

Abstract
Type III Neuregulin1 (Nrg1) isoforms are membrane-tethered proteins capable of participating in bidirectional juxtacrine signaling. Neuronal nicotinic acetylcholine receptors (nAChRs), which can modulate the release of a rich array of neurotransmitters, are differentially targeted to presynaptic sites. We demonstrate that Type III Nrg1 back signaling regulates the surface expression of α7 nAChRs along axons of sensory neurons. Stimulation of Type III Nrg1 back signaling induces an increase in axonal surface α7 nAChRs, which results from a redistribution of preexisting intracellular pools of α7 rather than from increased protein synthesis. We also demonstrate that Type III Nrg1 back signaling activates a phosphatidylinositol 3-kinase signaling pathway and that activation of this pathway is required for the insertion of preexisting α7 nAChRs into the axonal plasma membrane. These findings, in conjunction with prior results establishing that Type III Nrg1 back signaling controls gene transcription, demonstrate that Type III Nrg1 back signaling can regulate both short-and long-term changes in neuronal function.
Introduction
The Neuregulin1 (Nrg1) gene encodes an extremely important and diverse family of proteins that signal by binding to the ErbB family of receptor tyrosine kinases. Nrg1-ErbB signaling regulates neural development, glial growth, myelination, and the maintenance of synaptic connections in both the peripheral and central nervous system (Michailov et al., 2004; Taveggia et al., 2005; Chen et al., 2006; Lopez-Bendito et al., 2006). Nrg1 activation of ErbB signaling regulates the levels of several ion channels, including subtypes of neuronal nicotinic acetylcholine receptors (nAChRs; Yang et al., 1998; Liu et al., 2001; Kawai et al., 2002; Chang and Fischbach, 2006), N-methyl-d-aspartic acid receptors (Ozaki et al., 1997; Gu et al., 2005), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (Kwon et al., 2005; Li et al., 2007), and γ-amino butyric acidA receptors (Rieff et al., 1999; Okada and Corfas, 2004). Nrg1 has been implicated as a schizophrenia susceptibility gene, and the potential relationship between Nrg1 and circuits affected in schizophrenia underscores the importance of determining the molecular mechanisms of action of Nrg1 signaling in the developing and adult nervous systems (Stefansson et al., 2002).
The Nrg1 gene encodes over a dozen different protein products. Multiple promoters and alternative splicing leads to the synthesis of three (and perhaps as many as six) classes of proteins differing in their amino-terminal domains, as well as in the EGF-like domain (α or β), and/or in the length of the C termini (Falls, 2003). These differences in primary sequence translate into subtle and dramatic differences in signaling strategies. Although Types I and II Nrg1 are either directly secreted or released after constitutive cleavage, Type III Nrg1 remains membrane tethered as a result of its unique N-terminal transmembrane domain (Wang et al., 2001; Taveggia et al., 2005). Thus, Types I and II Nrg1 can participate in paracrine signaling, whereas Type III Nrg1 predominantly signals in a juxtacrine manner. Additionally, it has been shown that Type III Nrg1 also functions as a receptor, in that Type III Nrg1–ErbB interaction results in signaling within the Type III Nrg1–expressing cell (Bao et al., 2003, 2004).
The integrity of a neuron–neuron synapse is determined by several molecular interactions between pre- and postsynaptic cells. Throughout the central and peripheral nervous system, nAChRs function as cation-selective ligand-gated ion channels and play important roles in synaptic signaling (Berg and Conroy, 2002). There is increasing evidence for the importance of nAChRs in the modulation of transmitter release from presynaptic terminals (McGehee et al., 1995; MacDermott et al., 1999), neuronal survival (Dajas-Bailador et al., 2000; Mechawar et al., 2004), and postsynaptic signaling (Zhang et al., 1996; Zhang and Berg, 2007). Individual nAChR channels have unique biological functions that are determined by the pentameric combination of 11 distinct subunits: α2–α7, α9, α10, and β2–β4 (Rosenberg et al., 2002). Of particular interest are the α7* nAChRs (α7-containing nAChRs) because of their high permeability to calcium and abundant expression in both mammalian and avian peripheral nervous systems (Roth and Berg, 2003; Fucile et al., 2005).
The Type III isoform of Nrg1 was identified as the nerve-derived factor responsible for increasing acetylcholine-evoked currents in neurons and was shown to regulate expression of neuronal nAChR subunit mRNAs, including that of α7 (Yang et al., 1998). More recent findings in our laboratory reveal a requirement for presynaptic Type III Nrg1 in potentiation of glutamatergic transmission by nicotine in a central sensory motor–gating circuit (Du, C., C. Zhong, M. Hancock, D.A. Talmage, and L.W. Role. 2004. Society for Neuroscience 34th Annual Meeting; unpublished data). These results inspired us to investigate the molecular mechanisms underlying presynaptic Type III Nrg1's cell-autonomous effect on functional α7* nAChRs along axonal projections.
In this paper, we show that presynaptic Type III Nrg1 is required for normal levels of presynaptically targeted α7* nAChRs in primary sensory neurons. Sensory neurons from Type III Nrg1 mutant mice exhibit deficits of the surface expression of α7* nAChRs. Acute stimulation of Type III Nrg1 back signaling increases the number of α7* nAChRs on sensory axons from wild-type (WT), but not from homozygous Type III Nrg1, mutant embryos. These increases in axonal surface α7* nAChRs result from redistribution of preexisting intracellular pools of α7 rather than from increased α7 protein synthesis. Increases in surface pools of α7* nAChR are accomplished, at least in part, by local activation of a phosphatidylinositol 3-kinase (PtdIns 3K) signaling pathway, which in turn stimulates the membrane insertion of intracellular α7* nAChRs.
Results
Reduction of Type III Nrg1 expression results in decreased axonal α7 nAChR surface expression
Subsets of primary sensory neurons within the dorsal root ganglia (DRG) express Type III Nrg1 and α7 nAChR subunit mRNAs (Boyd et al., 1991; Wolpowitz et al., 2000; Genzen et al., 2001; Taveggia et al., 2005). In the following experiments, we examined the expression of α7* nAChRs and Type III Nrg1 on the surface of axons from sensory neurons of WT or Type III Nrg1−/− mouse embryos. Embryonic neurons were used for these experiments because mutant mice lacking Type III Nrg1 die at birth (Wolpowitz et al., 2000).
To label surface α7* nAChRs, primary sensory neurons were labeled with αBgTx-488 (Alexa 488–conjugated α-bungarotoxin) before fixation and permeabilization (Fig. 1 A). α-BgTx is an effective reagent for probing α7* nAChRs because of its high affinity for α7* nAChRs (Ravdin and Berg 1979). After fixation and permeabilization, we applied antibodies recognizing the cysteine-rich domain of Type III Nrg1 (red) or neurofilament (NF) proteins (blue; Fig. 1 A; Yang et al., 1998). In neurons from WT mice, axons were studded with distinct puncta of αBgTx-488 and Type III Nrg1 along the same sensory axons. Pretreatment with 5 μM methyllycaconitine or 1 μM of nicotine before addition of αBgTx-488 blocked surface labeling of α7* nAChRs, confirming the specificity of surface αBgTx-488 labeling (Fig. 1 D). We repeated this analysis using sensory neurons isolated from Type III Nrg1−/− embryos. Labeling with αBgTx-488 identified discrete clusters of α7* nAChRs on the axonal surface of mutant neurons (Fig. 1 A). However, when we quantified the number of these αBgTx-488 clusters we found that the mutants had ∼50% fewer clusters than WT neurons (WT, 6.47 ± 0.69 clusters/100 μm; Type III Nrg1−/−, 3.22 ± 0.59 clusters/100 μm; Fig. 1 B). Additionally, we found that the mutants had a 35% reduction in mean cluster area relative to WT neurons (WT, 0.26 ± 0.02 μm2; Type III Nrg1−/−, 0.17 ± 0.02 μm2; Fig. 1 C).
Figure 1.

Decreased surface expression of α7* nAChRs along axons of Type III Nrg1−/− sensory neurons. Sensory ganglia were extirpated from embryonic day (E) 14.5 WT or Type III Nrg1−/− embryos and plated as explants. After 2 d in vitro, neurons were labeled for surface α7* nAChRs with αBgTx-488 (green). Neurons were fixed, permeabilized, and stained for Type III Nrg1 (red) and NF protein (blue). (A) Representative micrographs of αBgTx-488 clusters along NF-positive axons of WT and Type III Nrg1−/− sensory neurons at 2 d in vitro. Type III Nrg1 staining was detected along WT (a) but not Type III Nrg1−/− (d) axons. Fewer surface αBgTx-488 clusters were detected along mutant axons (e) as compared with WT (b). Confocal images were acquired from a 100× oil objective. Bar, 5 μm. (B) Quantification of surface αBgTx-488 clusters per 100 μm of axonal length revealed an ∼50% reduction of clusters along mutant axons. The graph shows means ± SEM. Data were pooled from three independent experiments. Statistical significance determined by ANOVA. *, P < 0.001 (Statview). (C) Quantification of the surface αBgTx-488 cluster area. Loss of Type III Nrg1 expression resulted in an ∼35% reduction of αBgTx-488 cluster area. Data pooled from three independent experiments were analyzed using nonparametric statistics and presented as box plots (see Materials and methods). Statistical significance was determined by the Kolmogorov-Smirnov Test. *, P < 0.0001 (Statview). (D) Pretreatment of sensory neurons with 1 μM of nicotine (Nic; b and e) or 5 μM methyllycaconitine (MLA; c and f) prevents surface αBgTx-488 labeling along axons. Bar, 10 μm. (E) Immunoblot analysis of total α7 subunit protein levels in sensory neurons from E14.5 WT, Type III Nrg1+/−, and Type III Nrg1−/− embryos cultured for 2 d in vitro (a). MAPK1/2 probing in the bottom panel shows equal lysate loading. Immunoblots of total α7 subunit protein levels in brain extracts from WT or α7 nAChR−/− embryos are also shown (b). Glyceraldehyde 3-phosphate dehydrogenase probing in the bottom panel shows equal lysate loading (b). (F) In WT sensory neurons, nicotine application (1 μM for 1 min) resulted in an increased internal concentration of Ca2+ (indicated in pseudo color; described in Materials and methods). The white arrowhead highlights an axonal region affected by nicotine. Bar, 10 μm. (G) Changes in internal concentration of Ca2+ in response to application of nicotine in WT and mutant axons are plotted. Note that 100 nM αBgTx completely eliminated the response to nicotine in WT axons and that sensory axons from Type III Nrg1−/− animals did not respond to nicotine. Data are from two independent experiments. The graph shows means ± SEM. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P < 0.0001 (Statview).
Although there is a difference in α7* nAChR clusters expressed along axons of Type III Nrg1−/− sensory neurons, we did not detect an overall reduction of total α7 protein in mutant neurons (Fig. 1 E). This result, coupled with our findings in Fig. 1 (A–C), indicates that Type III Nrg1 specifically affects the surface expression of α7* nAChRs. The decrease in surface α7* nAChRs along axons of Type III Nrg1−/− neurons did not result from general deficits in endocytic recycling because these neurons retained WT rates of transferrin receptor recycling (Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200710037/DC1). We next looked to see whether Type III Nrg1−/− neurons have defects in axonal targeting of presynaptic proteins. We did not detect differences in the axonal targeting of synaptophysin or vesicular glutamate transporter 1 (vGlut1) along mutant axons (Fig. S1, C and D).
To demonstrate that the αBgTx-488 surface binding measurements reflected the presence of functional α7* nAChRs, we quantified internal concentrations of Ca2+ along sensory axons after a 1-min application of 1 μM of nicotine. In response to nicotine, we detected an increase in internal concentrations of Ca2+ within 1 min along axons of WT neurons (Fig. 1, F and G). When this experiment was repeated in the presence of 1 μM αBgTx, we did not detect an increase in intracellular concentrations of Ca2+, indicating that the nicotine-induced changes were mediated by functional α7* nAChRs. Nicotine application did not increase internal Ca2+ concentrations in Type III Nrg1−/− axons (Fig. 1 G), a result which is consistent with a significant decrease in surface α7* nAChRs in these neurons.
Type III Nrg1 back signaling increases surface expression of α7* nAChR clusters
Type III Nrg1 functions as a membrane-tethered bidirectional signaling molecule (Bao et al., 2003, 2004). To test the possibility that Type III Nrg1, acting as a receptor, contributes to the regulation of α7* nAChR levels on sensory axons, we treated WT or Type III Nrg1−/− sensory neurons with 2 nM of either the extracellular domain (ECD) of ErbB2 (B2-ECD; control) or ErbB4 (B4-ECD) for 24 h (Fig. 2, A and B). B4-ECD, but not B2-ECD, binds with high affinity to the EGF-like domain of Nrg1 (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200710037/DC1; Fitzpatrick et al., 1998; Bao et al., 2003). We visualized α7* nAChRs present on the surface of sensory axons by labeling with αBgTx-488 (green) before fixation (Fig. 2 A). Treating WT sensory neurons with B4-ECD for 24 h led to an increase in the number of αBgTx-488 clusters on the axonal surface (from 6.47 ± 0.69 clusters/100 μm in control cultures to 10.38 ± 1.59 clusters/100 μm; Fig. 2 B). When we repeated the experiment using sensory neurons from Type III Nrg1−/− embryos, there was no significant change in the number of αBgTx-488 clusters on mutant axons (from 3.07 ± 0.72 clusters/100 μm in control cultures to 2.74 ± 1.14 clusters/100 μm in B4-ECD treated). This effect did not result from general changes in the levels of surface proteins, as we did not detect a change in the axonal surface levels of TrkA in response to a 24-h B4-ECD treatment (Fig. S1, E and F).
Figure 2.

Type III Nrg1 back-signaling increases the surface expression of α7* nAChRs. E14.5 WT or Type III Nrg1−/− DRG explants were treated with B2-ECD (control) or B4-ECD for 24 h. Neurons were labeled for surface α7* nAChRs with αBgTx-488 (green), fixed, permeabilized, and labeled for NF protein (blue). (A) Representative micrographs of axons from WT (a and b) or Type III Nrg1−/− (c and d) sensory neurons under control (a and c) versus B4-ECD (b and d) conditions. B4-ECD treatment increased the number of surface αBgTx-488 clusters along NF-positive processes of WT neurons (b). Linescans of fluorescence intensity profile for αBgTx-488 staining along representative axons (see Materials and methods). Bar, 5 μm. (B) Quantification of surface αBgTx-488 clusters along NF-positive axons from WT versus Type III Nrg1−/− DRG explants treated with either B2-ECD (control) or B4-ECD for 24 h. In WT cultures, B4-ECD treatment induced an ∼1.6-fold increase in surface αBgTx clusters along NF-positive axons compared with the control. There was no detectable change in αBgTx clusters along axons of Type III Nrg1−/− neurons. The graph shows means ± SEM. Data were pooled from three independent experiments. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P < 0.03; **, P < 0.001 (Statview). (C) After 2 d in vitro, dissociated sensory neurons from E11 chick embryos were treated with B2-ECD (control) or B4-ECD for 1, 2, 6, or 12 h and labeled as described in A. Axonal surface αBgTx-488 clusters were quantified. Data were pooled from three independent experiments. The graph shows means ± SEM. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Statview). (D) Axonal-bound B4-ECD and αBgTx-488 were detected in puncta along axons treated with B4-ECD (b, d, and e). Sensory neurons from E14.5 WT mouse embryos were cultured for 2 d in vitro and treated with B2-ECD (control) or B4-ECD for 1 h. Before fixation, surface α7* nAChRs and axonal-bound B2-ECD (control) or B4-ECD were labeled with αBgTx-488 (green) and an antibody against the human Fc domain (anti-Fc; red), respectively. c and d and e are magnifications of the areas shown in dotted squares in a and b, respectively. Bar: (a and b) 5 μm; (c–e) 1 μm. (E) Sensory neurons from E11 chick embryos were treated with B2-ECD (control), B4-ECD, or soluble Nrg1β peptide (Nrg1-ECD) for 1 h. In parallel, neurons pretreated with an ErbB tyrosine kinase inhibitor (ErbB inh.) for 45 min were treated with B2-ECD, B4-ECD, or Nrg1-ECD for 1 h. Neurons were labeled as described in A, and surface αBgTx-488 clusters along axons were quantified. Data were pooled from three independent experiments. The graph shows means ± SEM. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P < 0.005; **, P < 0.01 (Statview).
We next asked how soon after B4-ECD treatment the increase in surface α7* nAChRs was seen. In sensory neurons treated with B4-ECD, we detected an increase in the surface expression of α7* nAChR clusters along axons within 1 h, with the maximal response occurring after 6 h (Fig. 2 C) and sustained for 24 h (Figs. 1 B, 2 B, and 3 A). Many of the surface α7* nAChR clusters induced by B4-ECD treatment were localized in close proximity to membrane-bound B4-ECD (Fig. 2 D).
Figure 3.

Type III Nrg1 back signaling increases the surface expression of α7* nAChRs in the absence of protein synthesis. Dissociated sensory neurons from E11 chick embryos were cultured for 2 d in vitro and treated with either B2-ECD (control) or B4-ECD for 1 or 24 h. (A) Quantification of surface or total pools of α7* nAChR by 125I-αBgTx radiolabeling in sensory neurons treated with either B2-ECD (control) or B4-ECD for 24 h. In response to a 24-h B4-ECD treatment, we detected an ∼2.7-fold increase in surface 125I-αBgTx binding compared with control conditions (B2-ECD [control], 1,339.15 ± 329.77 cpm; and B4-ECD, 3,562.81 ± 1,111.19 cpm). B4-ECD treatment did not induce a change in total 125I-αBgTx binding as compared with the control (B2-ECD [control], 11,159.74 ± 1,059.79 cpm; and B4-ECD, 12,258.85 ± 580.11 cpm). The graph shows means ± SEM. Data were pooled from three independent experiments with greater than or equal to three wells per condition per experiment. Statistical significance was determined by ANOVA. *, P < 0.05 (Statview). (B) Immunoblot analysis of total α7 subunit protein in sensory neurons treated with B2-ECD (control) or B4-ECD treatment for 24 h. In response to B4-ECD treatment, we did not detect a difference in total α7 subunit protein. NFM probing in bottom panel shows equivalent lysate loading. (C) Sensory neurons were treated with B2-ECD (control) or B4-ECD for 1 h. In parallel, neurons pretreated with CHX for 45 min were treated with B2-ECD or B4-ECD for 1 h. Neurons were labeled with αBgTx-488 (green), fixed, permeabilized, and colabeled for NF protein (blue). CHX treatment (c and d) did not affect either the basal number of αBgTx-488 clusters on control neurons (c) or the response to B4-ECD (d). Linescans of fluorescence intensity profiles of αBgTx-488 along representative axons (see Materials and methods) are shown. Bar, 5 μm. (D) Quantification of surface αBgTx-488 clusters along NF-labeled axons. B4-ECD treatment induced an ∼1.9-fold increase in surface αBgTx-488 clusters along axons, and B4-ECD treatment in the presence of CHX induced an ∼2.1-fold increase. Data were pooled from three independent experiments. The graph shows means ± SEM. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P = 0.01; **, P < 0.0001 (Statview). (E) Quantification of surface αBgTx-488 cluster area. B4-ECD treatment in the presence or absence of CHX induced an increase in αBgTx-488 cluster area. Data pooled from three independent experiments were analyzed using nonparametric statistics and presented as box plots (see Materials and methods). Statistical significance was determined by the Kolmogorov-Smirnov Test. *, P ≤ 0.0001 (Statview).
Prior studies have shown that activation of ErbB tyrosine kinase signaling can alter the surface expression of α7* nAChRs in neurons (Liu et al., 2001; Kawai et al., 2002; Chang and Fischbach, 2006). To verify that the increase in surface clusters we detected in response to B4-ECD treatment was mediated by Type III Nrg1 acting as a receptor, we assessed the response of sensory neurons treated with B4-ECD in the presence of 2 μM ErbB inh. (inhibitors of ErbB tyrosine kinase activity; PD 168393 and PD 158780) for 1 h (Fig. 2 E). We also assessed the effect on α7* nAChRs of acute activation of ErbB signaling by treating neurons with recombinant Nrg1β peptide (Nrg1-ECD, 10 ng/ml). In response to B4-ECD treatment, we detected an increase in surface α7* nAChR clusters along axons in the presence of ErbB inh. (ErbB inh., 12.31 ± 2.43 clusters/100-μm axonal length; and ErbB inh. + B4-ECD, 20.94 ± 2.37 clusters/100-μm axonal length; Fig. 2 E). Conversely, we did not detect an increase in axonal surface α7* nAChR clusters in response to Nrg1-ECD treatment (B2-ECD [control], 11.30 ± 1.74 clusters/100-μm axonal length; and Nrg1-ECD, 13.20 ± 2.72 clusters/100-μm axonal length; Fig. 2 E). Collectively, these results indicate that in sensory neurons, Type III Nrg1, acting as a receptor for B4-ECD, regulates the surface expression of α7* nAChR, whereas activation of ErbB tyrosine kinases does not.
Stimulation of Type III Nrg1 back signaling increases surface expression of α7* nAChRs in the absence of new protein synthesis
Stimulation of Type III Nrg1 back signaling elicits a transcriptional response mediated by the Nrg1 intracellular domain (Bao et al., 2003, 2004). To determine if Type III Nrg1–dependent regulation of axonal α7* nAChR levels resulted from Nrg1 intracellular domain–induced synthesis of α7 and/or changes in α7* nAChR surface expression along axons, we compared measures of total (surface and internal) neuronal 125I-αBgTx binding (Fig. 3 A) and total immunoreactive α7 nAChR protein (Fig. 3 B) in sensory neurons treated with B2-ECD (control) or B4-ECD for 24 h. We also asked whether B4-ECD treatment increased surface α7* nAChR levels in the presence of a protein synthesis inhibitor (Fig. 3, C–E).
Surface α7* nAChRs were quantified by 125I-αBgTx labeling in live in vitro preparations of chick sensory neurons and, in parallel, the total pool of α7* nAChRs was quantified by 125I-αBgTx binding after permeabilization with 0.5% saponin (Fig. 3 A). Under basal conditions, ∼12% of the total α7* nAChRs were on the surface, which is similar to prior results emphasizing the large internal pools of nAChRs in neurons (Cho et al., 2005). After a 24-h treatment with B4-ECD, we observed an ∼2.7-fold increase in surface 125I-αBgTx binding. In B4-ECD–stimulated neurons, ∼30% of the α7* nAChRs were at the surface, but the total pool of 125I-αBgTx–labeled sites was not changed. These results are consistent with Type III Nrg1 back signaling inducing a relocalization of α7* nAChRs from preexisting internal pools to clusters along the axonal surface. Results on increased surface versus total α7* nAChRs were confirmed by immunoblot assay (Fig. 3 B). These results indicate that in sensory neurons, Type III Nrg1, acting as a receptor, can increase axonal surface α7* nAChRs without significantly increasing the size of the total α7* nAChR pool.
To extend our examination of the idea that Type III Nrg1 back signaling stimulates insertion of preexisting α7* nAChRs into the axonal membrane, we repeated the B4-ECD stimulation of dispersed sensory neurons in the presence of the protein synthesis inhibitor cycloheximide (CHX; Fig. 3, C–E). Neurons were pretreated with 10 μg/ml CHX for 45 min and then stimulated for an additional hour with either B2-ECD (control) or B4-ECD. Neither the B4-ECD–induced increase in the number of surface αBgTx-488 clusters (Fig. 3 D) nor the increase in cluster area (Fig. 3 E) were affected by the CHX pretreatment (CHX, 9.72 ± 1.65 clusters/100-μm axonal length; CHX + B4-ECD, 19.96 ± 2.83 clusters/100-μm axonal length; CHX, 0.23 ± 0.02 μm2; and CHX + B4-ECD, 0.32 ± 0.01 μm2). Thus, the increase in surface α7* nAChRs resulted from a redistribution of preexisting receptors rather than from new receptor synthesis.
Type III Nrg1 back signaling increases surface expression of α7* nAChRs in the absence of endocytosis
Prior studies have shown that endocytosis can alter the surface expression of neurotransmitter receptors (Man et al., 2007; Xia et al., 2007). Using a pharmacological inhibitor of endocytosis, phenylarsine oxide (PAO; Hertel et al., 1985), we investigated whether B4-ECD–mediated increase in the surface expression of α7* nAChR clusters was caused by alterations in receptor endocytosis. We applied PAO to sensory neurons for 45 min before B2-ECD (control) or B4-ECD treatment. Although PAO did not significantly alter the number of surface α7* nAChR clusters along axons under control conditions, it significantly increased the area of the clusters (from 0.2 ± 0.01 μm2 under control conditions to 0.29 ± 0.03 μm2 after PAO treatment; Fig. 4 C). After B4-ECD treatment for 1 h, we detected an increase in both the number and area of surface α7* nAChR clusters along axons when endocytosis was blocked (Fig. 4 B, PAO, 16.03 ± 2.69 clusters/100-μm axonal length; and PAO + B4-ECD, 29.77 ± 3.41 clusters/100-μm axonal length; Fig. 4 C, PAO, 0.29 ± 0.03 μm2; and PAO + B4-ECD, 0.34 ± 0.04 μm2). Based on our findings, we conclude that the increase in surface α7* nAChRs results from an increase in membrane insertion rather than a decrease in endocytosis.
Figure 4.

Type III Nrg1 back signaling increases α7* nAChRs cluster area in the absence of endocytosis. Dissociated sensory neurons from E11 chick embryos were treated with B2-ECD (control) or B4-ECD for 1 h. In parallel, neurons were treated with PAO for 45 min and treated with B2-ECD or B4-ECD for an additional hour. Neurons were labeled with αBgTx-488 (green), fixed, permeabilized, and colabeled for NF protein (blue). (A) Representative micrographs of αBgTx-488 staining along NF-positive axons. B4-ECD treatment increased surface αBgTx-488 clusters in the presence of PAO (d). Linescans of fluorescence intensity profiles of αBgTx-488 along representative axons (see Materials and methods) are shown. Bar, 5 μm. (B) Quantification of surface αBgTx-488 clusters along NF-labeled axons. B4-ECD treatment in the presence and absence of PAO induced ∼1.7 and 1.9 increases in surface αBgTx-488 clusters along axons, respectively. Data were pooled from two independent experiments. The graph shows means ± SEM. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P < 0.03 (Statview). (C) Quantification of surface αBgTx-488 cluster area. B4-ECD treatment in the presence or absence of PAO induced an increase in αBgTx-488 cluster area. Data pooled from two independent experiments were analyzed using nonparametric statistics and presented as box plots (see Materials and methods). Statistical significance was determined by the Kolmogorov-Smirnov Test. *, P = 0.03; **, P = 0.0001; ***, P < 0.0001 (Statview).
Type III Nrg1 back signaling activates a PtdIns 3K signaling pathway
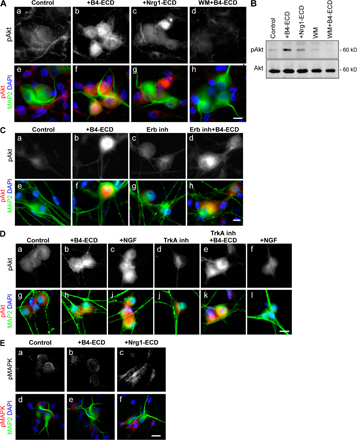
Our main objective in the following experiments was to test for signaling pathways activated downstream of Type III Nrg1 that might direct membrane targeting of α7* nAChRs. Because of the substantial evidence that PtdIns 3K signaling is a major regulator of membrane protein trafficking (Tengholm and Meyer, 2002; Man et al., 2003; Viard et al., 2004; Chae et al., 2005), we asked whether stimulation of Type III Nrg1 back signaling activated PtdIns 3K signaling. Sensory neurons were stimulated with B2-ECD (control) or B4-ECD for 5 min and then labeled with an antibody recognizing the PtdIns 3K product, phosphatidylinositol 3,4,5 trisphosphate (PIP3; Fig. 5 A). In neurons treated with B4-ECD, we detected an increase in PIP3 staining along tau-positive axons. This response was comparable to that seen after acute treatment with 100 ng/ml NGF and was completely blocked in cultures pretreated with 500 nM of the PtdIns 3K inhibitor wortmannin (WM).
Figure 5.

Type III Nrg1 back signaling activates the PtdIns 3K signaling pathway. (A) Dissociated sensory neurons from E11 chick embryos were treated for 5 min with B2-ECD (control), B4-ECD, 50 ng/ml NGF, or 10 ng/ml of soluble Nrg1β peptide (Nrg1-ECD). In parallel, neurons were treated with WM for 45 min before B4-ECD stimulation (WM + B4-ECD). Neurons were fixed, permeabilized, and costained for PIP3 (red) and tau protein (blue) to label axons. Both B4-ECD (g) and NGF (i) treatment induced puncta of PIP3 along tau-positive axons. Neither B4-ECD stimulation in the presence of WM (c and h) nor that of Nrg1-ECD (e and j) induced an increase in PIP3. Confocal images were obtained with a 40× oil objective. Bar, 10 μm. (B) Immunoblot analysis of phospho-Akt (Ser 473) in WT or Type III Nrg1−/− sensory neurons treated with either B2-ECD (control) or B4-ECD for 10 min. In WT neurons, B4-ECD treatment induced an approximately threefold increase in phospho-Akt, whereas no response was detected in mutant neurons. Total Akt in the bottom panel shows equal lysate loading. The bar graph represents phospho-Akt normalized to total Akt immunoreactive bands. Data are representative of three independent experiments. The graph shows means ± SEM. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P < 0.002 (Statview). (C and D) E14.5 WT (a and b) or Type III Nrg1−/− (c and d) DRG explants were treated with B2-ECD (control) or B4-ECD for 10 min. Surface-bound B4-ECD or B2-ECD were labeled with an antibody against the human Fc domain (anti-Fc; green) before fixation. Neurons were fixed, permeabilized, and stained for phospho-Akt (red) and NF protein (blue). B4-ECD treatment increased phospho-Akt along Fc-positive axons of WT neurons (b and D) but did not do so along axons of mutant neurons (d). Note the close proximity of anti-FC and phospho-Akt puncta in the high-power micrograph shown in e. The asterisk denotes an axon negative for both anti-Fc and phospho-Akt immunolabeling (c). A 63× oil objective was used (a–d). Confocal imaging was obtained with a 100× oil objective (D). Bar: (a–d)10 μm; (D) 5 μm. (E) Quantification of the average fluorescence intensity (AFI) of phospho-Akt along axons of WT or Type III Nrg1−/− sensory neurons treated with B2-ECD (control) or B4-ECD for 10 min or 1, 2, or 6 h (see Materials and methods). Along WT axons, B4-ECD treatment induced increases in phospho-Akt. Along axons of mutant neurons, we did not detect an increase in phospho-Akt in response to B4-ECD treatment. The graph shows means ± SEM. Data are from three independent experiments. Statistical significance was determined by ANOVA. *, P < 0.02.
As an additional test of whether Type III Nrg1 back signaling activates a PtdIns 3K signaling pathway, we measured the phosphorylation status of the PtdIns 3K effector kinase Akt in WT and Type III Nrg1−/− sensory neurons (Fig. 5, B–D; and Fig. S3, A–D, available at http://www.jcb.org/cgi/content/full/jcb.200710037/DC1). After a 10-min B4-ECD treatment, we detected phospho-Akt in puncta along B4-ECD–bound axons of WT neurons; however, we did not detect a change in phospho-Akt in axons from mutant explants (Fig. 5, C and D). The response to B4-ECD treatment was selective for the neurons and for the PtdIns 3K–Akt pathway. There was no activation of Akt in nonneuronal cells (Fig. S3 A) and there was no activation of MAPK in neurons (Fig. S3 E). Additionally, B4-ECD treatment was able to induce activation of Akt when ErbB kinase activity (Fig. S3 C) and NGF/TrkA signaling (Fig. S3 D) were blocked. Thus, we conclude that Type III Nrg1 back signaling rapidly and locally activates PtdIns 3K signaling.
PtdIns 3K signaling activated by Type III Nrg1 back signaling is required for increased α7 nAChR surface expression
We tested whether PtdIns 3K activation is required for Type III Nrg1–mediated changes in α7* nAChR surface expression (Fig. 6). We treated dispersed sensory neurons with either B2-ECD (control) or B4-ECD for 1 h in the presence of 500 nM of the PtdIns 3K inhibitor WM. Surface α7* nAChR clusters were labeled with αBgTx-488 (green), after which neurons were fixed and immunolabeled with an antibody against NF protein (blue; Fig. 6 A). In response to the B4-ECD treatment, we detected a significant increase in the number (Fig. 6 B) and area (Fig. 6 C) of αBgTx-488 clusters along NF-positive processes. WM treatment alone did not affect the number of αBgTx-488 clusters but did inhibit the response to B4-ECD, preventing the increase in the number of axonal surface α7*nAChRs (Fig. 6 B, B2-ECD [control], 9.08 ± 1.53 clusters/100-μm axonal length; B4-ECD, 17.41 ± 1.48 clusters/100-μm axonal length; WM, 9.58 ± 2.17 clusters/100-μm axonal length; and WM + B4-ECD, 10.75 ± 1.27 clusters/100-μm axonal length). Pretreatment with 5 μM of a small molecule inhibitor of Akt kinase activity (Akt inhibitor [Akt inh.]) also prevented the response to B4-ECD (Fig. 6 B, Akt inh., 10.33 ± 1.42 clusters/100-μm axonal length; and Akt inh. + B4-ECD, 8.48 ± 1.58 clusters/100-μm axonal length). Both WM and Akt inh. prevented the increase in surface αBgTx cluster area in response to B4-ECD treatment (Fig. 6 C, B2-ECD [control], 0.25 ± 0.02 μm2; B4-ECD, 0.35 ± 0.02 μm2; WM, 0.22 ± 0.02 μm2; WM + B4-ECD, 0.23 ± 0.02 μm2; Akt inh., 0.22 ± 0.02 μm2; and Akt inh. + B4-ECD, 0.25 ± 0.02 μm2). We conclude that Type III Nrg1 back signaling activates PtdIns 3K signaling and that PtdIns 3K/Akt activities are required for Type III Nrg1–induced targeting of α7* nAChRs to the axonal surface.
Figure 6.

PtdIns 3K—Akt signaling activated by Type III Nrg1 back signaling is required for increased α7* nAChR surface expression. Dissociated sensory neurons from E11 chick embryos were treated with B2-ECD (control) or B4-ECD for 1 h. In parallel, neurons were pretreated with WM or an Akt inh. for 45 min before treatment with B2-ECD or B4-ECD for an additional hour. Neurons were labeled for surface α7* nAChRs with αBgTx-488 (green), fixed, permeabilized, and costained for NF protein (blue). (A) Representative micrographs of αBgTx-488 staining along NF-positive axons. B4-ECD treatment increased surface αBgTx-488 clusters (b), which did not occur in the presence of WM (d). Linescans of fluorescence intensity profiles of αBgTx-488 along representative axons (see Materials and methods) are shown. Bar, 5 μm. (B) Quantification of surface αBgTx-488 clusters along sensory neuron axons represented in A. B4-ECD treatment induced an ∼1.9-fold increase of surface αBgTx-488 clusters but not in the presence of WM or Akt inh. Data were pooled from three independent experiments. The graph shows means ± SEM. Statistical significance was determined by ANOVA with post-hoc Fisher's PLSD test. *, P < 0.0001 (Statview). (C) Quantification of surface αBgTx-488 cluster area. B4-ECD treatment induced an increase in αBgTx-488 cluster area but not in the presence of WM or Akt inh. Data pooled from three independent experiments were analyzed using nonparametric statistics and presented as box plots (see Materials and methods). Statistical significance determined by the Kolmogorov-Smirnov Test. *, P = 0.0001 (Statview).
Discussion
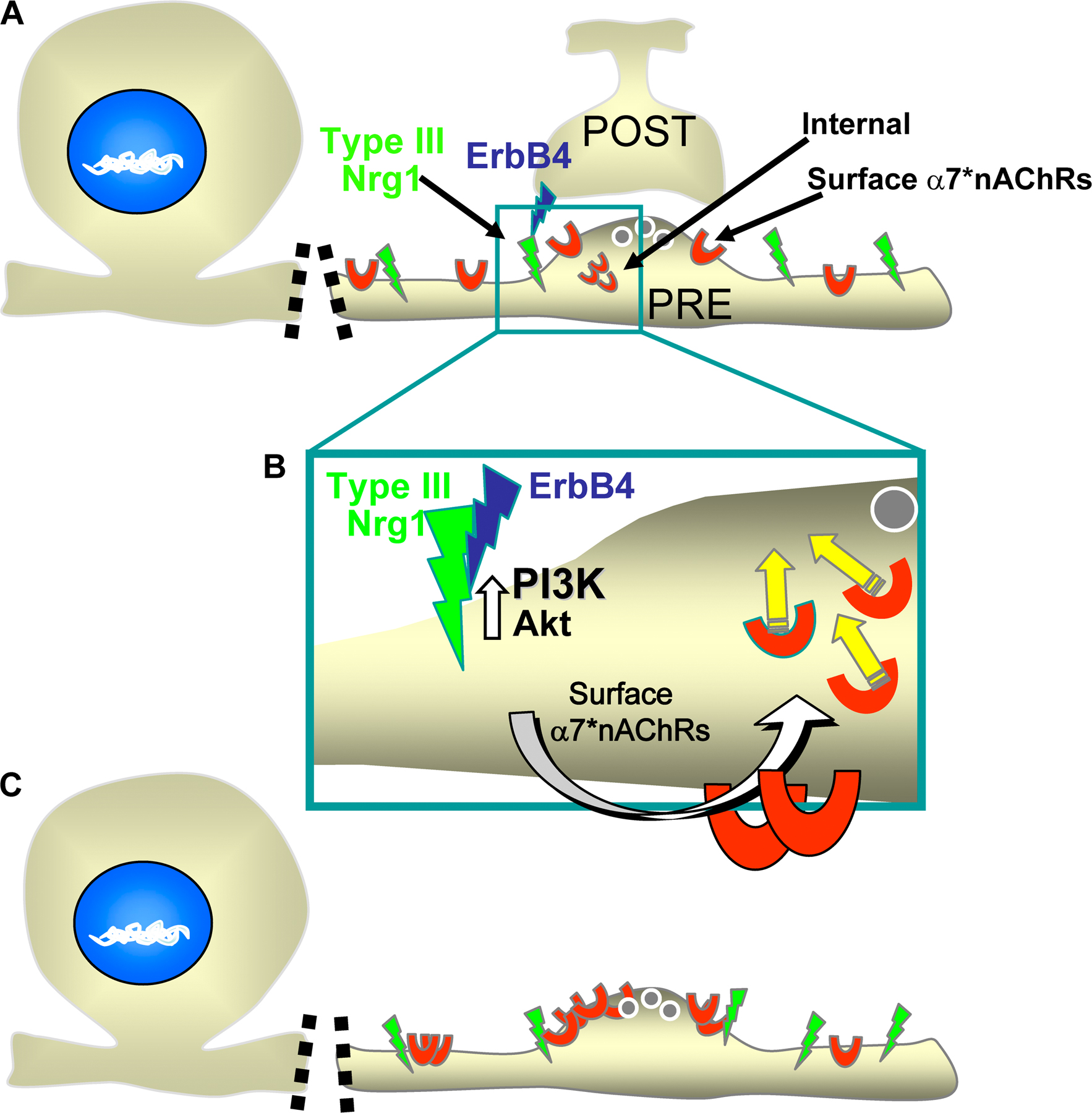
Nrg1-ErbB signaling plays an important role in synaptic plasticity, in part by regulating the levels of pre- and postsynaptic receptors and ion channels (Ozaki et al., 1997; Huang et al., 2000; Chae et al., 2005; Gu et al., 2005; Kwon et al., 2005; Bjarnadottir et al., 2007; Li et al., 2007; Role and Talmage, 2007; Woo et al., 2007), including nAChRs containing the α7 subunit (Yang et al., 1998; Liu et al., 2001; Kawai et al., 2002; Chang and Fischbach, 2006). In this study we demonstrate that Type III Nrg1, acting as a receptor for ErbB4, controls the insertion of α7* nAChRs into axonal membranes. Type III Nrg1 accomplishes this by activating a local PtdIns 3K signaling pathway. These findings expand the repertoire of identified mechanisms by which Type III Nrg1 bidirectional signaling contributes to the establishment and the maturation of functional presynaptic terminals (Bao et al., 2003, 2004). Our major findings supporting this conclusion are the following: genetic disruption of Type III Nrg1 affects surface α7* nAChR levels; Type III Nrg1 functioning as a receptor can regulate insertion of preformed α7* nAChRs into axonal surfaces; and Type III Nrg1 activates an axonal PtdIns 3K signaling pathway that mediates increased levels of surface α7* nAChRs.
Prior work has shown that presynaptic Type III Nrg1 is critical for establishing stable interactions with postsynaptic partners and, ultimately, for the survival of neuromuscular synapses as well as for glia development and myelination (Wolpowitz et al., 2000; Michailov et al., 2004; Taveggia et al., 2005; Chen et al., 2006). Our studies demonstrate that Type III Nrg1 acts as a presynaptic signaling receptor. Activation of presynaptic Type III Nrg1 signaling, either by postsynaptic ErbB4 (as shown in Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200710037/DC1), by the ErbB3 present on glia, or after depolarization (Bao et al., 2003, 2004), locally regulates the levels of presynaptic α7* nAChR surface expression. As a result, Type III Nrg1 back signaling, by regulating presynaptic α7* nAChRs, alters the magnitude of acetylcholine modulation of neurotransmitter release (MacDermott et al., 1999; Jones and Wonnacott, 2004).
We demonstrate that stimulating Type III Nrg1 activates a PtdIns 3K signaling pathway and that PtdIns 3K and Akt activity are required for increased surface expression of α7* nAChRs. At present, we do not know the molecular details of how Type III Nrg1 communicates with the PtdIns 3K signaling pathway or what Akt substrates regulate α7* nAChR trafficking. The intracellular domains of Type III Nrg1 lack known functional motifs that could mediate protein–protein interactions.
PtdIns 3K and its product PIP3 are ubiquitous signaling molecules that link cell surface receptors to intracellular downstream effectors. PtdIns 3K signaling regulates the trafficking of several cell surface proteins, such as glucose transporter 4 (Tengholm and Meyer, 2002), excitatory amino acid carrier 1 glutamate transporter (Davis et al., 1998), calcium channels (Viard et al., 2004), and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (Man et al., 2003). An increased understanding of the molecular mechanisms that regulate trafficking of these proteins is certain to inform our further studies on presynaptic α7* nAChR targeting.
We have used αBgTx binding to identify both surface and internal pools of α7* nAChRs. αBgTx binding requires at least partial assembly of α7 subunits into pentamers (Mitra et al., 2001). Because we do not see effects of Type III Nrg1 back signaling on αBgTx binding in permeabilized cells, it is unlikely that the early stages of assembling α7 subunits into functional receptors are targeted by back signaling. Assembly of α7* nAChRs and trafficking of α7* nAChRs through the exocytic pathway can be influenced by synaptic scaffolding proteins such as PICK1 (Baer et al., 2007), reversible palmitoylation (of α7* nAChRs by itself; Drisdel et al., 2004), tyrosine residues on unidentified proteins (Cho et al., 2005), and chronic nicotine (Marks et al., 1992). Any of these mechanisms might represent convergent targets with Type III Nrg1–PtdIns 3K signaling for influencing the rate of ER/Golgi to plasma membrane trafficking of α7* nAChRs.
Targeting nAChRs to presynaptic locales is an important part of establishing a plastic synapse. The α7* nAChRs in particular play a critical role in sustained modulation of neurotransmitter release (McGehee et al., 1995; Girod et al., 2000). Deficits in Type III Nrg1 signaling compromise the ability of nicotine to elicit sustained glutamatergic transmission at ventral hippocampal to nucleus accumbens synapses (Du, C., C. Zhong, M. Hancock, D.A. Talmage, and L.W. Role. 2004. Society for Neuroscience 34th Annual Meeting; unpublished data) and, based on the results presented here, we would predict a similar effect on nicotine/acetylcholine modulation of glutamate release from primary sensory afferents in the dorsal spinal cord (Genzen and McGehee, 2003). From the present data, we cannot say whether the requirement for Type III Nrg1 signaling is limited to developmental synaptogenesis or whether it continues to affect presynaptic α7* nAChR targeting to established synapses, contributing to the maintenance of synaptic plasticity throughout the lifespan. If Type III Nrg1 signaling does continue to regulate presynaptic levels of α7* nAChR or other receptors, then modulation of Type III Nrg1 signaling in sensory axons could contribute to adaptive physiological changes or to pathological changes in sensory transduction (Bermingham-McDonogh et al., 1997; Kerber et al., 2003).
There are interesting parallels between our findings and pathophysiological studies. Increasing evidence from genetic linkage studies has identified both the NRG1 (Stefansson et al., 2002; Harrison and Law, 2006) and the α7 nAChR gene CHRNA7 (Freedman et al., 2001; Leonard and Freedman, 2006) as susceptibility genes for schizophrenia. Postmortem studies have demonstrated decreased levels of αBgTx binding in the hippocampus of schizophrenic patients (Freedman et al., 1995; Breese et al., 2000), and decreased levels of α7 mRNA and αBgTx binding in dorsal lateral prefrontal cortex of schizophrenic brain are associated with risk alleles at the NRG1 locus (Mathew et al., 2007). It is worth noting that these risk alleles at the NRG1 locus also have been associated with increased expression of Type I and IV Nrg1 (Law et al., 2006). This raises the possibility that excess levels of soluble Nrg1 peptide disturbs the balance of bidirectional Nrg1-ErbB signaling that serves to ensure normal levels of functional α7* nAChRs.
Materials and methods
Animals and cell culture
DRG explants from E14.5 WT or Type III Nrg1−/− mouse embryos (Wolpowitz et al., 2000) were dissected and cleaned with forceps to remove connective tissue. DRG were plated on glass coverslips (precoated with 1 mg/ml poly-d-lysine and 100 μg/ml laminin) and cultured in MEM (Invitrogen) supplemented with 10% FBS, 4 mg/ml glucose, 2 mM l-glutamine, 50 U/ml each of penicillin and streptomycin (Invitrogen), and 40 ng/ml β-NGF (Harlan Bioproducts for Science) at 37°C for 2 d. DRG from E11 chick embryos were dispersed and cultured in DME media (Invitrogen) supplemented with 5% chick embryonic extract, 10% horse serum, 2 mM l-glutamine, 50 U/ml penicillin and streptomycin (Invitrogen), and 50 ng/ml β-NGF (Harlan Bioproducts for Science) at 37°C for 2 d. For PIP3 and phospho-Akt labeling and immunoblotting, culture media was replaced with serum-free media 4 h before soluble factor addition. For chick cultures, serum-free media consisted of DME, 1% BSA (Sigma-Aldrich), 2 mM l-glutamine (Invitrogen), insulin-transferrin-selenium supplement (Sigma-Aldrich) and 50 U/ml each of penicillin and streptomycin. For mouse cultures, serum-free media consisted of MEM, 1% BSA, 4 mg/ml glucose, 2 mM l-glutamine, insulin-transferrin-selenium supplement, and 50 U/ml each of penicillin and streptomycin.
Reagents
Where indicated, the following reagents were applied to the cells: 2 nM of soluble B2-ECD or B4-ECD (Bao et al., 2003), 50 ng/ml of recombinant HRG1-β1 EGF-like domain (R&D Systems), 50 ng/ml β-NGF (R&D Systems), 10 μg/ml CHX (Sigma-Aldrich), 5 μM phenylarsine oxide (Sigma-Aldrich), 500 nM WM (EMD), 5 μM Akt Inh. V (EMD), 2 μM each of PD 158780 and PD 168393 (EMD), and 200 nM TrkA inhibitor (EMD). Inhibitors were added to the media for 45 min before soluble factor addition.
Immunofluorescence
Cells were fixed with 4% PFA for 20 min at RT, permeabilized with 0.2% Triton X-100 (where indicated) for 5 min at RT, blocked with 10% NDS, and incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: anti-Type III Nrg1 (1:500; Yang et al., 1998), anti-NF (1:500; Sternberger Monoclonals, Inc.), anti-tau (1:250; Sigma-Aldrich), anti-human IgG (Fc-specific; 1:200; Sigma-Aldrich), anti-PI3,4,5P3 (1:50; Echelon Biosciences Inc.), anti–phospho-Akt (1:200; Cell Signaling Technology), anti-synaptophysin (1:500; Millipore); anti-vGlut1 (1:200; Synaptic Systems GmbH), anti-TrkA (1:5,000; gift from L.F. Reichardt, University of California, San Francisco, San Francisco, CA), anti-MAP2 (1:500; Millipore), and anti–phospho MAPK (1:1,000; Sigma-Aldrich). Cells were washed and incubated in secondary antibodies conjugated to Alexa 488 (1:500; Invitrogen), Alexa 594 (1:500; Invitrogen), or anti-mannobioside carbohydrate antibodies (1:50; Jackson ImmunoResearch Laboratories) for 1 h at RT. To label PI3,4,5P3, slips were treated with Avidin/Biotin Blocking kit (Vector Laboratories) before incubation in anti-PI3,4,5P3. The primary antibody signal was amplified by sequential binding with a biotinylated anti–mouse secondary (1:200; Jackson ImmunoResearch Laboratories) and NeutrAvidin–-rhodamine red (1:200; Invitrogen). Slips were mounted using VectaShield (Vector Laboratories), and images were captured using a microscope (Axio Imager; Carl Zeiss, Inc.) equipped with Plan-Apochromat objectives (20× with 0.8 NA or 63× oil with 1.4 NA), a charge-coupled device camera (Hamamatsu), and Metamorph software (Version 6.3r5; MDS Analytical Technologies). Confocal images were captured with an NLO Multiphoton (LSM 510; Carl Zeiss, Inc.) on a microscope (Axioskop2 FS; Carl Zeiss, Inc.) equipped with Plan-Neofluar objectives (40 or 100× oil with 1.3 NA) and a charge-coupled device camera. Brightness and contrast were adjusted using Photoshop software (Version 8.0; Adobe).
To label surface α7* nAChRs, live sensory neurons were incubated in αBgTx-488 (1:500; Invitrogen) for 15–20 min at 37°C. Cells were fixed and incubated in rabbit anti–Alexa 488 (1:250; Invitrogen) overnight at 4°C, followed by secondary antibody incubation. Surface cluster number and area were measured along NF-positive processes (≥10 μm from soma) using Metamorph software. The lengths of axonal processes were measured by manually tracing NF-positive processes. Control cultures in each experiment were used to define the threshold for measuring clusters at 50% maximum intensity and greater than or equal to four contiguous pixels (Kawai et al., 2002). To determine nonspecific binding, cultures were treated with 1 μM of nicotine or 5 μM methyllycaconitine before labeling. For each experiment, nonspecific labeling was ≤12% and was subtracted from all counts. Linescans with widths of 10 contiguous pixels were obtained using Metamorph software.
To quantify Akt activation along axons, neurons were stained with antibodies against phospho-Akt and NF to label axons. 15–20 axons per condition were analyzed in each experiment. The AFI of phospho-Akt along NF-positive processes was measured using MetaMorph software. The AFI of two to three randomly selected regions for each condition were measured as background AFI and subtracted from all measurements. To compare results between independent experiments, the mean AFI from different conditions in each independent experiment was normalized to the control AFI and is expressed as fold increase.
Ca2+ imaging
After 3 d in vitro, DRG explants were rinsed with HBSS, loaded with Fluo-3 Ca2+ binding dye (Invitrogen) dissolved in HBSS for 30 min at 37°C, and rinsed for 30 min at 37°C in HBSS. The coverslip was placed on a perfusion-equipped stage and perfused at a rate of 0.5 ml/min with HBSS containing 2 μM tetrodotoxin, 10 μM bicuculin, 50 μM d-(−)-2amino-5-phosphono-valeric acid and 20 μM CNQX (Sigma-Aldrich). Images were captured every 5 s using a spinning disc confocal microscope (DSU; Olympus) equipped with a UPlanSApo objective (60× water with 1.4 NA), electron-multiplying charge-coupled device camera (Hamamatsu), and Slidebook software (Version 4; Olympus). After acquiring images for 1 min, 1 μM of nicotine was focally applied by pressure ejection for 60 s and the explants were imaged for another 4 min. After a 1-μM nicotine application, fluorescence intensity within defined axonal regions was quantified with MetaMorph software.
125I-αBgTx binding assay
After 2 d in vitro, dispersed chick sensory neurons were treated with 2 nM of soluble B2-ECD or B4-ECD for 24 h. 6 nM 125I-αBgTx (2,000 Ci/μmol; GE Healthcare) was added directly to the media for the final 20 min. To measure total 125I-αBgTx binding, cultures were rinsed three times with PBS and permeabilized with 0.5% saponin in 0.2% FAF-BSA/PBS for 30 min before incubation with 6 nM 125I-αBgTx in 0.2% FAF-BSA/PBS for 2 h on ice. For both surface and total labeling, nonspecific binding was assessed by including 1 μM αBgTx (Sigma-Aldrich) during the labeling with 125I-αBgTx. After labeling, cells were washed and solubilized in 1 N NaOH, and bound 125I-αBgTx was quantified with a γ counter. Nonspecific binding was subtracted.
Immunoblotting
Neurons were lysed in RIPA buffer (Thermo Fisher Scientific) supplemented with 10 mM DTT, protease inhibitors (Sigma-Aldrich), and phosphatase inhibitors (Thermo Fisher Scientific). Lysates (phospho-Akt, 25 μg; α7, 40 μg) were separated on 10% SDS-PAGE gels and transferred to nitrocellulose filters. Filters were blocked in 5% milk in 0.1% Tween-20 TBS solution at RT for 2 h before overnight incubation in primary antibody solutions in 5% BSA or milk in Tween-20 TBS solution at 4°C (anti-α7 nAChR subunit [1:500; Santa Cruz Biotechnology, Inc.], anti-MAPK [1:5,000; Millipore], anti-glyceraldehyde 3-phosphate dehydrogenase [1:10,000; Millipore], anti–NF M [1:7,500; Millipore), anti–phospho-Akt [1:500; Cell Signaling Technology], and anti-Akt [1:1,000; Cell Signaling Technology]). Detection was performed using Alexa Fluor 680 (Invitrogen) or IRDye-800 (Rockland Immunochemicals) secondary antibodies and an Odyssey Infrared Imaging System (Version 2.1; LI-COR Biosciences).
Transferrin recycling
After 2–4 d in vitro, WT or Type III Nrg1−/− sensory neurons were serum starved for 25 min in serum-free MEM containing 0.5% BSA. Neurons were incubated with 10 μg/ml transferrin–Alexa 488 (Invitrogen) for 15 min in serum-free media, after which cells were rinsed and chased in complete media supplemented with 1 mg/ml of unlabeled transferrin (Sigma-Aldrich). During the chase period, live images of neurons were captured at 0.5–1-μm intervals every 2–4 min over a >20-min period using a spinning disc confocal microscope (DSU; Olympus) equipped with a UPlanSApo objective (60× water with 1.4 NA), electron-multiplying charge-coupled device camera, and Slidebook software. The AFI of transferrin–Alexa 488 in collapsed z series was measured using MetaMorph software. The AFI of two to three randomly selected regions for each condition was measured as background AFI and subtracted. The AFI from each time point was normalized to the AFI at t = 0 and is expressed as percentage of initial AFI.
Statistical significance
For normally distributed data, statistical significance was evaluated by ANOVA with a post-hoc Fischer's PLSD test for multiple comparisons (Statview; Adept Scientific). Non-normally distributed data were analyzed using nonparametric methods and are presented using box plots. The boxes include data points within the middle 50%. The bottom marks the twenty-fifth percentile, the middle line the fiftieth percentile, and the top the seventy-fifth percentile. Vertical lines mark the fifth and ninety-fifth percentiles (see Devay et al. [1999] for a detailed description of data presentation). Statistical significance was evaluated by the Kolmogorov-Smirnov test.
Online supplemental material
In Fig. S1, we show that endocytic recycling of transferrin (A and B) and the trafficking of presynaptic proteins vGlut1 and synaptophysin (C and D) are not impaired in Type III Nrg1−/− sensory neurons. Additionally, we show that activation of Type III Nrg1 back-signaling does not induce an increase in the surface expression of TrkA along axons (E and F). Fig. S2 shows soluble ErbB4-ECD (B4-ECD) bound to Type III Nrg1 puncta along WT axons only (A). B4-ECD treatment induces an increase in the axonal surface expression of α7*nAChRs in a dose-dependent manner (B). Fig. S3 shows that Type III Nrg1 back signaling activates Akt in neurons and not nonneuronal cells (A). Inhibition of ErbB (B) or TrkA (C) kinase activity does not prevent the B4-ECD-induced response. We also show that B4-ECD treatment does not activate MAPK in neurons (D). Fig. S4 consists a diagram summarizing a mechanism by which Type III Nrg1 back-signaling regulates the levels of α7*nAChRs along axons. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710037/DC1.
Supplementary Material
Acknowledgments
We thank M. Mertz for technical assistance with Ca2+ imaging, B.L. Sherman for mouse genotyping, M.A. Johnson for helpful comments on the manuscript, and L.F. Reichardt for the TrkA antibody.
This work was funded by grants from the National Institutes of Health (NS29071 to L.W. Role and D.A. Talmage; DA019941 to L.W. Role; and DK07328 to M.L. Hancock) and from the National Alliance for Research on Schizophrenia and Depression (Grable Distinguished Investigator Award to L.W. Role).
L.W. Role's present address is Department of Neurobiology and Behavior, State University of New York at Stony Brook, Stony Brook, NY 11794.
D.A. Talmage's present address is Department of Pharmacological Science and Center for Brain and Spinal Cord Research, State University of New York at Stony Brook, Stony Brook, NY 11794.
Abbreviations used in this paper: AFI, average fluorescence intensity; Akt inh., Akt inhibitor; a.u., arbitrary units; CHX, cycloheximide; DRG, dorsal root ganglia; E, embryonic day; ECD, extracellular domain; nAChR, nicotinic acetylcholine receptor; NF, neurofilament; Nrg1, Neuregulin1; PAO, phenylarsine oxide; PIP3, phosphatidylinositol 3,4,5 trisphosphate; PtdIns 3K, phosphatidylinositol 3-kinase; vGlut1, vesicular glutamate transporter 1; WM, wortmannin; WT, wild type.
References
- Baer, K., T. Burli, K.H. Huh, A. Wiesner, S. Erb-Vogtli, D. Gockeritz-Dujmovic, M. Moransard, A. Nishimune, M.I. Rees, J.M. Henley, et al. 2007. PICK1 interacts with alpha7 neuronal nicotinic acetylcholine receptors and controls their clustering. Mol. Cell. Neurosci. 35:339–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, J., D. Wolpowitz, L.W. Role, and D.A. Talmage. 2003. Back signaling by the Nrg-1 intracellular domain. J. Cell Biol. 161:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, J., H. Lin, Y. Ouyang, D. Lei, A. Osman, T.W. Kim, L. Mei, P. Dai, K.K. Ohlemiller, and R.T. Ambron. 2004. Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nat. Neurosci. 7:1250–1258. [DOI] [PubMed] [Google Scholar]
- Berg, D.K., and W.G. Conroy. 2002. Nicotinic alpha 7 receptors: synaptic options and downstream signaling in neurons. J. Neurobiol. 53:512–523. [DOI] [PubMed] [Google Scholar]
- Bermingham-McDonogh, O., Y.T. Xu, M.A. Marchionni, and S.S. Scherer. 1997. Neuregulin expression in PNS neurons: isoforms and regulation by target interactions. Mol. Cell. Neurosci. 10:184–195. [DOI] [PubMed] [Google Scholar]
- Bjarnadottir, M., D.L. Misner, S. Haverfield-Gross, S. Bruun, V.G. Helgason, H. Stefansson, A. Sigmundsson, D.R. Firth, B. Nielsen, R. Stefansdottir, et al. 2007. Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/− knock-outs compared with wild-type mice. J. Neurosci. 27:4519–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd, R.T., M.H. Jacob, A.E. McEachern, S. Caron, and D.K. Berg. 1991. Nicotinic acetylcholine receptor mRNA in dorsal root ganglion neurons. J. Neurobiol. 22:1–14. [DOI] [PubMed] [Google Scholar]
- Breese, C.R., M.J. Lee, C.E. Adams, B. Sullivan, J. Logel, K.M. Gillen, M.J. Marks, A.C. Collins, and S. Leonard. 2000. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 23:351–364. [DOI] [PubMed] [Google Scholar]
- Chae, K.S., M. Martin-Caraballo, M. Anderson, and S.E. Dryer. 2005. Akt activation is necessary for growth factor-induced trafficking of functional K(Ca) channels in developing parasympathetic neurons. J. Neurophysiol. 93:1174–1182. [DOI] [PubMed] [Google Scholar]
- Chang, Q., and G.D. Fischbach. 2006. An acute effect of neuregulin 1 beta to suppress alpha 7-containing nicotinic acetylcholine receptors in hippocampal interneurons. J. Neurosci. 26:11295–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S., M.O. Velardez, X. Warot, Z.X. Yu, S.J. Miller, D. Cros, and G. Corfas. 2006. Neuregulin 1-ErbB signaling is necessary for normal myelination and sensory function. J. Neurosci. 26:3079–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, C.H., W. Song, K. Leitzell, E. Teo, A.D. Meleth, M.W. Quick, and R.A. Lester. 2005. Rapid upregulation of alpha7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J. Neurosci. 25:3712–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador, F.A., P.A. Lima, and S. Wonnacott. 2000. The alpha7 nicotinic acetylcholine receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a Ca(2+) dependent mechanism. Neuropharmacology. 39:2799–2807. [DOI] [PubMed] [Google Scholar]
- Davis, K.E., D.J. Straff, E.A. Weinstein, P.G. Bannerman, D.M. Correale, J.D. Rothstein, and M.B. Robinson. 1998. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J. Neurosci. 18:2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devay, P., D.S. McGehee, C.R. Yu, and L.W. Role. 1999. Target-specific control of nicotinic receptor expression at developing interneuronal synapses in chick. Nat. Neurosci. 2:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisdel, R.C., E. Manzana, and W.N. Green. 2004. The role of palmitoylation in functional expression of nicotinic alpha7 receptors. J. Neurosci. 24:10502–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falls, D.L. 2003. Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res. 284:14–30. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, V.D., P.I. Pisacane, R.L. Vandlen, and M.X. Sliwkowski. 1998. Formation of a high affinity heregulin binding site using the soluble extracellular domains of ErbB2 with ErbB3 or ErbB4. FEBS Lett. 431:102–106. [DOI] [PubMed] [Google Scholar]
- Freedman, R., M. Hall, L.E. Adler, and S. Leonard. 1995. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol. Psychiatry. 38:22–33. [DOI] [PubMed] [Google Scholar]
- Freedman, R., S. Leonard, J.M. Gault, J. Hopkins, C.R. Cloninger, C.A. Kaufmann, M.T. Tsuang, S.V. Farone, D. Malaspina, D.M. Svrakic, et al. 2001. Linkage disequilibrium for schizophrenia at the chromosome 15q13-14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7). Am. J. Med. Genet. 105:20–22. [PubMed] [Google Scholar]
- Fucile, S., A. Sucapane, and F. Eusebi. 2005. Ca2+ permeability of nicotinic acetylcholine receptors from rat dorsal root ganglion neurones. J. Physiol. 565:219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzen, J.R., and D.S. McGehee. 2003. Short- and long-term enhancement of excitatory transmission in the spinal cord dorsal horn by nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA. 100:6807–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzen, J.R., W. Van Cleve, and D.S. McGehee. 2001. Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J. Neurophysiol. 86:1773–1782. [DOI] [PubMed] [Google Scholar]
- Girod, R., N. Barazangi, D. McGehee, and L.W. Role. 2000. Facilitation of glutamatergic neurotransmission by presynaptic nicotinic acetylcholine receptors. Neuropharmacology. 39:2715–2725. [DOI] [PubMed] [Google Scholar]
- Gu, Z., Q. Jiang, A.K. Fu, N.Y. Ip, and Z. Yan. 2005. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J. Neurosci. 25:4974–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, P.J., and A.J. Law. 2006. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol. Psychiatry. 60:132–140. [DOI] [PubMed] [Google Scholar]
- Hertel, C., S.J. Coulter, and J.P. Perkins. 1985. A comparison of catecholamine-induced internalization of β-adrenergic receptors and receptor-mediated endocytosis of epidermal growth factor in human astrocytoma cells. Inhibition by phenylarsine oxide. J. Biol. Chem. 260:12547–12553. [PubMed] [Google Scholar]
- Huang, Y.Z., S. Won, D.W. Ali, Q. Wang, M. Tanowitz, Q.S. Du, K.A. Pelkey, D.J. Yang, W.C. Xiong, M.W. Salter, and L. Mei. 2000. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 26:443–455. [DOI] [PubMed] [Google Scholar]
- Jones, I.W., and S. Wonnacott. 2004. Precise localization of α7 nicotinic receptors on glutamatergic axon terminals in the rat ventral tegmental area. J. Neurosci. 24:11244–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai, H., W. Zago, and D.K. Berg. 2002. Nicotinic alpha 7 receptor clusters on hippocampal GABAergic neurons: regulation by synaptic activity and neurotrophins. J. Neurosci. 22:7903–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber, G., R. Streif, F.W. Schwaiger, G.W. Kreutzberg, and G. Hager. 2003. Neuregulin-1 isoforms are differentially expressed in the intact and regenerating adult rat nervous system. J. Mol. Neurosci. 21:149–165. [DOI] [PubMed] [Google Scholar]
- Kwon, O.B., M. Longart, D. Vullhorst, D.A. Hoffman, and A. Buonanno. 2005. Neuregulin-1 reverses long-term potentiation at CA1 hippocampal synapses. J. Neurosci. 25:9378–9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, A.J., B.K. Lipska, C.S. Weickert, T.M. Hyde, R.E. Straub, R. Hashimoto, P.J. Harrison, J.E. Kleinman, and D.R. Weinberger. 2006. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc. Natl. Acad. Sci. USA. 103:6747–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard, S., and R. Freedman. 2006. Genetics of chromosome 15q13-q14 in schizophrenia. Biol. Psychiatry. 60:115–122. [DOI] [PubMed] [Google Scholar]
- Li, B., R.S. Woo, L. Mei, and R. Malinow. 2007. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron. 54:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., B. Ford, M.A. Mann, and G.D. Fischbach. 2001. Neuregulins increase alpha7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. J. Neurosci. 21:5660–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bendito, G., A. Cautinat, J.A. Sanchez, F. Bielle, N. Flames, A.N. Garratt, D.A. Talmage, L.W. Role, P. Charnay, O. Marin, and S. Garel. 2006. Tangential neuronal migration controls axon guidance: a role for neuregulin-1 in thalamocortical axon navigation. Cell. 125:127–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott, A.B., L.W. Role, and S.A. Siegelbaum. 1999. Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci. 22:443–485. [DOI] [PubMed] [Google Scholar]
- Man, H.Y., Q. Wang, W.Y. Lu, W. Ju, G. Ahmadian, L. Liu, S. D'Souza, T.P. Wong, C. Taghibiglou, J. Lu, et al. 2003. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 38:611–624. [DOI] [PubMed] [Google Scholar]
- Man, H.Y., Y. Sekine-Aizawa, and R.L. Huganir. 2007. Regulation of {alpha }-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. USA. 104:3579–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, M.J., J.R. Pauly, S.D. Gross, E.S. Deneris, I. Hermans-Borgmeyer, S.F. Heinemann, and A.C. Collins. 1992. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J. Neurosci. 12:2765–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew, S.V., A.J. Law, B.K. Lipska, M.I. Davila-Garcia, E.D. Zamora, S.N. Mitkus, R. Vakkalanka, R.E. Straub, D.R. Weinberger, J.E. Kleinman, and T.M. Hyde. 2007. {alpha }7 nicotinic acetylcholine receptor mRNA expression and binding in postmortem human brain are associated with genetic variation in Neuregulin 1. Hum. Mol. Genet. 16:2921–2932. [DOI] [PubMed] [Google Scholar]
- McGehee, D.S., M.J. Heath, S. Gelber, P. Devay, and L.W. Role. 1995. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 269:1692–1696. [DOI] [PubMed] [Google Scholar]
- Mechawar, N., A. Saghatelyan, R. Grailhe, L. Scoriels, G. Gheusi, M.M. Gabellec, P.M. Lledo, and J.P. Changeux. 2004. Nicotinic receptors regulate the survival of newborn neurons in the adult olfactory bulb. Proc. Natl. Acad. Sci. USA. 101:9822–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailov, G.V., M.W. Sereda, B.G. Brinkmann, T.M. Fischer, B. Haug, C. Birchmeier, L. Role, C. Lai, M.H. Schwab, and K.A. Nave. 2004. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 304:700–703. [DOI] [PubMed] [Google Scholar]
- Mitra, M., C.P. Wanamaker, and W.N. Green. 2001. Rearrangement of nicotinic receptor alpha subunits during formation of the ligand binding sites. J. Neurosci. 21:3000–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, M., and G. Corfas. 2004. Neuregulin1 downregulates postsynaptic GABAA receptors at the hippocampal inhibitory synapse. Hippocampus. 14:337–344. [DOI] [PubMed] [Google Scholar]
- Ozaki, M., M. Sasner, R. Yano, H.S. Lu, and A. Buonanno. 1997. Neuregulin-beta induces expression of an NMDA-receptor subunit. Nature. 390:691–694. [DOI] [PubMed] [Google Scholar]
- Ravdin, P.M., and D.K. Berg. 1979. Inhibition of neuronal acetylcholine sensitivity by alpha-toxins from Bungarus multicinctus venom. Proc. Natl. Acad. Sci. USA. 76:2072–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieff, H.I., L.T. Raetzman, D.W. Sapp, H.H. Yeh, R.E. Siegel, and G. Corfas. 1999. Neuregulin induces GABA(A) receptor subunit expression and neurite outgrowth in cerebellar granule cells. J. Neurosci. 19:10757–10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Role, L.W., and D.A. Talmage. 2007. Neurobiology: new order for thought disorders. Nature. 448:263–265. [DOI] [PubMed] [Google Scholar]
- Rosenberg, M.M., R.C. Blitzblau, D.P. Olsen, and M.H. Jacob. 2002. Regulatory mechanisms that govern nicotinic synapse formation in neurons. J. Neurobiol. 53:542–555. [DOI] [PubMed] [Google Scholar]
- Roth, A.L., and D.K. Berg. 2003. Large clusters of alpha7-containing nicotinic acetylcholine receptors on chick spinal cord neurons. J. Comp. Neurol. 465:195–204. [DOI] [PubMed] [Google Scholar]
- Stefansson, H., E. Sigurdsson, V. Steinthorsdottir, S. Bjornsdottir, T. Sigmundsson, S. Ghosh, J. Brynjolfsson, S. Gunnarsdottir, O. Ivarsson, T.T. Chou, et al. 2002. Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71:877–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia, C., G. Zanazzi, A. Petrylak, H. Yano, J. Rosenbluth, S. Einheber, X. Xu, R.M. Esper, J.A. Loeb, P. Shrager, et al. 2005. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 47:681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengholm, A., and T. Meyer. 2002. A PI3-kinase signaling code for insulin-triggered insertion of glucose transporters into the plasma membrane. Curr. Biol. 12:1871–1876. [DOI] [PubMed] [Google Scholar]
- Viard, P., A.J. Butcher, G. Halet, A. Davies, B. Nurnberg, F. Heblich, and A.C. Dolphin. 2004. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 7:939–946. [DOI] [PubMed] [Google Scholar]
- Wang, J.Y., S.J. Miller, and D.L. Falls. 2001. The N-terminal region of neuregulin isoforms determines the accumulation of cell surface and released neuregulin ectodomain. J. Biol. Chem. 276:2841–2851. [DOI] [PubMed] [Google Scholar]
- Wolpowitz, D., T.B. Mason, P. Dietrich, M. Mendelsohn, D.A. Talmage, and L.W. Role. 2000. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 25:79–91. [DOI] [PubMed] [Google Scholar]
- Woo, R.S., X.M. Li, Y. Tao, E. Carpenter-Hyland, Y.Z. Huang, J. Weber, H. Neiswender, X.-P. Dong, J. Wu, M. Gassmann, et al. 2007. Neuregulin-1 enhances depolarization-induced GABA release. Neuron. 54:599–610. [DOI] [PubMed] [Google Scholar]
- Xia, Y., S. Nawy, and R.C. Carroll. 2007. Activity-dependent synaptic plasticity in retinal ganglion cells. J. Neurosci. 27:12221–12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X., Y. Kuo, P. Devay, C. Yu, and L. Role. 1998. A cysteine-rich isoform of neuregulin controls the level of expression of neuronal nicotinic receptor channels during synaptogenesis. Neuron. 20:255–270. [DOI] [PubMed] [Google Scholar]
- Zhang, J., and D.K. Berg. 2007. Reversible inhibition of GABAA receptors by alpha7-containing nicotinic receptors on the vertebrate postsynaptic neurons. J. Physiol. 579:753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.W., J.S. Coggan, and D.K. Berg. 1996. Synaptic currents generated by neuronal acetylcholine receptors sensitive to alpha-bungarotoxin. Neuron. 17:1231–1240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}