic with suicidal tendencies, there is not much evidence to recommend SSRIs over placebo for the treatment of depressed alcoholics.
In sum, despite strong animal data that would support the use of SSRIs as a promising treatment for alcohol dependence, there is no evidence that they are of therapeutic benefit to a heterogeneous group of alcohol-dependent individuals. Notably, however, there is growing confirmation that SSRIs can improve the drinking outcomes of Type A-like or late-onset alcoholics. Rather than being a cause for discouragement, this finding might a) open up the possibility of identifying important bio-genetic or pharmacological mechanisms that underlie the alcoholism disease and b) improve understanding about which type of alcohol-dependent individual can benefit the most from specific serotonergic treatment. Further, there is no current evidence that providing SSRIs to depressed alcoholics without severe depressive symptoms and suicidal tendencies is of therapeutic benefit. Hence, what is clear is that clinicians should be cautious in prescribing SSRIs to alcohol-dependent individuals for the treatment of minor depressive or affective symptoms. Not only is this strategy unlikely to be a therapeutic benefit over placebo, and perhaps appropriate psychosocial management, but drinking outcomes can actually be worsened, especially if the alcohol-dependent individual is Type A-like or of late onset.
Serotonin-1 (5-HT1) partial receptor agonist
Preclinical studies have suggested that the 5-HT1A partial agonist, buspirone, may be effective at reducing ethanol consumption. Buspirone decreased volitional alcohol consumption from 60% to 30% in macaque monkeys, but there was considerable inter-individual variation [190]. In Sprague-Dawley rats, buspirone significantly reduced ethanol intake in animals induced to drink by repeated brainstem injection of tetrahydropapaveroline. In a group of medium alcohol-preferring rats, buspirone (0.0025–0.63 mg/kg) reduced, while buspirone (>2.5 mg/kg) increased, alcohol consumption without affecting water consumption [191]. While buspirone is a partial 5-HT1A agonist, the net effect of its repeated administration is to enhance 5-HT function via facilitation of the post-synaptic receptor, which is more sensitive than the autoreceptor, and down-regulation of autoreceptor function [192]. Nevertheless, this preclinical evidence would have been strengthened by operant studies examining the dose-response characteristics of buspirone as a function of ethanol concentration.
Buspirone has not been demonstrated to be an efficacious medication for the treatment of alcohol-dependent individuals without comorbidity. In a review of five published trials, buspirone was without a convincing effect in non-comorbid alcoholics; however, alcoholics with comorbid anxiety experienced some benefit [193,194]. Hence, buspirone’s anxiolytic effects might translate to those who also are dependent on alcohol.
In sum, there is no current evidence that would suggest a role for buspirone in the treatment of alcohol dependence without comorbid anxiety disorder.
Serotonin-2 (5-HT2) receptor antagonist
Preclinical studies have suggested that the 5-HT2 receptor antagonist, ritanserin, can reduce ethanol consumption in animals [195,196] cf. [197]. Also, the 5-HT2 antagonists, amperozide [198-201] and FG5974 [202,203], significantly suppress ethanol intake without affecting water consumption. The exact mechanism by which 5-HT2 receptor antagonists might reduce ethanol consumption is unknown. It has, however, been suggested that they might exert their effects by acutely substituting for alcohol’s pharmacobehavioral effects by facilitating burst firing in CMDA neurons [204], or by the suppression of dopamine neurotransmission following their chronic administration.
In the clinical setting, ritanserin is not an efficacious treatment for alcohol dependence. In a rigorously conducted, 12-week, multi-center clinical trial (N = 423) of ritanserin (2.5 or 5 mg/day) vs. placebo as an adjunct to weekly cognitive behavioral therapy, none of the ritanserin doses were superior to placebo [205]. In a later study using similar methodology, ritanserin (2.5, 5.0, or 10.0 mg/day) was not superior to placebo at improving drinking outcomes [206]. Although higher doses of ritanserin might be of therapeutic benefit, testing these doses is precluded by ritanserin’s potential to cause dose-dependent prolongation of the QTc interval on the electrocardiogram, thereby increasing the potential for life-threatening cardiac arrhythmias.
In sum, there is no clinical evidence that would support the use of ritanserin as a treatment for alcohol dependence.
Serotonin-3 (5-HT3) receptor antagonists
Preclinical studies provide strong support for the role of the 5-HT3 receptor in mediating alcohol’s important neurochemical effects, and for 5-HT3 receptor antagonists to be promising treatment for alcohol dependence.
In neurophysiological experiments, ethanol potentiates 5-HT3 receptor-mediated ion currents in NCB-20 neuroblastoma cells [207,208] and in human embryonic kidney 293 cells transfected with 5-HT3RA cDNA [209]. 5-HT3 receptor antagonists block these effects [210]. Thus, the 5-HT3 receptor is a site of action for ethanol in the brain [211,212].
Pharmacobehavioral studies show that many of alcohol’s reinforcing effects are mediated by 5-HT3 and dopamine interactions in the cortico-mesolimbic system [9,213-216].
5-HT3 receptor antagonists have three principal effects that demonstrate their ability to modulate ethanol consumption and related behaviors. First, 5-HT3 receptor antagonists suppress hyperlocomotion in the rat induced by dopamine or ethanol injection into the nucleus accumbens [217]. Second, 5-HT3 receptor antagonists inhibit DiMe-C7 (a neurokinin)-induced hyperlocomotion, which also is reduced by the dopamine antagonist, fluphenazine [218,219]. Third, 5-HT3 receptor antagonists reduce ethanol consumption in several animal models and across different species [191,213,220-228] cf. [229].
Human laboratory studies have generally supported a role for the 5-HT3 antagonist ondansetron in reducing preference and craving for alcohol. In two distinct experiments, Johnson and Cowen [214] and Johnson et al. [224] showed that ondansetron pretreatment attenuated low-dose alcohol-induced positive subjective effects (including the desire to drink). Swift et al. [230], using much higher alcohol and ondansetron doses, also discovered that ondansetron compared with placebo pretreatment reduced alcohol preference; however, a mixture of both stimulant and sedative interactions between ondansetron and alcohol also was observed. Whereas Doty et al. [231] did not find an effect of ondansetron on alcohol-induced mood, their experimental model of using a group rather than individual experimental setting could have decreased the sensitivity of their assessments.
Three clinical studies have provided evidence that ondansetron is a promising treatment for alcohol-dependent individuals, particularly those with an early-onset or Type B-like subtype.
First, in a 6-week, double-blind, placebo-controlled study of 71 non-severely alcohol-dependent males, Sellers et al. [232] observed that the 0.5-mg dose but not the 4-mg dose of ondansetron was associated with a non-significant trend (p = 0.06) toward a reduction in alcohol consumption. Post-hoc analysis that eliminated 11 subjects who consumed less than 10 drinks/drinking day rendered the difference in drinking outcomes between the ondansetron 0.5 mg and placebo groups to be significant statistically (p = 0.001). Despite the limitations of this initial trial, which included a relatively short treatment period, the inclusion of just males, and the small number of subjects, the results of this study provided general support for ondansetron’s promise in treating alcohol dependence. Also, these results show that ondansetron may exhibit a non-linear dose-response effect in the treatment of alcohol dependence.
Second, in a large-scale (N = 321), 12-week, randomized, double-blind clinical trial in which alcohol-dependent individuals received weekly cognitive behavioral therapy, Johnson et al. [233] showed that ondansetron (1, 4, and 16 μg/kg b.i.d.) was superior to placebo at improving drinking outcomes of those of the early onset or Type B-like subtype but not the late onset or Type A-like subtype. The self-reported decreases in alcohol consumption were corroborated by the concomitant reduction in carbohydrate-deficient transferrin level — a biomarker of transient alcohol consumption.
Third, Kranzler et al. [234] provided replication of the results by Johnson et al. [233] by showing that early-onset (Type B-like) alcoholics had a significantly greater improvement in drinking outcomes compared with their late-onset (Type A-like) counterparts following 8 weeks of ondansetron (4 μg/kg b.i.d.) treatment.
Intriguingly, these results demonstrate a differential effect of ondansetron treatment by subtype of alcohol-dependent individual. Indeed, the contrast is striking when compared with the effects of SSRIs on different subtypes of alcohol-dependent individuals as described above. Basically, early-onset or Type B-like alcoholics with apparent serotonergic deficiency respond best to a medication that blocks the 5-HT3 receptor, whereas late-onset or Type A-like alcoholics with apparently normal serotonergic function derive the most benefit from a medication that can increase 5-HT turnover and function. As mentioned earlier, Johnson [177] has proposed a bio-molecular explanation for these effects; however, other plausible possibilities exist. Although elaboration of this concept is beyond the scope of this review, the key feature is that polymorphic variation at the 5-HT transporter allele is affected differentially by the history of drinking behavior and, perhaps as a consequence, the expression of 5-HT turnover in these different polymorphic types modulates present drinking behavior. Further studies are needed to test this and other proposals that can explain the differential effect of various serotonergic agents among alcohol-dependent individuals of different subtype. Obviously, a molecular genetic explanation for this effect, if proven, may enable a pharmacogenetic approach to treatment whereby the appropriate medication can be provided to the particular subtype of alcohol-dependent individual who would benefit the most from such treatment.
In sum, preclinical data support an important role for 5-HT3 receptors in mediating alcohol’s important reinforcing effects associated with its abuse liability. Ondansetron is a promising medication for the treatment of early-onset or Type B-like alcohol dependence. Further studies are needed to determine whether treatment with various serotonergic agents can best be applied using a pharmacogenetic approach.
DOPAMINE
Dopamine receptor antagonists
CMDA neurons have been implicated as the principal pathway by which alcohol and most other abused drugs express their reinforcing effects associated with abuse liability [9,215,216]. Yet it has been difficult to show evidence that direct dopamine receptor antagonists have a role in the treatment of alcohol dependence. Presumably, direct opposition of dopamine pathways is associated with neuroadaptive changes that tend to reverse the initial effects of the blockade [128]. No traditional dopamine receptor blocker has been demonstrated to be an efficacious treatment for alcohol dependence. With the advent of atypical neuroleptics, there has been renewed interest in testing these medications as potential treatment for alcohol dependence. Indeed, medications such as aripiprazole and quetiapine are currently in clinical testing, and the results are awaited eagerly. Other medications that are selective for dopamine-3 receptor antagonism also are under development.
Dopamine receptor agonists
At low doses, dopamine-2/dopamine-3 agonists such as bromocriptine and 7-OH DPAT can reduce ethanol consumption in animals [235-237]. Although this might appear paradoxical to the dopamine theory of reinforcement for most abused drugs, it is possible that low-dose dopamine agonists preferentially augment autoreceptor function, thereby decreasing dopamine turnover.
Although an earlier report proposed that bromocriptine can decrease alcohol craving, subsequent studies have found no effect on alcohol drinking or related behaviors [238-240]. Nevertheless, perhaps due to the high addictive potential of dopamine agonists, this research approach has largely been abandoned in the clinical setting. Currently, dopamine receptor agonists do not hold promise as a treatment for alcohol dependence.
GABA-B RECEPTOR AGONIST — BACLOFEN
Animal studies have demonstrated that the GABAB receptor agonist, baclofen [beta-(4-chlorophenyl)-GABA], causes decreases in voluntary ethanol intake [241], the ethanol-deprivation effect [242], and morphine-induced stimulation of ethanol consumption [243].
Clinical trials have bolstered the findings of animal studies that suggest a role for baclofen in treating alcohol dependence. In an open-label, 4-week study, 9 alcohol-dependent men were given baclofen (up to 30 mg/day). Seven of the 9 subjects achieved abstinence, while the other 2 participants improved their self-reported drinking outcomes during the study period, according to self-reports corroborated by family members. Several objective biological markers of alcohol intake also showed significant reductions between the beginning and end of the study. Furthermore, craving, as measured by median Alcohol Craving Scale scores, decreased in the first study week and remained stable thereafter [244].
In a 4-week, randomized, placebo-controlled, double-blind clinical trial with 39 alcohol-dependent patients, 14 of 20 (70%) patients treated with baclofen (up to 30 mg/day) achieved abstinence, compared with 4 of 19 (21.1%) in the placebo group (p < 0.005). Baclofen treatment improved significantly drinking outcomes, state anxiety scores, and craving measures. Baclofen generally was well tolerated and had no apparent abuse liability. Adverse events, none of which were serious, consisted of nausea, vertigo, transient sleepiness, and abdominal pain [245].
These findings, which indicate that baclofen is safe and efficacious, with no addictive properties, suggest a potential role for baclofen in treating alcohol-dependent individuals. Additional studies of larger sample size and longer duration would help to establish the efficacy of baclofen in the treatment of alcohol-dependent individuals.
DISULFIRAM
Disulfiram is an FDA-approved medication that has been used for treating alcoholism since the 1940s and is perhaps still the most widely used such medication in the U.S. today. Its principal mode of action is as an aversive agent. Disulfiram inhibits aldehyde dehydrogenase and prevents the metabolism of alcohol’s primary metabolite, acetaldehyde. In turn, the accumulation of acetaldehyde in the blood causes unpleasant effects to occur if alcohol is ingested; these include sweating, headache, dyspnea, lowered blood pressure, flushing, sympathetic overactivity, palpitations, nausea, and vomiting. The association of these symptoms with drinking discourages further consumption of alcohol [246]. Serious side effects also have been reported, including hepatitis, hepatotoxicity, depression, and psychotic reactions [247,248]. Disulfiram also has been shown to reduce norepinephrine synthesis by inhibiting dopamine beta-hydroxylase [249], a mode of action that has been proposed to support early reports of its potential efficacy as a treatment for cocaine dependence. While a review of disulfiram’s potential effects on cocaine taking are outside the scope of this review, the reader is referred to recent studies by Petrakis et al. [249], Carroll et al. [250], and Baker et al. [251].
A 52-week, multi-site, randomized, controlled trial with 605 alcohol-dependent men found that disulfiram might help prevent relapse in compliant patients yet be ineffective at promoting continuous abstinence or a delay in the resumption of drinking [252].
Disulfiram has no significant effect on craving for alcohol. Hence, patients must be highly motivated to maintain disulfiram treatment, whereas those who wish to drink can simply stop taking the medication. The efficacy of disulfiram generally is limited to those who are highly compliant or who receive their medication under supervision — i.e., the type of alcohol-dependent individuals who might be likely to abstain on their own, without adjunctive pharmacotherapy. Including a supportive spouse or partner in a disulfiram treatment plan helps to improve outcome [246,253].
POTENTIAL TREATMENTS ON THE HORIZON
Cannabinoid-1 (CB1) receptor antagonists
Endocannabinoid receptors are found ubiquitously in the central nervous system, particularly in the cortex, hippocampus, basal ganglia, and cerebellum. Endogenous cannabinoids include anandamide and 2-arachidonylglycerol, which are metabolized by fatty acid amide hydrolase [254].
In C57BL/6J mice, cannabinoid-1 (CB1) receptor blockade reduced ethanol consumption to the amounts ingested by CB1 receptor null mutant mice [255]. Endocannabinoids may be involved in the neurochemical expression of susceptibility to the effects of ethanol. For instance, ethanol exposure can increase levels of brain 2-arachidonylglycerol and anandamide and down-regulate CB1 receptors [256,257]. In pharmacobehavioral studies, CB1 receptor antagonists suppress ethanol intake in rats with a chronic history of alcohol administration [258,259], reduce ethanol drinking in alcohol-preferring sP rats [260,261], and decrease operant responding and cue-induced reinstatement of ethanol consumption [262,263]. It is plausible, however, that an important method by which CB1 receptors influence ethanol taking is via their extensive connections to modulate other neuronal systems including monoamine pathways and their metabolism [264-266]. Figure 4 shows the interactions between CB1 and other neuronal systems [267].
Fig. 4.
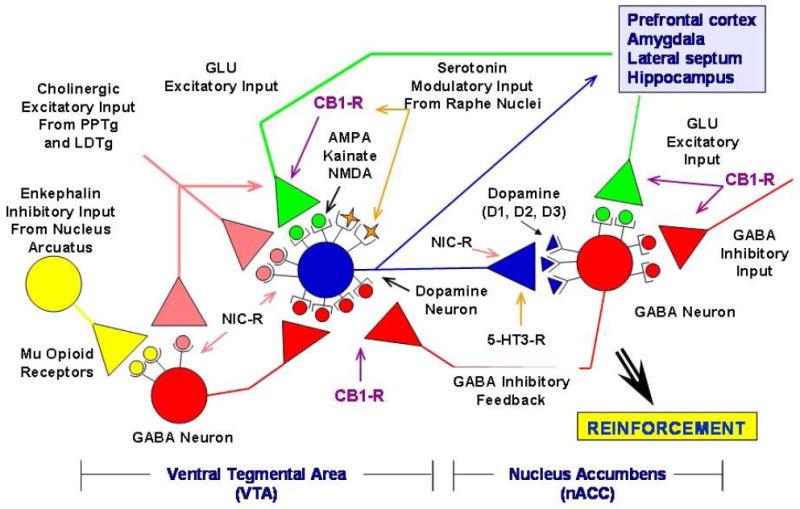
Neuronal pathways involved with the reinforcing effects of alcohol and other abused drugs. Cholinergic inputs that arise from the caudal part of the pedunculopontine tegmental nucleus (PPTg) and laterodorsal tegmental nucleus (LDTg) can stimulate ventral tegmental area (VTA) dopamine neurons. The VTA dopamine neuron projection to the nucleus accumbens (nACC) and cortex, the critical substrate for the reinforcing effects of abused drugs (including alcohol), is modulated by a variety of inhibitory [gamma-aminobutyric acid (GABA) and opioid] and excitatory [nicotinic (NIC-R), glutamate (GLU), and cannabinoid-1 receptor (CB1-R)] inputs. The GLU pathways include those that express alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), kainate, and N-methyl-D-aspartate (NMDA) receptors. Serotonin-3 receptors (5-HT3-R) also modulate dopamine release in the nACC. Adapted and embellished by Bankole A. Johnson, DSc, MD, PhD, from an original drawing by Dennis Twombly, PhD, at the National Institute on Alcohol Abuse and Alcoholism. Reprinted from the figure in Johnson [267]. Copyright © 2006, American Medical Association. All rights reserved.
In Europe, initial human studies of the effects of cannabinoid receptor blockade on the drinking outcomes of alcohol-dependent individuals have been completed, and the results are awaited eagerly. Nevertheless, the recent finding that the CB1 receptor antagonist (rimonabant) can increase mood disturbance and suicidality in smokers, which precluded the FDA from granting approval for that indication, might also impact the development of similar compounds for the treatment of alcohol dependence.
Other neurochemicals and small molecules
Presently, there are a host of other neurochemicals with potential benefit in treating alcohol dependence. At this stage, testing remains within the animal literature and other preclinical models, and it would, therefore, be beyond the scope of this review to discuss them in detail. These compounds include antagonists at mGluR5, mGluR2/3 agonists, stress-related neuropeptides such as corticotropin releasing factor antagonists and modulators of neuropeptide Y, and nociceptin (for a review, see Heilig and Egli [254]).
COMBINATION TREATMENTS
Combination treatments offer the promise of augmenting the effects of single medications by engaging multiple neuronal networks associated with the expression of alcohol’s reinforcing effects associated with its abuse liability. While this idea is alluring, medication combinations do create the potential for reduced compliance (due to the need to take additional tablets), heightened or new treatment emergent adverse events, or even inefficacy if the medications counteract one another.
Perhaps the best studied medication combination so far has been that of naltrexone and acamprosate. This combination has been proposed to be of potential added therapeutic benefit for three reasons. First, naltrexone, by its action on endogenous opioids, modulates CMDA activity, thereby reducing the reinforcing effects of alcohol [215,268]. Acamprosate modulates alcohol withdrawal-induced increases in extracellular glutamate in the cortico-mesolimbic system [91,269]. Thus, the combined effect of both naltrexone and acamprosate may be to modulate both the neurochemical effects responsible for triggering drinking and those associated with conditioned responses to drink even after a prolonged period of abstinence. Second, while naltrexone decreases positive craving for alcohol [55], acamprosate attenuates negative or conditioned craving post-drinking cessation [103]. It is, therefore, tempting to speculate that the combination of naltrexone and acamprosate would make it easier both to abstain and to prevent a “slip” from turning into a relapse. Third, acamprosate can increase blood levels of naltrexone, thereby augmenting its neurochemical effects [104,105].
In a European study, Kiefer et al. [270] showed that the combination of naltrexone and acamprosate was clinically additive at improving the drinking outcomes of alcohol-dependent individuals, but only the effect of the combination vs. acamprosate achieved statistical significance. Nevertheless, the recently completed COMBINE project in the U.S. did not find any therapeutic advantage to combining the two medications [62]. Hence, at present, it is not possible to advise practitioners to combine naltrexone and acamprosate. Further research may, however, provide a definitive answer as to the utility of the combination.
Mechanistically, there are many other medication combinations that are possible, some of which are being pursued. It is, however, noteworthy that preliminary clinical evidence suggests that the combination of ondansetron and naltrexone may result in added or synergistic therapeutic effects on alcohol drinking [271,272]. The results of definitive confirmatory trials are, however, awaited.
In sum, medication combinations may afford the opportunity to augment the treatment effects of single medications for the treatment of alcohol dependence. Such studies should, however, be conducted where there is a compelling pharmacological rationale for combining the medicines. This is because there also is the potential for reduced compliance, heightened or new treatment emergent adverse events, and inefficacy. Further, there are important issues that must be determined for all medication combinations, such as optimal dosing, sequencing of the medications, duration of treatment, and the increased complexity of managing such protocols.
CONCLUSIONS
Recently, there has been renewed interest in developing efficacious medicines for the treatment of alcohol dependence. Naltrexone and its depot formulations have demonstrated utility, but their therapeutic effect size is small. Despite FDA approval of acamprosate based upon the positive results of European studies, there has, as yet, not been a clear demonstration of its efficacy in U.S. studies. Even in the European studies, the therapeutic effect size of acamprosate is small. These discrepant findings might be the result of different populations of alcohol-dependent individuals, selection criteria, chronicity of the alcoholism disease, bio-molecular differences, different methodologies between U.S. and European studies, or sampling error due to the small effect size. For both naltrexone and acamprosate, research is ongoing to determine what type of alcohol-dependent individual benefits the most from using either medication. There also is the possibility that a pharmacogenetic approach may make it possible to improve the therapeutic outcome for those who receive naltrexone. At present, the combination of naltrexone and acamprosate cannot be recommended to be of therapeutic benefit, but this conclusion might change with future research. Topiramate is a promising medication for the treatment of alcohol dependence. Based on two studies, its therapeutic effect size appears to be in the medium range. Future research is needed to extend these results to other subpopulations of alcohol-dependent individuals. Serotonergic medications need to be administered with care to ensure that they are provided to the subtype of alcohol-dependent individual who will benefit the most from such treatment. While SSRIs benefit late-onset or Type A-like alcohol-dependent individuals, the 5-HT3 receptor antagonist, ondansetron, has efficacy in treating early-onset or Type B-like alcohol-dependent individuals. Molecular genetic studies are ongoing to understand the underpinnings of this differential response among various subtypes of alcoholic to different serotonergic agents. Although disulfiram is also FDA approved for the treatment of alcohol dependence, it is perhaps best utilized under supervised conditions. Given the explosion in neuroscientific ideas, the future holds promise for many new and efficacious medicines in the treatment of alcohol dependence.
Acknowledgments
I thank the National Institute on Alcohol Abuse and Alcoholism for its support through grants 7 R01 AA010522-12, 5 R01 AA012964-06, 5 R01 AA014628-03, 5 R01 AA013964-03, and 7 U10 AA011776-10; the National Institutes of Health for its support through University of Virginia General Clinical Research Center Grant M01 RR00847; the staff at the University of Virginia Center for Addiction Research and Education (CARE), and Robert H. Cormier, Jr. for his assistance with manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.U.S. Department of Health and Human Services. 10th Special Report to the US Congress on Alcohol and Health. Bethesda, MD: National Institute on Alcohol Abuse and Alcoholism; 2000. [Google Scholar]
- 2.McLellan AT, Lewis DC, O’Brien CP, Kleber HD. Drug dependence, a chronic medical illness: implications for treatment, insurance, and outcomes evaluation. JAMA. 2000;284:1689–95. doi: 10.1001/jama.284.13.1689. [DOI] [PubMed] [Google Scholar]
- 3.Swift RM. Drug therapy for alcohol dependence. N Engl J Med. 1999;340:1482–90. doi: 10.1056/NEJM199905133401907. [DOI] [PubMed] [Google Scholar]
- 4.Finney JW, Hahn AC, Moos RH. The effectiveness of inpatient and outpatient treatment for alcohol abuse: the need to focus on mediators and moderators of setting effects. Addiction. 1996;91:1773–96. [PubMed] [Google Scholar]
- 5.Johnson BA, Ait-Daoud N, Akhtar FZ, Ma JZ. Oral topiramate reduces the consequences of drinking and improves the quality of life of alcohol-dependent individuals. Arch Gen Psychiatry. 2004;61:905–12. doi: 10.1001/archpsyc.61.9.905. [DOI] [PubMed] [Google Scholar]
- 6.Johnson BA, DiClemente CC, Ait-Daoud N, Stoks SM. Brief Behavioral Compliance Enhancement Treatment (BBCET) manual. In: Johnson BA, Ruiz P, Galanter M, editors. Handbook of clinical alcoholism treatment. Baltimore, MD: Lippincott Williams & Wilkins; 2003. pp. 282–301. [Google Scholar]
- 7.Pettinati HM, Weiss RD, Miller WR, Donovan D, Ernst DB, Rounsaville BJ. COMBINE Monograph Series, Volume 2. Medical Management Treatment Manual: A Clinical Research Guide for Medically Trained Clinicians Providing Pharmacotherapy as Part of the Treatment for Alcohol Dependence (DHHS Publication No. 04-5289) Bethesda, MD: National Institute on Alcohol Abuse and Alcoholism; 2004. [Google Scholar]
- 8.Lee YK, Park SW, Kim YK, Kim DJ, Jeong J, Myrick H, et al. Effects of naltrexone on the ethanol-induced changes in the rat central dopaminergic system. Alcohol Alcohol. 2005;40:297–301. doi: 10.1093/alcalc/agh163. [DOI] [PubMed] [Google Scholar]
- 9.Hemby SE, Johnson BA, Dworkin SI. Neurobiological basis of drug reinforcement. In: Johnson BA, Roache JD, editors. Drug addiction and its treatment: nexus of neuroscience and behavior. Philadelphia: Lippincott-Raven; 1997. pp. 137–69. [Google Scholar]
- 10.Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res. 1983;277:119–27. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- 11.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–8. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matthews RT, German DC. Electrophysiological evidence for excitation of rat ventral tegmental area dopamine neurons by morphine. Neuroscience. 1984;11:617–25. doi: 10.1016/0306-4522(84)90048-4. [DOI] [PubMed] [Google Scholar]
- 13.Gianoulakis C. Alcohol-seeking behavior. The roles of the hypothalamic-pituitary-adrenal axis and the endogenous opioid system. Alcohol Health Res World. 1998;22:202–10. [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson BA, Ait-Daoud N. Neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Psychopharmacology. 2000;149:327–44. doi: 10.1007/s002130000371. [DOI] [PubMed] [Google Scholar]
- 15.Krystal JH, Madonick S, Perry E, Gueorguieva R, Brush L, Wray Y, et al. Potentiation of low dose ketamine effects by naltrexone: potential implications for the pharmacotherapy of alcoholism. Neuropsychopharmacology. 2006;31:1793–800. doi: 10.1038/sj.npp.1300994. [DOI] [PubMed] [Google Scholar]
- 16.Foster KL, McKay PF, Seyoum R, Milbourne D, Yin W, Sarma PV, et al. GABA(A) and opioid receptors of the central nucleus of the amygdala selectively regulate ethanol-maintained behaviors. Neuropsychopharmacology. 2004;29:269–84. doi: 10.1038/sj.npp.1300306. [DOI] [PubMed] [Google Scholar]
- 17.Matsuzawa S, Suzuki T, Misawa M, Nagase H. Roles of 5-HT3 and opioid receptors in the ethanol-induced place preference in rats exposed to conditioned fear stress. Life Sci. 1999;64:PL241–PL9. doi: 10.1016/s0024-3205(99)00144-7. [DOI] [PubMed] [Google Scholar]
- 18.Manzanares J, Ortiz S, Oliva JM, Perez-Rial S, Palomo T. Interactions between cannabinoid and opioid receptor systems in the mediation of ethanol effects. Alcohol Alcohol. 2005;40:25–34. doi: 10.1093/alcalc/agh112. [DOI] [PubMed] [Google Scholar]
- 19.Resch GE, Shridharani S, Millington WR, Garris DR, Simpson CW. Glycyl-glutamine in nucleus accumbens reduces ethanol intake in alcohol preferring (P) rats. Brain Res. 2005;1058:73–81. doi: 10.1016/j.brainres.2005.07.066. [DOI] [PubMed] [Google Scholar]
- 20.Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, et al. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol. 1994;45:330–4. [PubMed] [Google Scholar]
- 21.Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, et al. Mu-opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–8. [PubMed] [Google Scholar]
- 22.Olive MF, Koenig HN, Nannini MA, Hodge CW. Stimulation of endorphin neurotransmission in the nucleus accumbens by ethanol, cocaine, and amphetamine. J Neurosci. 2001;21(RC184):1–5. doi: 10.1523/JNEUROSCI.21-23-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marinelli PW, Quirion R, Gianoulakis C. A microdialysis profile of beta-endorphin and catecholamines in the rat nucleus accumbens following alcohol administration. Psychopharmacology. 2003;169:60–7. doi: 10.1007/s00213-003-1490-2. [DOI] [PubMed] [Google Scholar]
- 24.Marinelli PW, Quirion R, Gianoulakis C. An in vivo profile of beta-endorphin release in the arcuate nucleus and nucleus accumbens following exposure to stress or alcohol. Neuroscience. 2004;127:777–84. doi: 10.1016/j.neuroscience.2004.05.047. [DOI] [PubMed] [Google Scholar]
- 25.Zalewska-Kaszubska J, Gorska D, Dyr W, Czarnecka E. Effect of acute administration of ethanol on beta-endorphin plasma level in ethanol preferring and non-preferring rats chronically treated with naltrexone. Pharmacol Biochem Behav. 2006;85:155–9. doi: 10.1016/j.pbb.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 26.Altshuler HL, Phillips PE, Feinhandler DA. Alteration of ethanol self-administration by naltrexone. Life Sci. 1980;26:679–88. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- 27.Samson HH, Doyle TF. Oral ethanol self-administration in the rat: effect of naloxone. Pharmacol Biochem Behav. 1985;22:91–9. doi: 10.1016/0091-3057(85)90491-5. [DOI] [PubMed] [Google Scholar]
- 28.Froehlich JC, Zweifel M, Harts J, Lumeng L, Li TK. Importance of delta opioid receptors in maintaining high alcohol drinking. Psychopharmacology. 1991;103:467–72. doi: 10.1007/BF02244246. [DOI] [PubMed] [Google Scholar]
- 29.De Witte P. Naloxone reduces alcohol intake in a free-choice procedure even when both drinking bottles contain saccharin sodium or quinine substances. Neuropsychobiology. 1984;12:73–7. doi: 10.1159/000118113. [DOI] [PubMed] [Google Scholar]
- 30.Froehlich JC, Harts J, Lumeng L, Li TK. Naloxone attenuation of voluntary alcohol consumption. Alcohol Alcohol. 1987;(suppl 1):333–7. [PubMed] [Google Scholar]
- 31.Froehlich JC, Harts J, Lumeng L, Li TK. Naloxone attenuates voluntary ethanol intake in rats selectively bred for high ethanol preference. Pharmacol Biochem Behav. 1990;35:385–90. doi: 10.1016/0091-3057(90)90174-g. [DOI] [PubMed] [Google Scholar]
- 32.van Ree JM, Kornet M, Goosen C. Neuropeptides and alcohol addiction in monkeys. EXS. 1994;71:165–74. doi: 10.1007/978-3-0348-7330-7_17. [DOI] [PubMed] [Google Scholar]
- 33.Le AD, Poulos CX, Quan B, Chou S. The effects of selective blockade of delta and mu opiate receptors on ethanol consumption by C57B1/6 mice in a restricted access paradigm. Brain Res. 1993;630:330–2. doi: 10.1016/0006-8993(93)90672-a. [DOI] [PubMed] [Google Scholar]
- 34.Volpicelli JR, Davis MA, Olgin JE. Naltrexone blocks the post-shock increase of ethanol consumption. Life Sci. 1986;38:841–7. doi: 10.1016/0024-3205(86)90601-6. [DOI] [PubMed] [Google Scholar]
- 35.Heyser CJ, Moc K, Koob GF. Effects of naltrexone alone and in combination with acamprosate on the alcohol deprivation effect in rats. Neuropsychopharmacology. 2003;28:1463–71. doi: 10.1038/sj.npp.1300175. [DOI] [PubMed] [Google Scholar]
- 36.Kamdar NK, Miller SA, Syed YM, Bhayana R, Gupta T, Rhodes JS. Acute effects of naltrexone and GBR 12909 on ethanol drinking-in-the-dark in C57BL/6J mice. Psychopharmacology. 2007;192:207–17. doi: 10.1007/s00213-007-0711-5. [DOI] [PubMed] [Google Scholar]
- 37.Ross D, Hartmann RJ, Geller I. Ethanol preference in the hamster: effects of morphine sulfate and naltrexone, a long-acting morphine antagonist. Proc West Pharmacol Soc. 1976;19:326–30. [PubMed] [Google Scholar]
- 38.Berman RF, Lee JA, Olson KL, Goldman MS. Effects of naloxone on ethanol dependence in rats. Drug Alcohol Depend. 1984;13:245–54. doi: 10.1016/0376-8716(84)90065-6. [DOI] [PubMed] [Google Scholar]
- 39.Juarez J, Eliana Bde T. Alcohol consumption is enhanced after naltrexone treatment. Alcohol Clin Exp Res. 2007;31:260–4. doi: 10.1111/j.1530-0277.2006.00313.x. [DOI] [PubMed] [Google Scholar]
- 40.Williams KL, Broadbear JH, Woods JH. Noncontingent and response-contingent intravenous ethanol attenuates the effect of naltrexone on hypothalamic-pituitary-adrenal activity in rhesus monkeys. Alcohol Clin Exp Res. 2004;28:566–71. doi: 10.1097/01.alc.0000121655.48922.c4. [DOI] [PubMed] [Google Scholar]
- 41.Herz A. Endogenous opioid systems and alcohol addiction. Psychopharmacology. 1997;129:99–111. doi: 10.1007/s002130050169. [DOI] [PubMed] [Google Scholar]
- 42.Gianoulakis C, de Waele JP, Thavundayil J. Implication of the endogenous opioid system in excessive ethanol consumption. Alcohol. 1996;13:19–23. doi: 10.1016/0741-8329(95)02035-7. [DOI] [PubMed] [Google Scholar]
- 43.Grisel JE, Mogil JS, Grahame NJ, Rubinstein M, Belknap JK, Crabbe JC, et al. Ethanol oral self-administration is increased in mutant mice with decreased beta-endorphin expression. Brain Res. 1999;835:62–7. doi: 10.1016/s0006-8993(99)01384-0. [DOI] [PubMed] [Google Scholar]
- 44.Swift RM, Whelihan W, Kuznetsov O, Buongiorno G, Hsuing H. Naltrexone-induced alterations in human ethanol intoxication. Am J Psychiatry. 1994;151:1463–7. doi: 10.1176/ajp.151.10.1463. [DOI] [PubMed] [Google Scholar]
- 45.Davidson D, Swift R, Fitz E. Naltrexone increases the latency to drink alcohol in social drinkers. Alcohol Clin Exp Res. 1996;20:732–9. doi: 10.1111/j.1530-0277.1996.tb01679.x. [DOI] [PubMed] [Google Scholar]
- 46.Doty P, de Wit H. Effects of naltrexone pretreatment on the subjective and performance effects of ethanol in social drinkers. Behav Pharmacol. 1995;6:386–94. [PubMed] [Google Scholar]
- 47.King AC, Volpicelli JR, Frazer A, O’Brien CP. Effect of naltrexone on subjective alcohol response in subjects at high and low risk for future alcohol dependence. Psychopharmacology. 1997;129:15–22. doi: 10.1007/s002130050156. [DOI] [PubMed] [Google Scholar]
- 48.Krishnan-Sarin S, Krystal JH, Shi J, Pittman B, O’Malley SS. Family history of alcoholism influences naltrexone-induced reduction in alcohol drinking. Biol Psychiatry. 2007;62:694–7. doi: 10.1016/j.biopsych.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 49.McGeary JE, Monti PM, Rohsenow DJ, Tidey J, Swift R, Miranda RJ. Genetic moderators of naltrexone’s effects on alcohol cue reactivity. Alcohol Clin Exp Res. 2006;30:1288–96. doi: 10.1111/j.1530-0277.2006.00156.x. [DOI] [PubMed] [Google Scholar]
- 50.Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR, et al. A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology. 2003;28:1546–52. doi: 10.1038/sj.npp.1300219. [DOI] [PubMed] [Google Scholar]
- 51.O’Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B. Naltrexone and coping skills therapy for alcohol dependence: a controlled study. Arch Gen Psychiatry. 1992;49:881–7. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- 52.Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–80. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 53.Jaffe AJ, Rounsaville B, Chang G, Schottenfeld RS, Meyer RE, O’Malley SS. Naltrexone, relapse prevention, and supportive therapy with alcoholics: an analysis of patient treatment matching. J Consult Clin Psychol. 1996;64:1044–53. doi: 10.1037//0022-006x.64.5.1044. [DOI] [PubMed] [Google Scholar]
- 54.O’Malley SS, Jaffe AJ, Rode S, Rounsaville BJ. Experience of a “slip” among alcoholics treated with naltrexone or placebo. Am J Psychiatry. 1996;153:281–3. doi: 10.1176/ajp.153.2.281. [DOI] [PubMed] [Google Scholar]
- 55.Volpicelli JR, Watson NT, King AC, Sherman CE, O’Brien CP. Effect of naltrexone on alcohol “high” in alcoholics. Am J Psychiatry. 1995;152:613–5. doi: 10.1176/ajp.152.4.613. [DOI] [PubMed] [Google Scholar]
- 56.Bouza C, Angeles M, Munoz A, Amate JM. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction. 2004;99:811–28. doi: 10.1111/j.1360-0443.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 57.Srisurapanont M, Jarusuraisin N. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev. 2005;1:CD001867. doi: 10.1002/14651858.CD001867.pub2. [DOI] [PubMed] [Google Scholar]
- 58.Volpicelli JR, Rhines KC, Rhines JS, Volpicelli LA, Alterman AI, O’Brien CP. Naltrexone and alcohol dependence: role of subject compliance. Arch Gen Psychiatry. 1997;54:737–42. doi: 10.1001/archpsyc.1997.01830200071010. [DOI] [PubMed] [Google Scholar]
- 59.Litten RZ, Allen J, Fertig J. Pharmacotherapies for alcohol problems: a review of research with focus on developments since 1991. Alcohol Clin Exp Res. 1996;20:859–76. doi: 10.1111/j.1530-0277.1996.tb05264.x. [DOI] [PubMed] [Google Scholar]
- 60.Litten RZ, Allen JP. Advances in development of medications for alcoholism treatment. Psychopharmacology. 1998;139:20–33. doi: 10.1007/s002130050686. [DOI] [PubMed] [Google Scholar]
- 61.Croop RS, Faulkner EB, Labriola DF. The safety profile of naltrexone in the treatment of alcoholism. Results from a multicenter usage study. The Naltrexone Usage Study Group. Arch Gen Psychiatry. 1997;54:1130–5. doi: 10.1001/archpsyc.1997.01830240090013. [DOI] [PubMed] [Google Scholar]
- 62.Anton RF, O’Malley SS, Ciraulo DA, Cisler RA, Couper D, Donovan DM, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence — The COMBINE Study: a randomized controlled trial. JAMA. 2006;295:2003–17. doi: 10.1001/jama.295.17.2003. [DOI] [PubMed] [Google Scholar]
- 63.Gelernter J, Gueorguieva R, Kranzler HR, Zhang H, Cramer J, Rosenheck R, et al. Opioid receptor gene (OPRM1, OPRK1, and OPRD1) variants and response to naltrexone treatment for alcohol dependence: results from the VA Cooperative Study. Alcohol Clin Exp Res. 2007;31:555–63. doi: 10.1111/j.1530-0277.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- 64.Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci U S A. 1998;95:9608–13. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Befort K, Filliol D, Décaillot FM, Gavériaux-Ruff C, Hoehe MR, Kieffer BL. A single nucleotide polymorphic mutation in the human mu-opioid receptor severely impairs receptor signaling. J Biol Chem. 2001;276:3130–7. doi: 10.1074/jbc.M006352200. [DOI] [PubMed] [Google Scholar]
- 66.Beyer A, Koch T, Schröder H, Schulz S, Höllt V. Effect of the A118G polymorphism on binding affinity, potency and agonist-mediated endocytosis, desensitization, and resensitization of the human mu-opioid receptor. J Neurochem. 2004;89:553–60. doi: 10.1111/j.1471-4159.2004.02340.x. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Wang D, Johnson AD, Papp AC, Sadée W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem. 2005;280:32618–24. doi: 10.1074/jbc.M504942200. [DOI] [PubMed] [Google Scholar]
- 68.Gelernter J, Kranzler H, Cubells J. Genetics of two mu opioid receptor gene (OPRM1) exon I polymorphisms: population studies, and allele frequencies in alcohol- and drug-dependent subjects. Mol Psychiatry. 1999;4:476–83. doi: 10.1038/sj.mp.4000556. [DOI] [PubMed] [Google Scholar]
- 69.Zhang H, Luo X, Kranzler HR, Lappalainen J, Yang B-Z, Krupitsky E, et al. Association between two mu-opioid receptor gene (OPRM1) haplotype blocks and drug or alcohol dependence. Hum Mol Genet. 2006;15:807–19. doi: 10.1093/hmg/ddl024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.King AC, Schluger J, Gunduz M, Borg L, Perret G, Ho A, et al. Hypothalamic-pituitary-adrenocortical (HPA) axis response and biotransformation of oral naltrexone: preliminary examination of relationship to family history of alcoholism. Neuropsychopharmacology. 2002;26:778–88. doi: 10.1016/S0893-133X(01)00416-X. [DOI] [PubMed] [Google Scholar]
- 71.Monterosso JR, Flannery BA, Pettinati HM, Oslin DW, Rukstalis M, O’Brien CP, et al. Predicting treatment response to naltrexone: the influence of craving and family history. Am J Addict. 2001;10:258–68. doi: 10.1080/105504901750532148. [DOI] [PubMed] [Google Scholar]
- 72.Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 73.Lewis DH. Controlled release of bioactive agents from lactide/glycolide polymers. In: Chasin M, Langer R, editors. Biodegradable polymers as drug delivery systems. New York: Marcel Dekker; 1990. pp. 1–41. [Google Scholar]
- 74.Johnson BA, Ait-Daoud N, Aubin H-J, van den Brink W, Guzzetta R, Loewy J, et al. A pilot evaluation of the safety and tolerability of repeat dose administration of long-acting injectable naltrexone (Vivitrex®) in patients with alcohol dependence. Alcohol Clin Exp Res. 2004;28:1356–61. doi: 10.1097/01.alc.0000139823.30096.52. [DOI] [PubMed] [Google Scholar]
- 75.Garbutt JC, Kranzler HR, O’Malley SS, Gastfriend DR, Pettinati HM, Silverman BL, et al. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA. 2005;293:1617–25. doi: 10.1001/jama.293.13.1617. [DOI] [PubMed] [Google Scholar]
- 76.Kranzler HR, Wesson DR, Billot L. Drug Abuse Sciences Naltrexone Depot Study Group. Naltrexone depot for treatment of alcohol dependence: a multicenter, randomized, placebo-controlled clinical trial. Alcohol Clin Exp Res. 2004;28:1051–9. doi: 10.1097/01.alc.0000130804.08397.29. [DOI] [PubMed] [Google Scholar]
- 77.Galloway GP, Koch M, Cello R, Smith DE. Pharmacokinetics, safety, and tolerability of a depot formulation of naltrexone in alcoholics: an open-label trial. BMC Psychiatry. 2005;5:18. doi: 10.1186/1471-244X-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conigrave KM, Degenhardt LJ, Whitfield JB, Saunders JB, Helander A, Tabakoff B, et al. CDT, GGT, and AST as markers of alcohol use: the WHO/ISBRA collaborative project. Alcohol Clin Exp Res. 2002;26:332–9. [PubMed] [Google Scholar]
- 79.Kranzler HR, Modesto-Lowe V, Nuwayser ES. Sustained-release naltrexone for alcoholism treatment: a preliminary study. Alcohol Clin Exp Res. 1998;22:1074–9. [PubMed] [Google Scholar]
- 80.Alim TN, Tai B, Chiang CN, Green T, Rosse RB, Lindquist T, et al. Tolerability study of a depot form of naltrexone substance abusers [abstract] In: Harris LS, editor. Problems of Drug Dependence 1994: Proceedings of the 56th Annual Scientific Meeting, The College on Problems of Drug Dependence, Inc., Volume II. NIDA Res Monogr. Vol. 153. 1995. p. 253. [Google Scholar]
- 81.Heishman SJ, Francis-Wood A, Keenan RM, Chiang CN, Terrill JB, Tai B, et al. Safety and pharmacokinetics of a new formulation of naltrexone [abstract] In: Harris LS, editor. Problems of Drug Dependence 1993: Proceedings of the 55th Annual Scientific Meeting, The College on Problems of Drug Dependence, Inc., Volume II. NIDA Res Monogr. Vol. 141. 1994. p. 82. [Google Scholar]
- 82.Comer SD, Collins ED, Kleber HD, Nuwayser ES, Kerrigan JH, Fischman MW. Depot naltrexone: long-lasting antagonism of the effects of heroin in humans. Psychopharmacology. 2002;159:351–60. doi: 10.1007/s002130100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zieglgansberger W, Hauser C, Wetzel C, Putzke J, Siggins GR, Spanagel R. Actions of acamprosate on neurons of the central nervous system. In: Soyka M, editor. Acamprosate in relapse prevention of alcoholism. Berlin: Springer; 1996. pp. 65–70. [Google Scholar]
- 84.De Witte P, Bachteler D, Spanagel R. Acamprosate: preclinical data. In: Spanagel R, Mann KF, editors. Drugs for relapse prevention of alcoholism. Basel, Switzerland: Birkhäuser Verlag; 2005. pp. 73–83. [Google Scholar]
- 85.De Witte P, Littleton J, Parot P, Koob G. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19:517–37. doi: 10.2165/00023210-200519060-00004. [DOI] [PubMed] [Google Scholar]
- 86.Harris BR, Prendergast MA, Gibson DA, Rogers DT, Blanchard JA, Holley RC, et al. Acamprosate inhibits the binding and neurotoxic effects of trans-ACPD, suggesting a novel site of action at metabotropic glutamate receptors. Alcohol Clin Exp Res. 2002;26:1779–93. doi: 10.1097/01.ALC.0000042011.99580.98. [DOI] [PubMed] [Google Scholar]
- 87.Boismare F, Daoust M, Moore N, Saligaut C, Lhuintre JP, Chretien P, et al. A homotaurine derivative reduces the voluntary intake of ethanol by rats: are cerebral GABA receptors involved? Pharmacol Biochem Behav. 1984;21:787–9. doi: 10.1016/s0091-3057(84)80020-9. [DOI] [PubMed] [Google Scholar]
- 88.Le Magnen J, Tran G, Durlach J. Lack of effects of Ca-acetyl homotaurinate on chronic and acute toxicities of ethanol in rats. Alcohol. 1987;4:103–8. doi: 10.1016/0741-8329(87)90006-1. [DOI] [PubMed] [Google Scholar]
- 89.Czachowski CL, Legg BH, Samson HH. Effects of acamprosate on ethanol-seeking and self-administration in the rat. Alcohol Clin Exp Res. 2001;25:344–50. [PubMed] [Google Scholar]
- 90.Escher T, Mittleman G. Schedule-induced alcohol drinking: non-selective effects of acamprosate and naltrexone. Addict Biol. 2006;11:55–63. doi: 10.1111/j.1369-1600.2006.00004.x. [DOI] [PubMed] [Google Scholar]
- 91.Dahchour A, De Witte P, Bolo N, Nédélec JF, Muzet M, Durbin P, et al. Central effects of acamprosate: part 1. Acamprosate blocks the glutamate increase in the nucleus accumbens microdialysate in ethanol withdrawn rats. Psychiatry Res. 1998;82:107–14. doi: 10.1016/s0925-4927(98)00016-x. [DOI] [PubMed] [Google Scholar]
- 92.Rossetti ZL, Carboni S. Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. Eur J Pharmacol. 1995;283:177–83. doi: 10.1016/0014-2999(95)00344-k. [DOI] [PubMed] [Google Scholar]
- 93.Gewiss M, Heidbreder C, Opsomer L, Durbin P, De Witte P. Acamprosate and diazepam differentially modulate alcohol-induced behavioural and cortical alterations in rats following chronic inhalation of ethanol vapour. Alcohol Alcohol. 1991;26:129–37. doi: 10.1093/oxfordjournals.alcalc.a045093. [DOI] [PubMed] [Google Scholar]
- 94.Spanagel R, Putzke J, Stefferl A, Schobitz B, Zieglgansberger W. Acamprosate and alcohol: II. Effects on alcohol withdrawal in the rat. Eur J Pharmacol. 1996;305:45–50. doi: 10.1016/0014-2999(96)00175-6. [DOI] [PubMed] [Google Scholar]
- 95.Bolo N, Nédélec JF, Muzet M, De Witte P, Dahchour A, Durbin P, et al. Central effects of acamprosate: part 2. Acamprosate modifies the brain in-vivo proton magnetic resonance spectrum in healthy young male volunteers. Psychiatry Res. 1998;82:115–27. doi: 10.1016/s0925-4927(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 96.Putzke J, Spanagel R, Tolle TR, Zieglgansberger W. The anti-craving drug acamprosate reduces c-fos expression in rats undergoing ethanol withdrawal. Eur J Pharmacol. 1996;317:39–48. doi: 10.1016/s0014-2999(96)00696-6. [DOI] [PubMed] [Google Scholar]
- 97.Littleton J. Acamprosate in alcohol dependence: how does it work? Addiction. 1995;90:1179–88. doi: 10.1046/j.1360-0443.1995.90911793.x. [DOI] [PubMed] [Google Scholar]
- 98.Krystal JH, Petrakis IL, Krupitsky E, Schutz C, Trevisan L, D’Souza DC. NMDA receptor antagonism and the ethanol intoxication signal: from alcoholism risk to pharmacotherapy. Ann N Y Acad Sci. 2003;1003:176–84. doi: 10.1196/annals.1300.010. [DOI] [PubMed] [Google Scholar]
- 99.Sinclair JD, Li TK. Long and short alcohol deprivation: effects on AA and P alcohol-preferring rats. Alcohol. 1989;6:505–9. doi: 10.1016/0741-8329(89)90059-1. [DOI] [PubMed] [Google Scholar]
- 100.Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A. 1992;89:2046–50. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Spanagel R, Holter SM, Allingham K, Landgraf R, Zieglgansberger W. Acamprosate and alcohol: I. Effects on alcohol intake following alcohol deprivation in the rat. Eur J Pharmacol. 1996;305:39–44. doi: 10.1016/0014-2999(96)00174-4. [DOI] [PubMed] [Google Scholar]
- 102.Wolffgramm J, Heyne A. From controlled drug intake to loss of control: the irreversible development of drug addiction in the rat. Behav Brain Res. 1995;70:77–94. doi: 10.1016/0166-4328(95)00131-c. [DOI] [PubMed] [Google Scholar]
- 103.Spanagel R, Zieglgansberger W. Anti-craving compounds for ethanol: new pharmacological tools to study addictive processes. Trends Pharmacol Sci. 1997;18:54–9. [PubMed] [Google Scholar]
- 104.Mason BJ, Goodman AM, Dixon RM, Hameed MH, Hulot T, Wesnes K, et al. A pharmacokinetic and pharmacodynamic drug interaction study of acamprosate and naltrexone. Neuropsychopharmacology. 2002;27:596–606. doi: 10.1016/S0893-133X(02)00368-8. [DOI] [PubMed] [Google Scholar]
- 105.Johnson BA, O’Malley SS, Ciraulo DA, Roache JD, Chambers RA, Sarid-Segal O, et al. Dose-ranging kinetics and behavioral pharmacology of naltrexone and acamprosate, both alone and combined, in alcohol-dependent subjects. J Clin Psychopharmacol. 2003;23:281–93. doi: 10.1097/01.jcp.0000084029.22282.bb. [DOI] [PubMed] [Google Scholar]
- 106.Ooteman W, Koeter MW, Verheul R, Schippers GM, van den Brink W. The effect of naltrexone and acamprosate on cue-induced craving, autonomic nervous system and neuroendocrine reactions to alcohol-related cues in alcoholics. Eur Neuropsychopharmacol. 2007;17:558–66. doi: 10.1016/j.euroneuro.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 107.Mann K, Lehert P, Morgan MY. The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol Clin Exp Res. 2004;28:51–63. doi: 10.1097/01.ALC.0000108656.81563.05. [DOI] [PubMed] [Google Scholar]
- 108.Kranzler HR, Van Kirk J. Efficacy of naltrexone and acamprosate for alcoholism treatment: a meta-analysis. Alcohol Clin Exp Res. 2001;25:1335–41. [PubMed] [Google Scholar]
- 109.Chick J, Lehert P, Landron F Plinius Maior Society. Does acamprosate improve reduction of drinking as well as aiding abstinence? J Psychopharmacol. 2003;17:397–402. doi: 10.1177/0269881103174017. [DOI] [PubMed] [Google Scholar]
- 110.Mason BJ, Goodman AM, Chabac S, Lehert P. Effect of oral acamprosate on abstinence in patients with alcohol dependence in a double-blind, placebo-controlled trial: the role of patient motivation. J Psychiatr Res. 2006;40:383–93. doi: 10.1016/j.jpsychires.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 111.Verheul R, Lehert P, Geerlings PJ, Koeter MW, van den Brink W. Predictors of acamprosate efficacy: results from a pooled analysis of seven European trials including 1485 alcohol-dependent patients. Psychopharmacology. 2005;178:167–73. doi: 10.1007/s00213-004-1991-7. [DOI] [PubMed] [Google Scholar]
- 112.Nagy J. Renaissance of NMDA receptor antagonists: do they have a role in the pharmacotherapy for alcoholism? IDrugs. 2004;7:339–50. [PubMed] [Google Scholar]
- 113.Kotlinska J, Bochenski M, Danysz W. N-methyl-D-aspartate and group I metabotropic glutamate receptors are involved in the expression of ethanol-induced sensitization in mice. Behav Pharmacol. 2006;17:1–8. doi: 10.1097/01.fbp.0000181600.95405.c7. [DOI] [PubMed] [Google Scholar]
- 114.Bisaga A, Evans SM. Acute effects of memantine in combination with alcohol in moderate drinkers. Psychopharmacology. 2004;172:16–24. doi: 10.1007/s00213-003-1617-5. [DOI] [PubMed] [Google Scholar]
- 115.Krupitsky EM, Neznanova O, Masalov D, Burakov AM, Didenko T, Romanova T, et al. Effect of memantine on cue-induced alcohol craving in recovering alcohol-dependent patients. Am J Psychiatry. 2007;164:519–23. doi: 10.1176/ajp.2007.164.3.519. [DOI] [PubMed] [Google Scholar]
- 116.Evans SM, Levin FR, Brooks DJ, Garawi F. A pilot double-blind treatment trial of memantine for alcohol dependence. Alcohol Clin Exp Res. 2007;31:775–82. doi: 10.1111/j.1530-0277.2007.00360.x. [DOI] [PubMed] [Google Scholar]
- 117.Gibbs JW, Sombati S, DeLorenzo RJ, Coulter DA. Cellular actions of topiramate: blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia. 2000;41(suppl 1):S10–6. doi: 10.1111/j.1528-1157.2000.tb02164.x. [DOI] [PubMed] [Google Scholar]
- 118.Skradski S, White HS. Topiramate blocks kainate-evoked cobalt influx into cultured neurons. Epilepsia. 2000;41(suppl 1):S45–7. doi: 10.1111/j.1528-1157.2000.tb02171.x. [DOI] [PubMed] [Google Scholar]
- 119.Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003;23:7069–74. doi: 10.1523/JNEUROSCI.23-18-07069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate enhances GABA-mediated chloride flux and GABA-evoked chloride currents in murine brain neurons and increases seizure threshold. Epilepsy Res. 1997;28:167–79. doi: 10.1016/s0920-1211(97)00045-4. [DOI] [PubMed] [Google Scholar]
- 121.White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia. 2000;41(suppl 1):S17–20. [PubMed] [Google Scholar]
- 122.Zhang X, Velumian AA, Jones OT, Carlen PL. Modulation of high-voltage-activated calcium channels in dentate granule cells by topiramate. Epilepsia. 2000;41(suppl 1):S52–60. doi: 10.1111/j.1528-1157.2000.tb02173.x. [DOI] [PubMed] [Google Scholar]
- 123.Taverna S, Sancini G, Mantegazza M, Franceschetti S, Avanzini G. Inhibition of transient and persistent Na+ current fractions by the new anticonvulsant topiramate. J Pharmacol Exp Ther. 1999;288:960–8. [PubMed] [Google Scholar]
- 124.Herrero AI, Del Olmo N, Gonzalez-Escalada JR, Solis JM. Two new actions of topiramate: inhibition of depolarizing GABA(A)-mediated responses and activation of a potassium conductance. Neuropharmacology. 2002;42:210–20. doi: 10.1016/s0028-3908(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 125.Dodgson SJ, Shank RP, Maryanoff BE. Topiramate as an inhibitor of carbonic anhydrase isoenzymes. Epilepsia. 2000;41(suppl 1):S35–9. doi: 10.1111/j.1528-1157.2000.tb06047.x. [DOI] [PubMed] [Google Scholar]
- 126.Shank RP, Gardocki JF, Streeter AJ, Maryanoff BE. An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia. 2000;41(suppl 1):S3–9. [PubMed] [Google Scholar]
- 127.Johnson BA. Progress in the development of topiramate for treating alcohol dependence: from a hypothesis to a proof-of-concept study. Alcohol Clin Exp Res. 2004;28:1137–44. doi: 10.1097/01.alc.0000134533.96915.08. [DOI] [PubMed] [Google Scholar]
- 128.Johnson BA. Recent advances in the development of treatments for alcohol and cocaine dependence: focus on topiramate and other modulators of GABA or glutamate function. CNS Drugs. 2005;19:873–96. doi: 10.2165/00023210-200519100-00005. [DOI] [PubMed] [Google Scholar]
- 129.Gabriel KI, Cunningham CL. Effects of topiramate on ethanol and saccharin consumption and preferences in C57BL/6J mice. Alcohol Clin Exp Res. 2005;29:75–80. doi: 10.1097/01.alc.0000150014.79657.64. [DOI] [PubMed] [Google Scholar]
- 130.Nguyen SA, Malcolm R, Middaugh LD. Topiramate reduces ethanol consumption by C57BL/6 mice. Synapse. 2007;61:150–6. doi: 10.1002/syn.20350. [DOI] [PubMed] [Google Scholar]
- 131.Lynch WJ, Johnson BA. Effects of topiramate on alcohol consumption in the rat. Alcohol Clin Exp Res. 2007;31(s2):261A. abstract. [Google Scholar]
- 132.Farook JM, Morrell DJ, Lewis B, Littleton JM, Barron S. Topiramate (Topamax®) reduces conditioned abstinence behaviours and handling-induced convulsions (HIC) after chronic administration of alcohol in Swiss-Webster mice. Alcohol Alcohol. doi: 10.1093/alcalc/agm047. in press. [DOI] [PubMed] [Google Scholar]
- 133.Johnson BA, Mann K, Willenbring ML, Litten RZ, Swift RM, Lesch OM, et al. Challenges and opportunities for medications development in alcoholism: an international perspective on collaborations between academia and industry. Alcohol Clin Exp Res. 2005;29:1528–40. doi: 10.1097/01.alc.0000174690.63787.fc. [DOI] [PubMed] [Google Scholar]
- 134.Johnson BA, Ait-Daoud N, Bowden CL, DiClemente CC, Roache JD, Lawson K, et al. Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet. 2003;361:1677–85. doi: 10.1016/S0140-6736(03)13370-3. [DOI] [PubMed] [Google Scholar]
- 135.Ma JZ, Ait-Daoud N, Johnson BA. Topiramate reduces the harm of excessive drinking: implications for public health and primary care. Addiction. 2006;101:1561–8. doi: 10.1111/j.1360-0443.2006.01576.x. [DOI] [PubMed] [Google Scholar]
- 136.Miranda R, Monti P, Swift R, MacKillop J, Tidey J, Gwaltney C, et al. Effects of topiramate on alcohol cue reactivity and the subjective effects of drinking. Poster presentation at the 45th Annual Meeting of the American College of Neuropsychopharmacology; Hollywood, Florida. December 6, 2006. [Google Scholar]
- 137.Johnson BA, Rosenthal N, Capece JA, Wiegand F, Mao L, Beyers K, et al. Topiramate for the treatment of alcohol dependence: results of a multi-site trial. New Research poster presentation at the 160th Annual Meeting of the American Psychiatric Association; San Diego, California. May 22, 2007. [Google Scholar]
- 138.Johnson & Johnson Pharmaceutical Research & Development. Investigator’s Brochure: Topiramate (RWJ-17021-000) 10. Dec, 2005. [Google Scholar]
- 139.Biton V, Edwards KR, Montouris GD, Sackellares JC, Harden CL, Kamin M, et al. Topiramate titration and tolerability. Ann Pharmacother. 2001;35:173–9. doi: 10.1345/aph.10093. [DOI] [PubMed] [Google Scholar]
- 140.Myers RD, Veale WL. Alcohol preference in the rat: reduction following depletion of brain serotonin. Science. 1968;160:1469–71. doi: 10.1126/science.160.3835.1469. [DOI] [PubMed] [Google Scholar]
- 141.Nachman M, Lester D, Le Magnen J. Alcohol aversion in the rat: behavioral assessment of noxious drug effects. Science. 1970;168:1244–6. doi: 10.1126/science.168.3936.1244. [DOI] [PubMed] [Google Scholar]
- 142.Daoust M, Chretien P, Moore N, Saligaut C, Lhuintre JP, Boismare F. Isolation and striatal (3H) serotonin uptake: role in the voluntary intake of ethanol by rats. Pharmacol Biochem Behav. 1985;22:205–8. doi: 10.1016/0091-3057(85)90378-8. [DOI] [PubMed] [Google Scholar]
- 143.Geller I. Effects of para-chlorophenylalanine and 5-hydroxytryptophan on alcohol intake in the rat. Pharmacol Biochem Behav. 1973;1:361–5. doi: 10.1016/0091-3057(73)90130-5. [DOI] [PubMed] [Google Scholar]
- 144.Gill K, Amit Z, Koe BK. Treatment with sertraline, a new serotonin uptake inhibitor, reduces voluntary ethanol consumption in rats. Alcohol. 1988;5:349–54. doi: 10.1016/0741-8329(88)90019-5. [DOI] [PubMed] [Google Scholar]
- 145.Gill K, Filion Y, Amit Z. A further examination of the effects of sertraline on voluntary ethanol consumption. Alcohol. 1988;5:355–8. doi: 10.1016/0741-8329(88)90020-1. [DOI] [PubMed] [Google Scholar]
- 146.Zabik JE, Binkerd K, Roache JD. Serotonin and ethanol aversion in the rat. In: Naranjo CA, Sellers EM, editors. Research advances in new psychopharmacological treatments for alcoholism: proceedings of the symposium; Toronto. 4-5 October 1984; Amsterdam: Excerpta Medica; 1985. pp. 87–105. [Google Scholar]
- 147.McBride WJ, Murphy JM, Lumeng L, Li TK. Serotonin and ethanol preference. Recent Dev Alcohol. 1989;7:187–209. doi: 10.1007/978-1-4899-1678-5_10. [DOI] [PubMed] [Google Scholar]
- 148.Boyce-Rustay JM, Wiedholz LM, Millstein RA, Carroll J, Murphy DL, Daws LC, et al. Ethanol-related behaviors in serotonin transporter knockout mice. Alcohol Clin Exp Res. 2006;30:1957–65. doi: 10.1111/j.1530-0277.2006.00241.x. [DOI] [PubMed] [Google Scholar]
- 149.Blundell JE, Latham CJ. Behavioural pharmacology of feeding. In: Silverstone T, editor. Drugs and appetite. London: Academic Press; 1982. pp. 41–80. [Google Scholar]
- 150.Blundell JE. Serotonin and appetite. Neuropharmacology. 1984;23:1537–51. doi: 10.1016/0028-3908(84)90098-4. [DOI] [PubMed] [Google Scholar]
- 151.Maurel S, De Vry J, Schreiber R. Comparison of the effects of the selective serotonin-reuptake inhibitors fluoxetine, paroxetine, citalopram and fluvoxamine in alcohol-preferring cAA rats. Alcohol. 1999;17:195–201. doi: 10.1016/s0741-8329(98)00046-9. [DOI] [PubMed] [Google Scholar]
- 152.Gill K, Amit Z. Serotonin uptake blockers and voluntary alcohol consumption. A review of recent studies. Recent Dev Alcohol. 1989;7:225–48. doi: 10.1007/978-1-4899-1678-5_12. [DOI] [PubMed] [Google Scholar]
- 153.Gottfries CG. Influence of depression and antidepressants on weight. Acta Psychiatr Scand Suppl. 1981;290:353–6. doi: 10.1111/j.1600-0447.1981.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 154.Simpson RJ, Lawton DJ, Watt MH, Tiplady B. Effect of zimelidine, a new antidepressant, on appetite and body weight. Br J Clin Pharmacol. 1981;11:96–8. doi: 10.1111/j.1365-2125.1981.tb01112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Leander JD. Fluoxetine suppresses palatability-induced ingestion. Psychopharmacology. 1987;91:285–7. doi: 10.1007/BF00518178. [DOI] [PubMed] [Google Scholar]
- 156.Stellar JR, Stellar E. The neurobiology of motivation and reward. New York: Springer-Verlag; 1985. [Google Scholar]
- 157.Wurtman JJ, Wurtman RJ. Fenfluramine and fluoxetine spare protein consumption while suppressing caloric intake by rats. Science. 1977;198:1178–80. doi: 10.1126/science.929195. [DOI] [PubMed] [Google Scholar]
- 158.Wurtman JJ, Wurtman RJ. Drugs that enhance central serotoninergic transmission diminish elective carbohydrate consumption by rats. Life Sci. 1979;24:895–903. doi: 10.1016/0024-3205(79)90339-4. [DOI] [PubMed] [Google Scholar]
- 159.Li ET, Anderson GH. 5-Hydroxytryptamine control of meal to meal composition chosen by rats. Fed Proc. 1983;42:542–8. [Google Scholar]
- 160.Heisler LK, Kanarek RB, Gerstein A. Fluoxetine decreases fat and protein intakes but not carbohydrate intake in male rats. Pharmacol Biochem Behav. 1997;58:767–73. doi: 10.1016/s0091-3057(97)00036-1. [DOI] [PubMed] [Google Scholar]
- 161.Heisler LK, Kanarek RB, Homoleski B. Reduction of fat and protein intakes but not carbohydrate intake following acute and chronic fluoxetine in female rats. Pharmacol Biochem Behav. 1999;63:377–85. doi: 10.1016/s0091-3057(99)00021-0. [DOI] [PubMed] [Google Scholar]
- 162.Smith GP. The physiology of the meal. In: Silverstone T, editor. Drugs and appetite. London; Academic Press: 1982. [Google Scholar]
- 163.Fantino M. Role of sensory input in the control of food intake. J Auton Nerv Syst. 1984;10:347–58. doi: 10.1016/0165-1838(84)90032-8. [DOI] [PubMed] [Google Scholar]
- 164.Wise RA, Raptis L. Effects of pre-feeding on food-approach latency and food consumption speed in food deprived rats. Physiol Behav. 1985;35:961–3. doi: 10.1016/0031-9384(85)90266-5. [DOI] [PubMed] [Google Scholar]
- 165.Haraguchi M, Samson HH, Tolliver GA. Reduction in oral ethanol self-administration in the rat by the 5-HT uptake blocker fluoxetine. Pharmacol Biochem Behav. 1990;35:259–62. doi: 10.1016/0091-3057(90)90236-b. [DOI] [PubMed] [Google Scholar]
- 166.Gulley JM, McNamara C, Barbera TJ, Ritz MC, George FR. Selective serotonin reuptake inhibitors: effects of chronic treatment on ethanol-reinforced behavior in mice. Alcohol. 1995;12:177–81. doi: 10.1016/0741-8329(94)00079-s. [DOI] [PubMed] [Google Scholar]
- 167.Murphy JM, Waller MB, Gatto GJ, McBride WJ, Lumeng L, Li TK. Effects of fluoxetine on the intragastric self-administration of ethanol in the alcohol preferring P line of rats. Alcohol. 1988;5:283–6. doi: 10.1016/0741-8329(88)90066-3. [DOI] [PubMed] [Google Scholar]
- 168.Naranjo CA, Sellers EM, Roach CA, Woodley DV, Sanchez-Craig M, Sykora K. Zimelidine-induced variations in alcohol intake by nondepressed heavy drinkers. Clin Pharmacol Ther. 1984;35:374–81. doi: 10.1038/clpt.1984.46. [DOI] [PubMed] [Google Scholar]
- 169.Naranjo CA, Sellers EM, Sullivan JT, Woodley DV, Kadlec K, Sykora K. The serotonin uptake inhibitor citalopram attenuates ethanol intake. Clin Pharmacol Ther. 1987;41:266–74. doi: 10.1038/clpt.1987.27. [DOI] [PubMed] [Google Scholar]
- 170.Naranjo CA, Sellers EM. Serotonin uptake inhibitors attenuate ethanol intake in problem drinkers. Recent Dev Alcohol. 1989;7:255–66. doi: 10.1007/978-1-4899-1678-5_13. [DOI] [PubMed] [Google Scholar]
- 171.Naranjo CA, Kadlec KE, Sanhueza P, Woodley-Remus D, Sellers EM. Fluoxetine differentially alters alcohol intake and other consummatory behaviors in problem drinkers. Clin Pharmacol Ther. 1990;47:490–8. doi: 10.1038/clpt.1990.62. [DOI] [PubMed] [Google Scholar]
- 172.Naranjo CA, Poulos CX, Bremner KE, Lanctot KL. Citalopram decreases desirability, liking, and consumption of alcohol in alcohol-dependent drinkers. Clin Pharmacol Ther. 1992;51:729–39. doi: 10.1038/clpt.1992.85. [DOI] [PubMed] [Google Scholar]
- 173.Gorelick DA, Paredes A. Effect of fluoxetine on alcohol consumption in male alcoholics. Alcohol Clin Exp Res. 1992;16:261–5. doi: 10.1111/j.1530-0277.1992.tb01373.x. [DOI] [PubMed] [Google Scholar]
- 174.Naranjo CA, Bremner KE, Lanctot KL. Effects of citalopram and a brief psycho-social intervention on alcohol intake, dependence and problems. Addiction. 1995;90:87–99. doi: 10.1046/j.1360-0443.1995.9018712.x. [DOI] [PubMed] [Google Scholar]
- 175.Kabel DI, Petty F. A placebo-controlled, double-blind study of fluoxetine in severe alcohol dependence: adjunctive pharmacotherapy during and after inpatient treatment. Alcohol Clin Exp Res. 1996;20:780–4. doi: 10.1111/j.1530-0277.1996.tb01686.x. [DOI] [PubMed] [Google Scholar]
- 176.Kranzler HR, Burleson JA, Korner P, Del Boca FK, Bohn MJ, Brown J, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152:391–7. doi: 10.1176/ajp.152.3.391. [DOI] [PubMed] [Google Scholar]
- 177.Johnson BA. Serotonergic agents and alcoholism treatment: rebirth of the subtype concept—an hypothesis. Alcohol Clin Exp Res. 2000;24:1597–601. [PubMed] [Google Scholar]
- 178.Cloninger CR. Neurogenetic adaptive mechanisms in alcoholism. Science. 1987;236:410–6. doi: 10.1126/science.2882604. [DOI] [PubMed] [Google Scholar]
- 179.Buydens-Branchey L, Branchey MH, Noumair D. Age of alcoholism onset. I. Relationship to psychopathology. Arch Gen Psychiatry. 1989;46:225–30. doi: 10.1001/archpsyc.1989.01810030031004. [DOI] [PubMed] [Google Scholar]
- 180.Linnoila M, Virkkunen M. Biologic correlates of suicidal risk and aggressive behavioral traits. J Clin Psychopharmacol. 1992;12(2 suppl):19S–20S. [PubMed] [Google Scholar]
- 181.Linnoila M, De Jong J, Virkkunen M. Family history of alcoholism in violent offenders and impulsive fire setters. Arch Gen Psychiatry. 1989;46:613–6. doi: 10.1001/archpsyc.1989.01810070039006. [DOI] [PubMed] [Google Scholar]
- 182.Kranzler HR, Burleson JA, Brown J, Babor TF. Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res. 1996;20:1534–41. doi: 10.1111/j.1530-0277.1996.tb01696.x. [DOI] [PubMed] [Google Scholar]
- 183.Pettinati HM, Volpicelli JR, Kranzler HR, Luck G, Rukstalis MR, Cnaan A. Sertraline treatment for alcohol dependence: interactive effects of medication and alcoholic subtype. Alcohol Clin Exp Res. 2000;24:1041–9. [PubMed] [Google Scholar]
- 184.Dundon W, Lynch KG, Pettinati HM, Lipkin C. Treatment outcomes in type A and B alcohol dependence 6 months after serotonergic pharmacotherapy. Alcohol Clin Exp Res. 2004;28:1065–73. doi: 10.1097/01.alc.0000130974.50563.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chick J, Aschauer H, Hornik K. Investigators’ Group. Efficacy of fluvoxamine in preventing relapse in alcohol dependence: a one-year, double-blind, placebo-controlled multicentre study with analysis by typology. Drug Alcohol Depend. 2004;74:61–70. doi: 10.1016/j.drugalcdep.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 186.Cornelius JR, Salloum IM, Ehler JG, Jarrett PJ, Cornelius MD, Perel JM, et al. Fluoxetine in depressed alcoholics: a double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54:700–5. doi: 10.1001/archpsyc.1997.01830200024004. [DOI] [PubMed] [Google Scholar]
- 187.Kranzler HR, Mueller T, Cornelius J, Pettinati HM, Moak D, Martin PR, et al. Sertraline treatment of co-occurring alcohol dependence and major depression. J Clin Psychopharmacol. 2006;26:13–20. doi: 10.1097/01.jcp.0000194620.61868.35. [DOI] [PubMed] [Google Scholar]
- 188.Mason BJ, Kocsis JH, Ritvo EC, Cutler RB. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence or absence of major depression. JAMA. 1996;275:761–7. [PubMed] [Google Scholar]
- 189.McGrath PJ, Nunes EV, Stewart JW, Goldman D, Agosti V, Ocepek-Welikson K, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53:232–40. doi: 10.1001/archpsyc.1996.01830030054009. [DOI] [PubMed] [Google Scholar]
- 190.Collins DM, Myers RD. Buspirone attenuates volitional alcohol intake in the chronically drinking monkey. Alcohol. 1987;4:49–56. doi: 10.1016/0741-8329(87)90060-7. [DOI] [PubMed] [Google Scholar]
- 191.Meert TF. Effects of various serotonergic agents on alcohol intake and alcohol preference in Wistar rats selected at two different levels of alcohol preference. Alcohol Alcohol. 1993;28:157–70. [PubMed] [Google Scholar]
- 192.Blier P, de Montigny C. Modification of 5-HT neuron properties by sustained administration of the 5-HT1A agonist gepirone: electrophysiological studies in the rat brain. Synapse. 1987;1:470–80. doi: 10.1002/syn.890010511. [DOI] [PubMed] [Google Scholar]
- 193.Bruno F. Buspirone in the treatment of alcoholic patients. Psychopathology. 1989;22(suppl 1):49–59. doi: 10.1159/000284626. [DOI] [PubMed] [Google Scholar]
- 194.Malec TS, Malec EA, Dongier M. Efficacy of buspirone in alcohol dependence: a review. Alcohol Clin Exp Res. 1996;20:853–8. doi: 10.1111/j.1530-0277.1996.tb05263.x. [DOI] [PubMed] [Google Scholar]
- 195.Meert TF, Awouters F, Niemegeers CJ, Schellekens KH, Janssen PA. Ritanserin reduces abuse of alcohol, cocaine, and fentanyl in rats. Pharmacopsychiatry. 1991;24:159–63. doi: 10.1055/s-2007-1014461. [DOI] [PubMed] [Google Scholar]
- 196.Myers RD, Lankford M, Bjork A. Selective reduction by the 5-HT antagonist amperozide of alcohol preference induced in rats by systemic cyanamide. Pharmacol Biochem Behav. 1992;43:661–7. doi: 10.1016/0091-3057(92)90392-s. [DOI] [PubMed] [Google Scholar]
- 197.Svensson L, Fahlke C, Hard E, Engel JA. Involvement of the serotonergic system in ethanol intake in the rat. Alcohol. 1993;10:219–24. doi: 10.1016/0741-8329(93)90039-q. [DOI] [PubMed] [Google Scholar]
- 198.Myers RD, Lankford MF. Suppression of alcohol preference in high alcohol drinking rats: efficacy of amperozide versus naltrexone. Neuropsychopharmacology. 1996;14:139–49. doi: 10.1016/0893-133X(95)00081-N. [DOI] [PubMed] [Google Scholar]
- 199.Myers RD, Lankford M. Action of the 5-HT2A antagonist amperozide on alcohol-induced poikilothermia in rats. Pharmacol Biochem Behav. 1998;59:91–5. doi: 10.1016/s0091-3057(97)00330-4. [DOI] [PubMed] [Google Scholar]
- 200.Biggs TA, Myers RD. Naltrexone and amperozide modify chocolate and saccharin drinking in high alcohol-preferring P rats. Pharmacol Biochem Behav. 1998;60:407–13. doi: 10.1016/s0091-3057(97)00598-4. [DOI] [PubMed] [Google Scholar]
- 201.Overstreet DH, McArthur RA, Rezvani AH, Post C. Selective inhibition of alcohol intake in diverse alcohol-preferring rat strains by the 5-HT2A antagonists amperozide and FG 5974. Alcohol Clin Exp Res. 1997;21:1448–54. [PubMed] [Google Scholar]
- 202.Roberts AJ, McArthur RA, Hull EE, Post C, Koob GF. Effects of amperozide, 8-OH-DPAT, and FG 5974 on operant responding for ethanol. Psychopharmacology. 1998;137:25–32. doi: 10.1007/s002130050589. [DOI] [PubMed] [Google Scholar]
- 203.Lankford MF, Bjork AK, Myers RD. Differential efficacy of serotonergic drugs FG5974, FG5893, and amperozide in reducing alcohol drinking in P rats. Alcohol. 1996;13:399–404. doi: 10.1016/0741-8329(96)00061-4. [DOI] [PubMed] [Google Scholar]
- 204.Ugedo L, Grenhoff J, Svensson TH. Ritanserin, a 5-HT2 receptor antagonist, activates midbrain dopamine neurons by blocking serotonergic inhibition. Psychopharmacology. 1989;98:45–50. doi: 10.1007/BF00442004. [DOI] [PubMed] [Google Scholar]
- 205.Johnson BA, Jasinski DR, Galloway GP, Kranzler H, Weinreib R, Anton RF, et al. Ritanserin in the treatment of alcohol dependence—a multi-center clinical trial. Psychopharmacology. 1996;128:206–15. doi: 10.1007/s002130050126. [DOI] [PubMed] [Google Scholar]
- 206.Wiesbeck GA, Weijers HG, Chick J, Naranjo CA, Boening J. Ritanserin in relapse prevention in abstinent alcoholics: results from a placebo-controlled double-blind international multicenter trial. Ritanserin in Alcoholism Work Group. Alcohol Clin Exp Res. 1999;23:230–5. doi: 10.1111/j.1530-0277.1999.tb04105.x. [DOI] [PubMed] [Google Scholar]
- 207.Lovinger DM, White G. Ethanol potentiation of 5-hydroxytryptamine3 receptor-mediated ion current in neuroblastoma cells and isolated adult mammalian neurons. Mol Pharmacol. 1991;40:263–70. [PubMed] [Google Scholar]
- 208.Zhou Q, Lovinger DM. Pharmacologic characteristics of potentiation of 5-HT3 receptors by alcohols and diethyl ether in NCB-20 neuroblastoma cells. J Pharmacol Exp Ther. 1996;278:732–40. [PubMed] [Google Scholar]
- 209.Lovinger DM, Zhou Q. Alcohols potentiate ion current mediated by recombinant 5-HT3RA receptors expressed in a mammalian cell line. Neuropharmacology. 1994;33:1567–72. doi: 10.1016/0028-3908(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 210.Lovinger DM. Inhibition of 5-HT3 receptor-mediated ion current by divalent metal cations in NCB-20 neuroblastoma cells. J Neurophysiol. 1991;66:1329–37. doi: 10.1152/jn.1991.66.4.1329. [DOI] [PubMed] [Google Scholar]
- 211.Lovinger DM. Ethanol potentiates ion current mediated by 5-HT3 receptors on neuroblastoma cells and isolated neurons. Alcohol Alcohol Suppl. 1991;1:181–5. [PubMed] [Google Scholar]
- 212.Lovinger DM. 5-HT3 receptors and the neural actions of alcohols: an increasingly exciting topic. Neurochem Int. 1999;35:125–30. doi: 10.1016/s0197-0186(99)00054-6. [DOI] [PubMed] [Google Scholar]
- 213.Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 214.Johnson BA, Cowen PJ. Alcohol-induced reinforcement: dopamine and 5-HT3 receptor interactions in animals and humans. Drug Dev Res. 1993;30:153–69. [Google Scholar]
- 215.Koob GF. Neural mechanisms of drug reinforcement. Ann N Y Acad Sci. 1992;654:171–91. doi: 10.1111/j.1749-6632.1992.tb25966.x. [DOI] [PubMed] [Google Scholar]
- 216.Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–92. [PubMed] [Google Scholar]
- 217.Bradbury AJ, Costall B, Domeney AM, Naylor RJ. Laterality of dopamine function and neuroleptic action in the amygdala in the rat. Neuropharmacology. 1985;24:1163–70. doi: 10.1016/0028-3908(85)90149-2. [DOI] [PubMed] [Google Scholar]
- 218.Hagan RM, Jones BJ, Jordan CC, Tyers MB. Effect of 5-HT3 receptor antagonists on responses to selective activation of mesolimbic dopaminergic pathways in the rat. Br J Pharmacol. 1990;99:227–32. doi: 10.1111/j.1476-5381.1990.tb14685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Eison AS, Iversen SD, Sandberg BE, Watson SP, Hanley MR, Iversen LL. Substance P analog, DiMe-C7: evidence for stability in rat brain and prolonged central actions. Science. 1982;215:188–90. doi: 10.1126/science.6171884. [DOI] [PubMed] [Google Scholar]
- 220.Costall B, Domeney AM, Naylor RJ, Tyers MB. Effects of the 5-HT3 receptor antagonist, GR38032F, on raised dopaminergic activity in the mesolimbic system of the rat and marmoset brain. Br J Pharmacol. 1987;92:881–94. doi: 10.1111/j.1476-5381.1987.tb11394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hodge CW, Samson HH, Lewis RS, Erickson HL. Specific decreases in ethanol- but not water-reinforced responding produced by the 5-HT3 antagonist ICS 205-930. Alcohol. 1993;10:191–6. doi: 10.1016/0741-8329(93)90034-l. [DOI] [PubMed] [Google Scholar]
- 222.Fadda F, Garau B, Marchei F, Colombo G, Gessa GL. MDL 72222, a selective 5-HT3 receptor antagonist, suppresses voluntary ethanol consumption in alcohol-preferring rats. Alcohol Alcohol. 1991;26:107–10. doi: 10.1093/oxfordjournals.alcalc.a045088. [DOI] [PubMed] [Google Scholar]
- 223.Rodd-Henricks ZA, McKinzie DL, Li T-K, Crile RS, Murphy JM, McBride WJ. Intracranial self-administration of ethanol into the posterior VTA of Wistar rats is mediated by 5-HT3 receptors. Alcohol Clin Exp Res. 1999;23(suppl 5):49A. abstract. [Google Scholar]
- 224.Johnson BA, Campling GM, Griffiths P, Cowen PJ. Attenuation of some alcohol-induced mood changes and the desire to drink by 5-HT3 receptor blockade: a preliminary study in healthy male volunteers. Psychopharmacology. 1993;112:142–4. doi: 10.1007/BF02247375. [DOI] [PubMed] [Google Scholar]
- 225.Dyr W, Kostowski W. Evidence that the amygdala is involved in the inhibitory effects of 5-HT3 receptor antagonists on alcohol drinking in rats. Alcohol. 1995;12:387–91. doi: 10.1016/0741-8329(95)00023-k. [DOI] [PubMed] [Google Scholar]
- 226.Sellers EM, Higgins GA, Tompkins DM, Romach MK. Serotonin and alcohol drinking. NIDA Res Monogr. 1992;119:141–5. [PubMed] [Google Scholar]
- 227.McBride WJ, Li TK. Animal models of alcoholism: neurobiology of high alcohol-drinking behavior in rodents. Crit Rev Neurobiol. 1998;12:339–69. doi: 10.1615/critrevneurobiol.v12.i4.40. [DOI] [PubMed] [Google Scholar]
- 228.Tomkins DM, Le AD, Sellers EM. Effect of the 5-HT3 antagonist ondansetron on voluntary ethanol intake in rats and mice maintained on a limited access procedure. Psychopharmacology. 1995;117:479–85. doi: 10.1007/BF02246222. [DOI] [PubMed] [Google Scholar]
- 229.Beardsley PM, Lopez OT, Gullikson G, Flynn D. Serotonin 5-HT3 antagonists fail to affect ethanol self-administration of rats. Alcohol. 1994;11:389–95. doi: 10.1016/0741-8329(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 230.Swift RM, Davidson D, Whelihan W, Kuznetsov O. Ondansetron alters human alcohol intoxication. Biol Psychiatry. 1996;40:514–21. doi: 10.1016/0006-3223(95)00432-7. [DOI] [PubMed] [Google Scholar]
- 231.Doty P, Zacny JP, de Wit H. Effects of ondansetron pretreatment on acute responses to ethanol in social drinkers. Behav Pharmacol. 1994;5:461–9. doi: 10.1097/00008877-199408000-00007. [DOI] [PubMed] [Google Scholar]
- 232.Sellers EM, Toneatto T, Romach MK, Somer GR, Sobell LC, Sobell MB. Clinical efficacy of the 5-HT3 antagonist ondansetron in alcohol abuse and dependence. Alcohol Clin Exp Res. 1994;18:879–85. doi: 10.1111/j.1530-0277.1994.tb00054.x. [DOI] [PubMed] [Google Scholar]
- 233.Johnson BA, Roache JD, Javors MA, DiClemente CC, Cloninger CR, Prihoda TJ, et al. Ondansetron for reduction of drinking among biologically predisposed alcoholic patients: a randomized controlled trial. JAMA. 2000;284:963–71. doi: 10.1001/jama.284.8.963. [DOI] [PubMed] [Google Scholar]
- 234.Kranzler HR, Pierucci-Lagha A, Feinn R, Hernandez-Avila C. Effects of ondansetron in early- versus late-onset alcoholics: a prospective, open-label study. Alcohol Clin Exp Res. 2003;27:1150–5. doi: 10.1097/01.ALC.0000075547.77464.76. [DOI] [PubMed] [Google Scholar]
- 235.Russell RN, McBride WJ, Lumeng L, Li TK, Murphy JM. Apomorphine and 7-OH DPAT reduce ethanol intake of P and HAD rats. Alcohol. 1996;13:515–9. doi: 10.1016/0741-8329(95)00062-3. [DOI] [PubMed] [Google Scholar]
- 236.Mason GA, Rezvani AH, Grady DR, Garbutt JC. The subchronic effects of the TRH analog TA-0910 and bromocriptine on alcohol preference in alcohol-preferring rats: development of tolerance and cross-tolerance. Alcohol Clin Exp Res. 1994;18:1196–201. doi: 10.1111/j.1530-0277.1994.tb00104.x. [DOI] [PubMed] [Google Scholar]
- 237.Weiss F, Mitchiner M, Bloom FE, Koob GF. Free-choice responding for ethanol versus water in alcohol preferring (P) and unselected Wistar rats is differentially modified by naloxone, bromocriptine, and methysergide. Psychopharmacology. 1990;101:178–86. doi: 10.1007/BF02244123. [DOI] [PubMed] [Google Scholar]
- 238.Dongier M, Vachon L, Schwartz G. Bromocriptine in the treatment of alcohol dependence. Alcohol Clin Exp Res. 1991;15:970–7. doi: 10.1111/j.1530-0277.1991.tb05197.x. [DOI] [PubMed] [Google Scholar]
- 239.Naranjo CA, George SR, Bremner KE. Novel neuropharmacological treatments of alcohol dependence. Clin Neuropharmacol. 1992;15(suppl 1 Pt A):74A–5A. doi: 10.1097/00002826-199201001-00040. [DOI] [PubMed] [Google Scholar]
- 240.Powell BJ, Campbell JL, Landon JF, Liskow BI, Thomas HM, Nickel EJ, et al. A double-blind, placebo-controlled study of nortriptyline and bromocriptine in male alcoholics subtyped by comorbid psychiatric disorders. Alcohol Clin Exp Res. 1995;19:462–8. doi: 10.1111/j.1530-0277.1995.tb01532.x. [DOI] [PubMed] [Google Scholar]
- 241.Colombo G, Agabio R, Carai MA, Lobina C, Pani M, Reali R, et al. Ability of baclofen in reducing alcohol intake and withdrawal severity: I—preclinical evidence. Alcohol Clin Exp Res. 2000;24:58–66. [PubMed] [Google Scholar]
- 242.Colombo G, Serra S, Brunetti G, Vacca G, Carai MA, Gessa GL. Suppression by baclofen of alcohol deprivation effect in Sardinian alcohol-preferring (sP) rats. Drug Alcohol Depend. 2003;70:105–8. doi: 10.1016/s0376-8716(02)00333-2. [DOI] [PubMed] [Google Scholar]
- 243.Colombo G, Serra S, Vacca G, Gessa GL, Carai MA. Suppression by baclofen of the stimulation of alcohol intake induced by morphine and WIN 55,212-2 in alcohol-preferring rats. Eur J Pharmacol. 2004;492:189–93. doi: 10.1016/j.ejphar.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 244.Addolorato G, Caputo F, Capristo E, Colombo G, Gessa GL, Gasbarrini G. Ability of baclofen in reducing alcohol craving and intake: II—Preliminary clinical evidence. Alcohol Clin Exp Res. 2000;24:67–71. [PubMed] [Google Scholar]
- 245.Addolorato G, Caputo F, Capristo E, Domenicali M, Bernardi M, Janiri L, et al. Baclofen efficacy in reducing alcohol craving and intake: a preliminary double-blind randomized controlled study. Alcohol Alcohol. 2002;37:504–8. doi: 10.1093/alcalc/37.5.504. [DOI] [PubMed] [Google Scholar]
- 246.Ait-Daoud N, Johnson BA. Medications for the treatment of alcoholism. In: Johnson BA, Ruiz P, Galanter M, editors. Handbook of clinical alcoholism treatment. Baltimore, MD: Lippincott Williams & Wilkins; 2003. pp. 119–30. [Google Scholar]
- 247.Sidmak Laboratories, Inc. Antabuse [package insert] East Hanover, Nj: Sidmak Laboratories, Inc; 2001. [Google Scholar]
- 248.O’Shea B. Disulfiram revisited. Hosp Med. 2000;61:849–51. doi: 10.12968/hosp.2000.61.12.1483. [DOI] [PubMed] [Google Scholar]
- 249.Petrakis IL, Carroll KM, Nich C, Gordon LT, McCance-Katz EF, Frankforter T, et al. Disulfiram treatment for cocaine dependence in methadone-maintained opioid addicts. Addiction. 2000;95:219–28. doi: 10.1046/j.1360-0443.2000.9522198.x. [DOI] [PubMed] [Google Scholar]
- 250.Carroll KM, Fenton LR, Ball SA, Nich C, Frankforter TL, Shi J, et al. Efficacy of disulfiram and cognitive behavior therapy in cocaine-dependent outpatients: a randomized placebo-controlled trial. Arch Gen Psychiatry. 2004;61:264–72. doi: 10.1001/archpsyc.61.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Baker JR, Jatlow P, McCance-Katz EF. Disulfiram effects on responses to intravenous cocaine administration. Drug Alcohol Depend. 2007;87:202–9. doi: 10.1016/j.drugalcdep.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fuller RK, Branchey L, Brightwell DR, Derman RM, Emrick CD, Iber FL, et al. Disulfiram treatment of alcoholism. A Veterans Administration cooperative study. JAMA. 1986;256:1449–55. [PubMed] [Google Scholar]
- 253.Anton RF. Pharmacologic approaches to the management of alcoholism. J Clin Psychiatry. 2001;62(suppl 20):11–7. [PubMed] [Google Scholar]
- 254.Heilig M, Egli M. Pharmacological treatment of alcohol dependence: target symptoms and target mechanisms. Pharmacol Ther. 2006;111:855–76. doi: 10.1016/j.pharmthera.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 255.Wang L, Liu J, Harvey-White J, Zimmer A, Kunos G. Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci U S A. 2003;100:1393–8. doi: 10.1073/pnas.0336351100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Caille S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci. 2007;27:3695–702. doi: 10.1523/JNEUROSCI.4403-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Basavarajappa BS, Hungund BL. Chronic ethanol increases the cannabinoid receptor agonist anandamide and its precursor N-arachidonoylphosphatidylethanolamine in SK-N-SH cells. J Neurochem. 1999;72:522–8. doi: 10.1046/j.1471-4159.1999.0720522.x. [DOI] [PubMed] [Google Scholar]
- 258.Lallemand F, De Witte P. SR147778, a CB1 cannabinoid receptor antagonist, suppresses ethanol preference in chronically alcoholized Wistar rats. Alcohol. 2006;39:125–34. doi: 10.1016/j.alcohol.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 259.Rodriguez de Fonseca F, Roberts AJ, Bilbao A, Koob GF, Navarro M. Cannabinoid receptor antagonist SR141716A decreases operant ethanol self administration in rats exposed to ethanol-vapor chambers. Zhongguo Yao Li Xue Bao. 1999;20:1109–14. [PubMed] [Google Scholar]
- 260.Gessa GL, Serra S, Vacca G, Carai MA, Colombo G. Suppressing effect of the cannabinoid CB1 receptor antagonist, SR147778, on alcohol intake and motivational properties of alcohol in alcohol-preferring sP rats. Alcohol Alcohol. 2005;40:46–53. doi: 10.1093/alcalc/agh114. [DOI] [PubMed] [Google Scholar]
- 261.Colombo G, Vacca G, Serra S, Carai MA, Gessa GL. Suppressing effect of the cannabinoid CB1 receptor antagonist, SR 141716, on alcohol’s motivational properties in alcohol-preferring rats. Eur J Pharmacol. 2004;498:119–23. doi: 10.1016/j.ejphar.2004.07.069. [DOI] [PubMed] [Google Scholar]
- 262.Economidou D, Mattioli L, Cifani C, Perfumi M, Massi M, Cuomo V, et al. Effect of the cannabinoid CB1 receptor antagonist SR-141716A on ethanol self-administration and ethanol-seeking behaviour in rats. Psychopharmacology. 2006;183:394–403. doi: 10.1007/s00213-005-0199-9. [DOI] [PubMed] [Google Scholar]
- 263.Cippitelli A, Bilbao A, Hansson AC, del Arco I, Sommer W, Heilig M, et al. Cannabinoid CB1 receptor antagonism reduces conditioned reinstatement of ethanol-seeking behavior in rats. Eur J Neurosci. 2005;21:2243–51. doi: 10.1111/j.1460-9568.2005.04056.x. [DOI] [PubMed] [Google Scholar]
- 264.Steffens M, Feuerstein TJ. Receptor-independent depression of DA and 5-HT uptake by cannabinoids in rat neocortex—involvement of Na(+)/K(+)-ATPase. Neurochem Int. 2004;44:529–38. doi: 10.1016/j.neuint.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 265.Moranta D, Esteban S, Garcia-Sevilla JA. Differential effects of acute cannabinoid drug treatment, mediated by CB1 receptors, on the in vivo activity of tyrosine and tryptophan hydroxylase in the rat brain. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:516–24. doi: 10.1007/s00210-004-0921-x. [DOI] [PubMed] [Google Scholar]
- 266.Tzavara ET, Perry KW, Rodriguez DE, Bymaster FP, Nomikos GG. The cannabinoid CB(1) receptor antagonist SR141716A increases norepinephrine outflow in the rat anterior hypothalamus. Eur J Pharmacol. 2001;426:R3–4. doi: 10.1016/s0014-2999(01)01228-6. [DOI] [PubMed] [Google Scholar]
- 267.Johnson BA. New weapon to curb smoking: no more excuses to delay treatment. Arch Intern Med. 2006;166:1547–50. doi: 10.1001/archinte.166.15.1547. [DOI] [PubMed] [Google Scholar]
- 268.Hemby SE, Co C, Koves TR, Smith JE, Dworkin SI. Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology. 1997;133:7–16. doi: 10.1007/s002130050365. [DOI] [PubMed] [Google Scholar]
- 269.Dahchour A, De Witte P. Effects of acamprosate on excitatory amino acids during multiple ethanol withdrawal periods. Alcohol Clin Exp Res. 2003;27:465–70. doi: 10.1097/01.ALC.0000056617.68874.18. [DOI] [PubMed] [Google Scholar]
- 270.Kiefer F, Jahn H, Tarnaske T, Helwig H, Briken P, Holzbach R, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry. 2003;60:92–9. doi: 10.1001/archpsyc.60.1.92. [DOI] [PubMed] [Google Scholar]
- 271.Johnson BA, Ait-Daoud N, Prihoda TJ. Combining ondansetron and naltrexone effectively treats biologically predisposed alcoholics: from hypotheses to preliminary clinical evidence. Alcohol Clin Exp Res. 2000;24:737–42. [PubMed] [Google Scholar]
- 272.Ait-Daoud N, Johnson BA, Javors M, Roache JD, Zanca NA. Combining ondansetron and naltrexone treats biological alcoholics: corroboration of self-reported drinking by serum carbohydrate deficient transferrin, a biomarker. Alcohol Clin Exp Res. 2001;25:847–9. [PubMed] [Google Scholar]