

Abstract
Telomeres are maintained by three DNA-binding proteins (telomeric repeat binding factor 1 [TRF1], TRF2, and protector of telomeres 1 [POT1]) and several associated factors. One factor, TRF1-interacting protein 2 (TIN2), binds TRF1 and TRF2 directly and POT1 indirectly. Along with two other proteins, TPP1 and hRap1, these form a soluble complex that may be the core telomere maintenance complex. It is not clear whether subcomplexes also exist in vivo. We provide evidence for two TIN2 subcomplexes with distinct functions in human cells. We isolated these two TIN2 subcomplexes from nuclear lysates of unperturbed cells and cells expressing TIN2 mutants TIN2-13 and TIN2-15C, which cannot bind TRF2 or TRF1, respectively. In cells with wild-type p53 function, TIN2-15C was more potent than TIN2-13 in causing telomere uncapping and eventual growth arrest. In cells lacking p53 function, TIN2-15C was more potent than TIN2-13 in causing telomere dysfunction and cell death. Our findings suggest that distinct TIN2 complexes exist and that TIN2-15C–sensitive subcomplexes are particularly important for cell survival in the absence of functional p53.
Introduction
Telomeres are the repetitive DNA sequences and specialized proteins that cap the ends of linear eukaryotic chromosomes and protect them from degradation or fusion by DNA repair processes. Telomere integrity and length maintenance are essential for prolonged cell proliferation and are thought to play important roles in suppressing aging and cancer (Blackburn, 2000; Rodier et al., 2005).
Telomere length is generally maintained by telomerase, a reverse transcription that adds telomeric DNA repeats to chromosome ends. Telomere length homeostasis also depends on proteins that act at telomeres in cis to control the recruitment or access of telomerase (Smogorzewska and de Lange, 2004). Most human cells do not express telomerase. Because DNA replication machineries cannot fully copy DNA 3′ ends, such cells lose telomeric DNA with each S phase. When telomeres become critically short, the cells enter a permanent growth-arrested state termed senescence (Rodier et al., 2005). Both telomerase-expressing and telomerase-negative cells use a host of proteins to ensure a proper protective telomeric structure.
The precise structure of mammalian telomeres is not known. However, a “t-loop” structure, in which the 3′ overhang loops back and invades the telomeric DNA duplex, has been inferred by electron microscopy and biochemical experiments (Griffith et al., 1999). The t-loop model explains how telomeric ends are protected from recognition by DNA repair machineries. This protection is sometimes termed capping. Telomeres can become uncapped when critically short, presumably too short to form a t-loop, or when certain telomeric proteins are defective.
Several telomere-associated proteins are known to be important for telomere length regulation and capping (Smogorzewska and de Lange, 2004; Rodier et al., 2005). These include the direct telomeric DNA-binding proteins telomeric repeat binding factor 1 (TRF1), TRF2, and protector of telomeres 1 (POT1), proteins that associate with these telomeric DNA binding actors (e.g., TRF1-interacting protein 2 [TIN2], hRap1, and tankyrases), and a variety of proteins involved in other processes, such as DNA repair and recombination. Of the direct DNA-binding proteins, TRF1 binds double-stranded telomeric DNA and is an important regulator of telomere length (van Steensel and de Lange, 1997). In contrast, TRF2, which also binds double-stranded telomeric DNA, is more important for telomere capping (van Steensel et al., 1998; Karlseder et al., 1999; Smogorzewska and De Lange, 2002). POT1 binds the single-stranded 3′ overhang and is likely a terminal regulator of telomere length and end protection (Baumann and Cech, 2001).
TIN2 is an important telomere-associated protein because it binds both TRF1 (Kim et al., 1999) and TRF2 (Kim et al., 2004; Liu et al., 2004a; Ye et al., 2004a) and indirectly interacts with POT1 via the intermediary protein TPP1 (also termed pTOP [Liu et al., 2004b], PIP1 [Ye et al., 2004b], and TINT1 [Houghtaling et al., 2004]). TIN2 participates in the regulation of telomere length through its interactions with TRF1 (Kim et al., 1999) and TPP1 (Houghtaling et al., 2004; Liu et al., 2004b; Ye et al., 2004b). In addition, TIN2 appears to be a critical component in forming telomere complexes that function in end protection (Kim et al., 2004).
The functions of the three telomeric DNA-binding proteins (TRF1, TRF2, and POT1) are very likely coordinated. Perturbations of either TRF1 or TRF2 or their associated proteins POT1, hRap1, or TIN2 influence both telomere length and capping (van Steensel and de Lange, 1997; Kim et al., 1999, 2004; Loayza and De Lange, 2003; Iwano et al., 2004; Yang et al., 2005). These observations suggest that TRF1, TRF2, POT1, and TIN2 may function in the same pathway. Consistent with this idea, six proteins copurified in a large molecular weight complex (Liu et al., 2004a; Ye et al., 2004a; O'Connor et al., 2006). This complex may be the core molecular machinery that regulates mammalian telomeres. However, gel filtration identified a TRF2–hRap1 complex that also contains TIN2 and POT1 but not TRF1 (Ye et al., 2004a). Furthermore, when TRF1 is removed from telomeres, TIN2 and TPP1 remain at telomeres via increased association with TRF2 (Houghtaling et al., 2004). And, although POT1 was shown to be associated with TRF1 (Loayza and De Lange, 2003), POT1 and TRF2 also form a complex with telomeric DNA, and POT1 overexpression protects against loss of telomeric single-stranded DNA caused by expression of a dominant-negative TRF2 (DN-TRF2; Yang et al., 2005). Thus, there may be distinct telomeric complexes that participate in maintaining telomere length and capping.
It is not yet clear whether there is a single TIN2 complex that always contains TRF1, TRF2, and TPP1/POT1 and their interacting proteins or whether TIN2 forms multiple complexes, some of which contain TRF1, whereas others contain TRF2 (Houghtaling et al., 2004; Kim et al., 2004; Ye et al., 2004a). Furthermore, although it is hypothesized that telomeric complexes respond to telomere shortening, it is not known which telomere complexes are important for telomere capping, cellular senescence, and cell death. Here, we find that at least two major TIN2 complexes can be identified by immunoprecipitation and gel filtration of cell lysates and that one of these is crucial for cellular senescence and cell survival in the absence of p53.
Results
TIN2 depletion disrupts TRF1 and TRF2 and causes cell death in the absence of p53 function
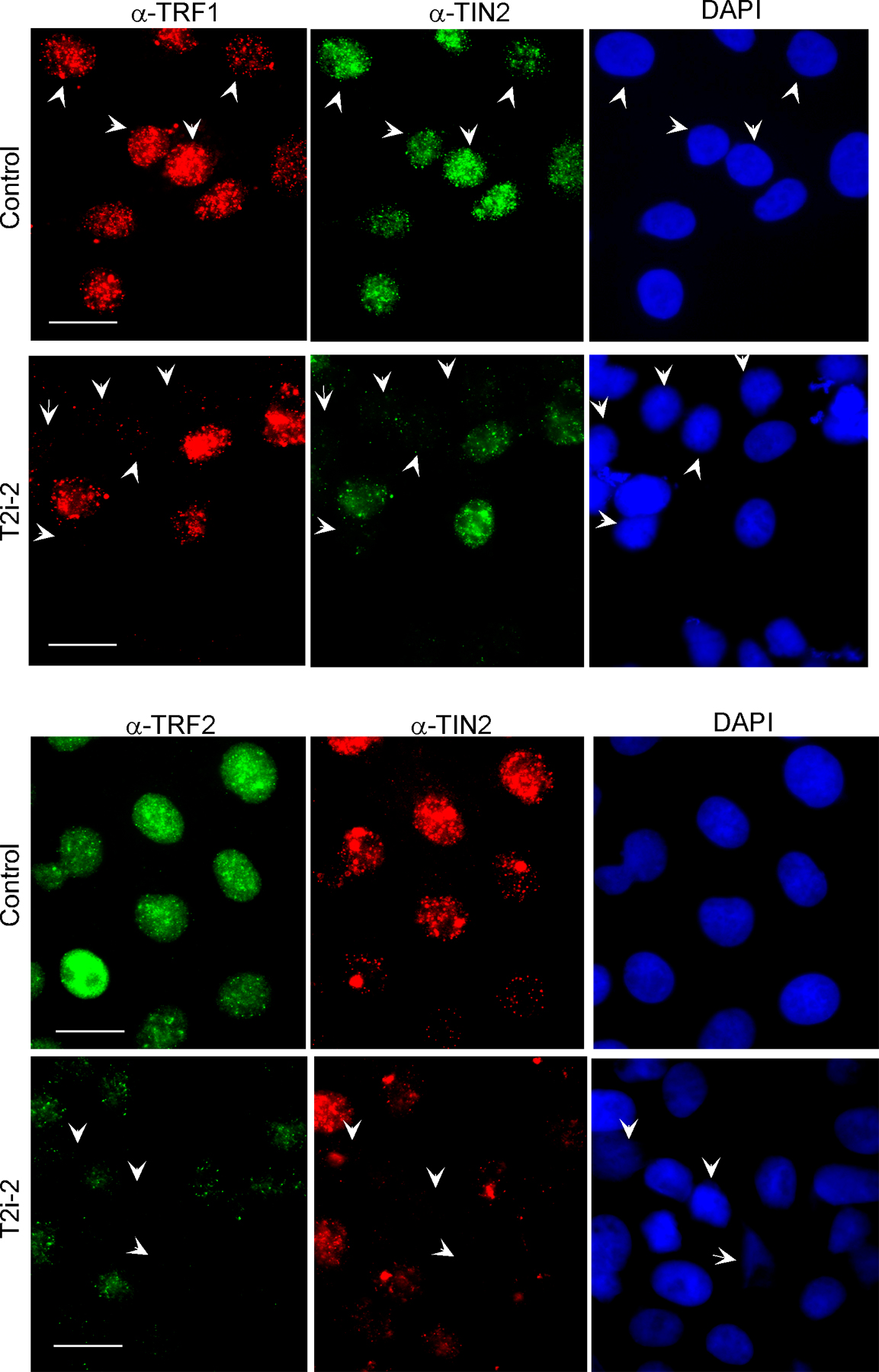
To understand the effects of TIN2 on cell fate and telomeric complex integrity, we used RNAi to ablate TIN2 expression. We expressed short hairpin RNAs (shRNAs; Brummelkamp et al., 2002) complementary to three regions of the TIN2 mRNA (T2i-1, T2i-2, and T2i-3) in HT1080 human fibrosarcoma cells. Two of the shRNAs, T2i-2 and T2i-3, reduced TIN2 protein levels by 70–80% (Fig. 1 a). T2i-2 and T2i-3 (not depicted) also reduced TRF1 and TRF2 protein levels (Fig. 1 b), which is consistent with the reported degradation of TRF1 upon removal from telomeres (Chang et al., 2003). T2i-2 also reduced focal immunostaining, which is indicative of telomeric localization of TIN2, TRF1, and TRF2 (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200710028/DC1). Thus, loss of TIN2 completely disrupted telomeres, as determined by the loss of TRF1 and TRF2 protein levels and telomeric occupancy.
Figure 1.

Effects of TIN2 ablation on TRF1, TRF2, and cell viability. (a) RNAi reduces TIN2 expression. HT1080 cells were transiently transfected with pSuper vectors expressing shRNAs corresponding to a nonexpressed (nonspecific [N/S]) mRNA or one of three distinct regions in the TIN2 mRNA (T2i-1, T2i-2, and T2i-3) as described in Materials and methods. Transfection efficiency varied from 60 to 70%. Cell lysates were analyzed 48 h later by Western blotting for TIN2 expression with α-tubulin used as a loading control. TIN2 protein levels were reduced to 23 and 24% of control levels by T2i-2 and T2i-3, respectively. Control cells expressed the N/S shRNA. (b)TIN2 reduction decreases TRF1 and TRF2 protein expression. Lysates from HT1080 cells transfected with the N/S (control) or T2i-2 pSuper vectors described above were analyzed for TIN2, TRF1, TRF2, and α-tubulin by Western blotting. The protein band intensities were analyzed as described in panel a. TIN2 and TRF2 protein levels were reduced to 16 and 13%, respectively, of control levels. (c) TIN2 reduction induces apoptosis in human tumor cells. HT1080 cells were transiently transfected with pSuper vectors containing no insert (vector) or N/S (control), T2i-1, T2i-2, or T2i-3 shRNAs and analyzed 48 h later for apoptosis as described in Materials and methods. Transfection efficiency was 60–70%. Where indicated, the caspase inhibitor ZVAD (100 μM) was added 8 h after transfection. 200 cells were analyzed for apoptosis in three independent experiments. Error bars represent the standard deviation. (d) TIN2 reduction induces apoptosis in normal human cells. Normal human fibroblasts (HCA2 and WI-38) were transiently cotransfected with pSuper vectors containing no insert (vector) or T2i-1 or T2i-2 shRNAs, and a lenti-GFP vector at a ratio of 10:1. 48 h later, GFP positive cells were analyzed for apoptosis as described in Materials and methods. Where indicated, WI-38 cells were first infected with a retrovirus expressing GSE-22, selected, and then infected with the pSuper and GFP vectors. 200 cells were analyzed in three experiments. Error bars represent the standard deviation. (e) GSE-22 inactivates p53 activity in normal cells. WI38 cells were infected with a retrovirus expressing GSE-22 or an insertless virus, selected, plated, and irradiated with a 10-Gy x ray. The cells were fixed 19 h later and immunostained for p21 and p53. Bars, ∼10 μM.
After the loss of telomeric TIN2, TRF1, and TRF2, T2i-2 and T2i-3 induced caspase-dependent cell death, which is indicative of apoptosis (Fig. 1 c). Cell death was not observed when cells expressed an insertless vector, a nonspecific shRNA or T2i-1, which did not reduce TIN2 expression (Fig. 1, a and c). T2i-2 also induced apoptosis in primary human fibroblasts (strains HCA2 and WI-38; Fig. 1 d). Strikingly, inactivation of p53 by GSE-22, a short peptide that disrupts p53 tetramerization and causes accumulation of monomeric p53 in these cells (Gudkov et al., 1993), failed to rescue WI-38 cells from T2i-2–induced apoptosis (Fig. 1 d). Loss of p53 activity was confirmed by the absence of detectable p21 immunostaining and enhanced p53 immunostaining in x-irradiated GSE-22–expressing but not control cells (Fig. 1 e). Thus, TIN2 was essential for TRF1 and TRF2 stability and occupancy at telomeres as well as the viability of normal and tumor-derived human cells, regardless of p53 status. This result suggests that the loss of TIN2 promotes a seriously aberrant telomeric structure that induces cell death even in the absence of p53, which is similar to the effects of telomere disruption caused by expression of a mutant telomerase template RNA (Li et al., 2004).
TIN2 subcomplexes are formed in cells
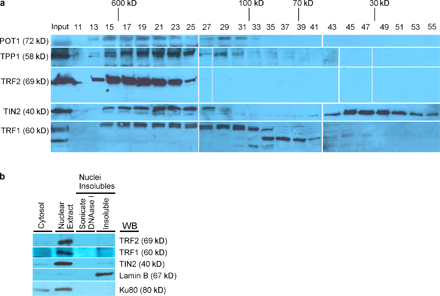
To gain insight into physiologically relevant TIN2 complexes, we prepared nuclear extracts from HeLa cells using a modified Dignam protocol (Dignam et al., 1983). The majority of endogenous TRF1 is extracted by this high salt (>0.3 M KCl) method (Okabe et al., 2004). To detect TIN2 complexes, the extracts were dialyzed into buffer approximating physiological ionic strength and then fractionated by size-exclusion chromatography. The resulting fractions were analyzed by Western blotting (Fig. 2, a and b).
Figure 2.

TIN2-mediated complexes. (a) Endogenous TIN2 complexes in HeLa nuclear lysate. Telomeric proteins in HeLa nuclear extracts (2. 5 ml) were fractionated on a Sepharcryl S-300 HR column and immunobloted for the indicated proteins. 20 μl aliquots of the indicated fractions were loaded. Eluting fractions were collected from the column after the void volume (No. 13). Molecular sizes indicated were derived from a standard curve based on the elution standards as described in Materials and methods. (b) Western blotting signals in Fig. 2 a were quantified with the Multi-Gauge program. Relative band intensities for each protein were plotted. (c) TIN2-complexes. We prepared lysates from HT1080 cells that transiently expressed V5-tagged POT1 and stably expressed Flag-TIN2 (lanes 2, 4, 6, and 8) or HA-TRF1 (lanes 1 and 10), or both Flag-TIN2 and HA-TRF1 (lanes 3, 5, 7, 9, and 11; Kim et al., 1999). We isolated TIN2 complexes using Flag, V5, and HA antibodies and analyzed the lysates (10%, input) and immunoprecipitates (IP) for the indicated proteins by Western blotting (WB). (d) Proposed TIN2 complexes. Complex A, TRF1–TIN2; complex B, TIN2–TRF2/hRap1–TPP1/POT1; complex C, TRF1–TIN2–TRF2/hRap1–TPP1/POT1.
We detected TIN2 across a broad range of molecular mass, from ≤1.5 MD to ∼100 kD in fractions 13–33 (Fig. 2 a). TRF1 eluted in two distinct peaks, coeluting with TIN2 in fractions 15–17 and 27–33. All six telomere-associated proteins (TRF1, TIN2, TRF2, hRap1, POT1, and TPP1) cofractionated in fractions 15–17, which is consistent with the large complex termed shelterin (de Lange, 2005) and referred to here as complex C. We also observed a protein complex containing TIN2, TRF2, hRap1, POT1, and TPP1 but lacking TRF1 (Fig. 2 a, fractions 19–23), which is consistent with previous studies (Ye et al., 2004a; O'Connor et al., 2006). We term this complex complex B. The complex B we detected here differs somewhat from that previously reported (termed fraction 1, isolated by a two-step chromatographic protocol; O'Connor et al., 2006) in which TPP1 was only marginally detectable. Our one-step protocol, in contrast, showed that TPP1 was a prominent component of complex B. We quantified the signals from the Western blots to illustrate the elution profiles of the six telomeric proteins (Fig. 2 b).
Under our experimental conditions, we observed an additional telomere protein complex (complex A) containing significant amounts of TIN2 and TRF1 in the lower molecular mass range (Fig. 2 a, fractions 27–33). Fractions 27–31 also contained low levels of a TPP1–POT1 complex, as previously reported (Xin et al., 2007). However, in contrast to our findings (Fig. 2 a), these studies (Liu et al., 2004a; Ye et al., 2004a; O'Connor et al., 2006) did not detect complex A in the same molecular mass range, possibly because of the lower salt used for extraction or different gel-filtration conditions. Complex A does not appear to arise from larger complexes (e.g., complex C) as a consequence of high salt extraction because there is no evidence of dissociated or degraded monomeric proteins (TRF2, POT1, TPP1, and hRap1) in the low molecular weight fractions (Fig. 2a, fractions 35–49). This result is most consistent with a bona fide TRF1–TIN2 complex (complex A) existing in vivo. As expected, we detected degraded TIN2 in fractions 57–65. We confirmed the absence of monomeric proteins (and the presence of degraded protein) using twice the amount of sample and a different gel filtration column (Superdex S-200, 600–10 kD) that is better at fractionating low–molecular weight proteins (Fig. S2 a, available at http://www.jcb.org/cgi/content/full/jcb.200710028/DC1). This fractionation showed that low molecular ranges (Fig. S2 a, fractions 29–55) contained only degradation products of TIN2 and TRF1. TRF1 is degraded when dissociated from telomeres (Chang et al., 2003). Moreover, the high salt nuclear extracts contained >90% of the TRF1, TRF2, and TIN2, which suggests that most of these proteins were released from telomeres (Fig. S2 b). These results suggest that complex A (TRF1–TIN2) might be degraded upon dissociation from telomeres by high salt (0.5 M); in lower salt (0.3–0.4 M), this complex is not released from telomeres and is therefore not detected.
Our chromatographic results are consistent with the existence of three heteromeric complexes (complexes A, B, and C), although, taken alone, the results are also consistent with each of the proteins being assembled into homomeric complexes of approximately equal size.
To discriminate between these possibilities and more directly probe telomeric protein assemblies without high salt extraction, we overexpressed epitope-tagged POT1, TIN2, or TRF1 (V5-tagged POT1, Flag-tagged TIN2, and HA-tagged TRF1) in HT1080 cells. We prepared cell lysates using physiological ionic strength (0.15 M NaCl), which does not liberate endogenous TRF1 and TIN2 as determined by Western blotting (unpublished data). We expected anti-V5 to immunoprecipitate complexes B and C, anti-HA to precipitate complexes A and C, and anti-Flag to precipitate all three complexes.
The major complex precipitated by HA antibodies (Fig. 2 c, lanes 10 and 11) was complex A (TRF1–TIN2). A minor complex containing TRF1, TIN2, and TPP1 was also detectable. These findings suggest that most TRF1 proteins formed a complex with TIN2 under the immunoprecipitation conditions of RIPA buffer and physiological salt.
V5 antibodies precipitated a major complex of POT1, TPP1, TIN2, TRF2, and hRap1 (complex B) regardless of whether TRF1 was overexpressed and a minor complex containing TRF1, TIN2, TPP1, POT1, TRF2, and hRap1 (complex C). Because POT1 does not interact directly with TRF2 (Loayza and De Lange, 2003), this result suggests that POT1/TPP1 (Xin et al., 2007) forms a more stable complex with TIN2–TRF2/hRap1 than with TIN2–TRF1.
When Flag-TIN2 was the precipitating protein, Flag antibodies (Fig. 2 c, lanes 6 and 7) precipitated more complex B in the absence of overexpressed TRF1 than in its presence. This result suggests that TRF1 and TRF2 compete with TIN2 to form two separable complexes, A and B. In addition, we detected a small amount of complex C in lysates from cells that overexpressed TRF1 and TIN2. In the Discussion, we propose possible explanations for the relatively minor abundance of complex C after immunoprecipitation compared with nuclear extraction.
Together, the fractionation and immunoprecipitation data indicate the existence of distinct TIN2 complexes in cells. For simplicity, we term these complexes as follows: TRF1–TIN2, complex A; TIN2–TRF2/hRap1–TPP1/POT1, complex B; TRF1–TIN2–TRF2/hRap1–TPP1/POT1, complex C (Fig. 2 d).
TIN2 mutants have distinct effects on TIN2 complexes
TIN2-13 and TIN2-15C are TIN2 truncation mutants with different binding capabilities (Fig. 3 a). TIN2-15C lacks TRF1 binding (Kim et al., 1999). It retains the TRF2-binding domain but interacts less strongly than wild-type TIN2 with TRF2 (Kim et al., 2004). Immunoprecipitation of lysates from cells expressing epitope-tagged TIN2-15C and POT1 showed that TIN2-15C retains TPP1/POT1 binding (Fig. 3 b). Reciprocally, TIN2-13 retains TRF1 binding (Kim et al., 1999) but lacks TRF2 (Kim et al., 2004) and TPP1/POT1 (Ye et al., 2004b) binding. We used these mutants to identify TIN2 complexes in cells and determine their role in the cell death caused by complete loss of TIN2 in p53-deficient cells.
Figure 3.

TIN2 complexes disrupted by TIN2 mutants. (a) TIN2 deletion mutants. Wild type TIN2 (aa 1–354) showing N-terminal (N-term), TRF1-interaction (TRF1-Int), and C-terminal (C-term) domains, and deletion mutants TIN2-13 (aa 180–354) and TIN2-15C (aa 1–257). (b) Interaction of TIN2-15C with POT1/TPP1. Lysates from HT1080 cells that transiently expressed Myc-TIN2-15C, V5-POT1, and HA-TRF1 were precipitated using anti-Myc or V5 antibodies. Unprecipitated lysates (10%) and the immune precipitates (IP) were analyzed for POT1-V5, TIN2-15C, TRF2, and HA-TRF1 by Western blotting (WB). (c) TIN2–TRF2–POT1 complexes disrupted by TIN2-15C but not TIN2-13. Lysates from HT1080 cells transiently expressing wild-type TIN2 (WT-TIN2), control vector, TIN2-13, or TIN2-15C and V5-POT1 were immunoprecipitated (IP) by anti-V5. The lysates (15%, input) and immune precipitates were analyzed for TIN2, V5, TRF2, and α-tubulin by Western blotting (WB; lanes 1–8). The band indicated by an asterisk is a degraded WT-TIN2 product. (d) TRF1–TIN2 complexes disrupted by TIN2-13 but not TIN2-15C. Lysates from HT1080 cells transiently expressing WT-TIN2, TIN2-13, TIN2-15C, and HA-TRF1 were immunoprecipitated (IP) by anti-HA. The lysates (15%, input) and precipitates were analyzed for TIN2, TRF2, HA, and α-tubulin by Western blotting (WB; lanes 9–18). The precipitating heavy chain is indicated (IgG). The band indicated by an asterisk is a degraded WT-TIN2 product.
We first tested the effects of TIN2 mutants on complex A, B, and C formation. Based on their composition (Fig. 2), we predicted that TIN2-13, which lacks TPP1 and TRF2 binding (Liu et al., 2004b; Ye et al., 2004b), would perturb complexes A and C, whereas TIN2-15C, which lacks TRF1 binding, would perturb complexes B and C (Fig. 3 a). We expressed V5-POT1 plus wild-type TIN2, TIN2-13, or TIN2-15C in HT1080 cells and immunoprecipitated cell lysates us)ing V5 antibodies (Fig. 3 c, lanes 5–8).
When TIN2-13 was expressed, V5 antibodies did not precipitate TIN2-13, and the level of endogenous TIN2 was not altered in the precipitate (note that anti-V5 precipitated less POT1 in Fig. 3 c, lane 6; compare lanes 6 and 7). This result indicates that V5-POT1 mainly precipitated TIN2 B complexes and that TIN2-13 does not disrupt complex B, as predicted. This result also confirms the inability of TIN2-13 to interact with TRF2 (Kim et al., 2004) and TPP1/POT1 (Ye et al., 2004b).
In contrast, when TIN2-15C was expressed, V5 antibodies precipitated TIN2-15C, and the presence of TIN2-15C reduced the level of endogenous TIN2 in the precipitate (Fig. 3 c, lane 8). Furthermore, TIN2-15C expression reduced the level of TRF2 in the precipitate (Fig. 3 c, compare lanes 6 and 8), indicating that TIN2-15C inhibits the TIN2–TRF2 interaction. This result suggests that TIN2-15C disrupts complex B formation, as predicted, and is consistent with the finding that TIN2-15C reduces TRF2 stability in cells and dissociates TRF2 from telomeres (Kim et al., 2004). TIN2-15C or DN-TRF2 slightly reduced (84 and 89%, respectively) the length of the 3′ single-stranded telomeric overhang in normal HCA2 cells (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200710028/DC1). Inactivation of TRF2 by DN-TRF2 or TRF2 deletion reduced or slightly increased the 3′ overhang length in cells that were deficient in p53 or in both p53 and ligase IV (van Steensel et al., 1998; Celli and de Lange, 2005). Thus, TIN2-15C and DN-TRF2 may have similar effects on 3′ overhang length regulation in normal cells.
We also expressed HA-TRF1 plus wild-type TIN2, TIN2-13, or TIN2-15C and immunoprecipitated lysates using HA antibodies (Fig. 3 d, lanes 13–16). TIN2-13 reduced the binding of endogenous TIN2 to HA-TRF1 (Fig. 3 d, compare lanes 13 and 15), but TIN2-15C did not disrupt this interaction (compare lanes 13 and 16). Thus, as predicted, TIN2-13 disrupted TRF1–TIN2 complexes (complex A) but TIN2-15C did not.
We conclude that the TIN2 mutants TIN2-13 and TIN2-15C disrupt different TIN2 complexes, at least by immunoprecipitation analysis, and thus favor the formation of different telomeric complexes when expressed in cells.
Effects of TIN2 mutants in cells
To determine the biological effects of the TIN2 complexes, we expressed GFP (control), TIN2-13, or TIN2-15C in primary human fibroblasts (strain HCA2). We also analyzed these cells after p53 inactivation, achieved by expressing GSE-22 (Gudkov et al., 1993) or a p53 shRNA (sh-p53).
We first tested presenescent or replicatively senescent cells with or without p53 inactivation and measured apoptotic cell death using a sensitive cumulative assay (Fig. 4 a; Goldstein et al., 2005). We define presenescent cultures as those that retain >70% of their replicative life span and thus contain cells with mostly long, functional telomeres. In contrast to the robust cell death caused by complete loss of TIN2 (Fig. 1, c and d), TIN2-13 or TIN2-15C caused little or no cell death when expressed in presenescent or senescent cells with normal p53 function (Fig. 4 a). However, when p53 was inactivated, the fraction of apoptotic cells increased in both cell populations (Fig. 4 a). Notably, coexpression of TIN2-13 and GSE-22 elevated apoptosis only about twofold above the levels in control cells (expressing GFP and GSE-22), whereas coexpression of TIN2-15C and GSE-22 increased cell death 8- to 10-fold above control levels (Fig. 4 a). Coexpression of TIN2 mutants with sh-p53 gave very similar results (Fig. 4 a). These findings suggest that disruption of the TIN2–TRF2 or TIN2–TPP1/POT1 interaction (by TIN2-15C) has more severe consequences for cell survival than disruption of the TRF1–TIN2 interaction (by TIN2-13). They also indicate that disruption of telomeric complexes B and C (by TIN2-15C) is more deleterious than disruption of complex A and C (by TIN2-13). Moreover, the cell death caused by disrupting these complexes does not require p53 function.
Figure 4.

Effects of TIN2 mutants in presenescent and senescent cells. (a) Cell death caused by TIN2-15C in presenescent cells. Where indicated, presenescent (P) HCA2 cells were first infected with a retrovirus expressing GSE-22 and a lentivirus expressing sh-p53 and selected for 2–3 d. Cells were then infected with lentiviruses expressing GFP, TIN2-13, or TIN2-15C. Where indicated, senescent (S) HCA2 cells were first infected with lentiviruses expressing GFP, TIN2-13, or TIN2-15C and then infected with a lentivirus expressing GSE-22. Cells were scored for apoptotic death by assessing release of cytochrome c from mitochondria as described previously (Goldstein et al., 2005). 300 cells were scored in two or three independent experiments. Error bars represent the standard deviation. (b) Effects of TIN2-15C on senescent cells reactivated by GSE-22. 5 × 104 senescent HCA2 cells were infected with lentiviruses expressing GFP (control), TIN2-13, TIN2-15C, or DN-TRF2 and then infected with lenti-GSE-22. Colonies were stained at the indicated days after infection. (c) Expression levels of TIN2 mutants and DN-TRF2. Lysates from presenescent cells infected with the indicated lentiviruses were analyzed for TIN2 and TRF2 by Western blotting (WB). (d) Cell death induced by TIN2-15C in p53-positive and -negative cancer cells. p53-positive (breast MCF-7 and prostate LN-Cap) cancer cells and p53-negative (breast MDA-MB-231 and MDA-MB-157, prostate PPC-1, and fibrosarcoma HT1080) cancer cells were transiently transfected with pIRES2-eGFP vectors expressing no insert (vector), TIN2-13, or TIN2-15C. 48 h later, the cells were analyzed for GFP fluorescence. GFP-positive cells were scored for cell death by collapse of the mitochondrial membrane potential as described previously (Davalos and Campisi, 2003). 200 GFP-positive cells were scored in two or three independent experiments for each transfection. Error bars represent the standard deviation.
Late generation mice lacking both telomerase and p53 progress very few generations relative to telomerase deficiency alone, which suggests the existence of a telomere-dependent cell death pathway that does not require p53 (Chin et al., 1999). To further test the idea that disruption of different telomeric complexes differentially affects the survival of cells that lack p53 function, we used lentiviral vectors to express TIN2 mutants and GSE-22 in replicatively senescent HCA2 fibroblasts. These senescent cells have several short, dysfunctional telomeres (Zou et al., 2004), similar to those in late-generation telomerase-deficient mice. Moreover, because HCA2 cells express low levels of p16 (unpublished data), which inhibits the proliferation of cells that lack p53 function, p53 inactivation causes senescent HCA2 cells to resume proliferation, as reported for other low p16-expressing human fibroblast strains (Beausejour et al., 2003). We measured the ability of senescent HCA2 cells to form colonies after p53 inactivation and disruption of telomere complexes by TIN2 mutants. We compared the effects of TIN2 mutants to the effect of DN-TRF2, which uncaps telomeres and induces p53-dependent cell death (Fig. 4 b; Karlseder et al., 1999).
Senescent cells readily formed colonies upon expression of GSE-22, as expected (Beausejour et al., 2003). Subsequent expression of GFP (control), TIN2-13, or DN-TRF2 had no effect on colony formation after 12 (Fig. 4 b) or 30 d (not depicted). In contrast, subsequent expression of TIN2-15C initially had no effect on cell proliferation (for 3–10 d; not depicted) but then dramatically reduced colony formation. The differences between TIN2-15C and TIN2-13 or DN-TRF2 in supporting colony formation could not be explained by differences in protein expression levels because TIN2-13 and DN-TRF2 were expressed more robustly than TIN2-15C (Fig. 4 c).
Together, these findings suggest that p53-dependent cell growth requires both TIN2 A and B complexes. In contrast, the growth of cells that lack p53 function does not require TIN2 A complexes, which are disrupted by TIN2-13 (Fig. 3), but does require TIN2 B complexes, which are disrupted by TIN2-15C (Fig. 3). DN-TRF2, which lacks the TRF2 DNA-binding domain and causes p53-dependent cell death (Karlseder et al., 1999), is clearly not equivalent to TIN2-15C in biological activity, possibly because it does not affect POT1 function in complex B (see Discussion).
We confirmed the selective sensitivity of p53-deficient cells to TIN2-15C using several human cancer cell lines. These included HT1080 fibrosarcoma cells (which were reported to undergo no cell death upon expression of DN-TRF2; Karlseder et al., 1999), MDA-MB-231, and MDA-MB-157 breast cancer cells, and PPC-1 prostate cancer cells. We used a transfected a pIRES2-eGFP vector (Kim et al., 2004) and measured apoptotic cell death. TIN2-15C expression induced significant cell death within 48 h in all these p53-deficient cancer cells (Fig. 4 d), whereas TIN2-13 was two- to eightfold less effective, depending on the cell line (Fig. 4 d). Compared with normal cells in which p53 was inactivated by GSE-22, apoptosis was more robust in the cancer cells, being evident as early as 18 h after transfection, presumably because cancer cells harbor multiple mutations that disrupt cell growth and survival pathways. The TIN2 mutants caused much less cell death in p53-positive cancer cells (MCF-7 and LN-Cap; Fig. 4d). In all cases, TIN2-15C, and to a lesser extent TIN2-13, caused substantial cell death in p53-deficient cells (Fig. 4 d) but very little cell death in cells with wild-type p53 (Fig. 4 a).
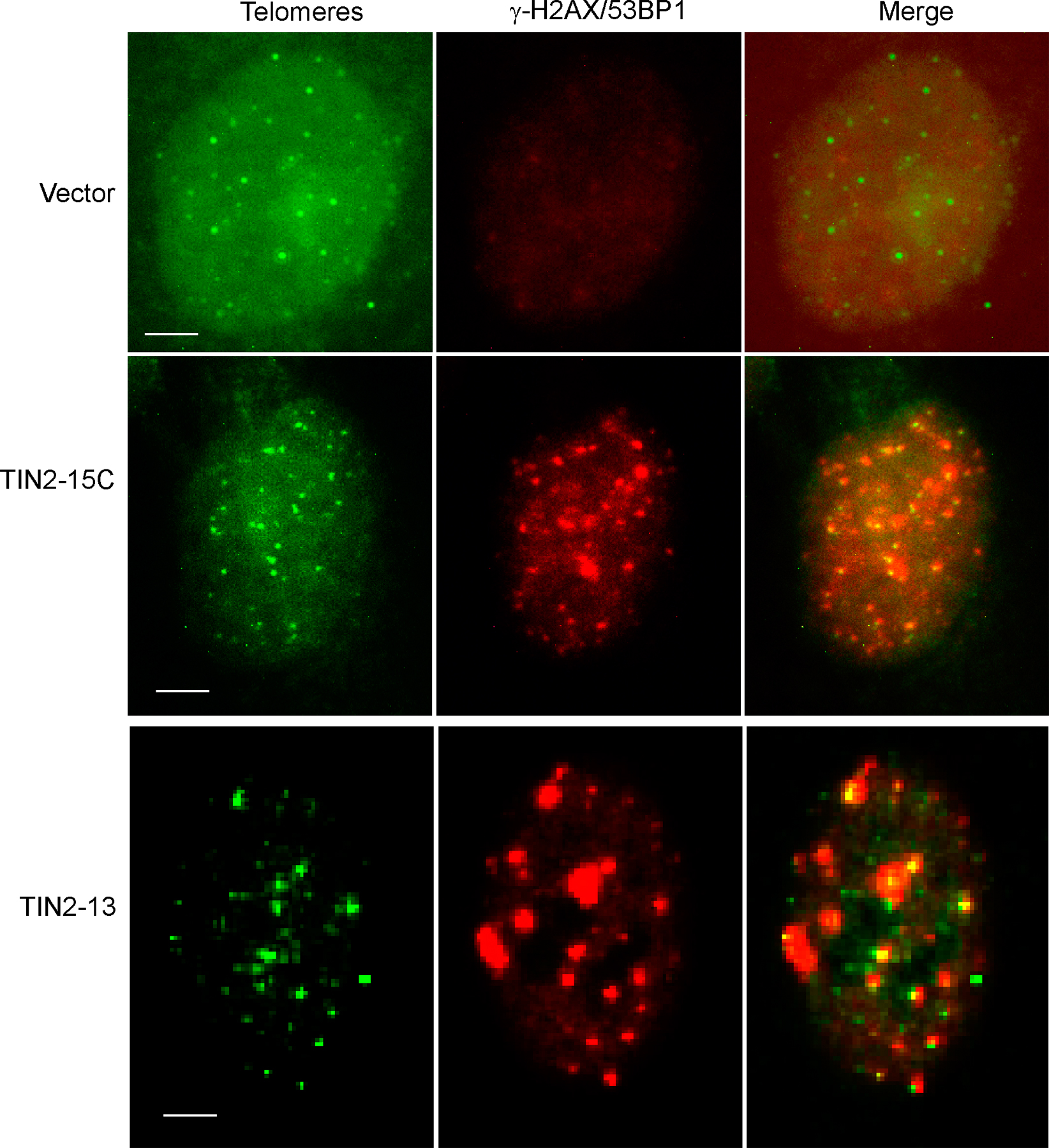
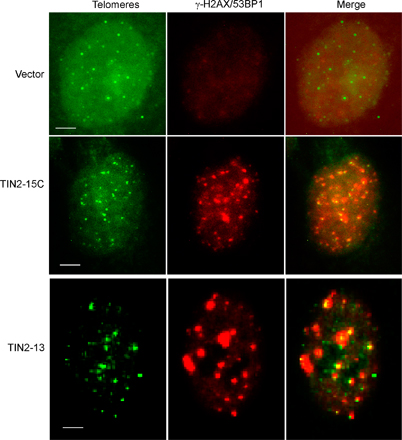
Normal cells responded to TIN2-15C by undergoing senescence. TIN2-15C, much more than TIN2-13, retarded cell proliferation (Fig. 5 a) and induced senescence-associated β-galactosidase (Fig. 5 b). Both mutants also induced 53BP1/γH2AX foci (d'Adda di Fagagna et al., 2003; Takai et al., 2003). Most of these foci (60–90%) colocalized with a telomeric DNA probe (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200710028/DC1), indicating that they marked dysfunctional telomeres. The TIN2 mutants induced 53BP1 foci in presenescent cells as well as replicatively senescent cells, which already have several telomeric 53BP1 foci. In all cases, TIN2-15C induced many more 53BP1 foci than TIN2-13 (Fig. 5 c), which suggests that TIN2-15C is more potent than TIN2-13 at uncapping telomeres. These results also suggest that B complexes are more important than A complexes for protecting cells from cell death.
Figure 5.

Effects of TIN2 mutants in normal cells. (a) TIN2-15C suppresses proliferation of normal cells. We infected presenescent HCA2 cells with lentiviruses expressing GFP, wild-type (wt) TIN2, TIN2-13, or TIN2-15C and determined the cell number the indicated number of days thereafter. Plotted are cumulative cell numbers versus days in culture. (b) TIN2-15C induces cellular senescence. HCA2 cells were infected with lenti-GFP or lenti-TIN2-15C and assessed 10 d later for senescence-associated β-galactosidase as described previously (Dimri et al., 1995). Replicatively senescent HCA2 cells were used as positive controls. Bars, ∼10 μm. (c) TIN2-13 and TIN2-15C induce damage foci in presenescent and senescent cells. Presenescent (P) and senescent (S) HCA2 cells were infected with lenti-TIN2-13 or lenti-TIN2-15C and stained 48 h later for expression of the mutant proteins and γH2AX or 53BP1 foci. 50–80 cells were scored in two or three independent experiments for each transfection. Error bars represent the standard deviation.
Chromosomal abnormalities induced by TIN2 mutants
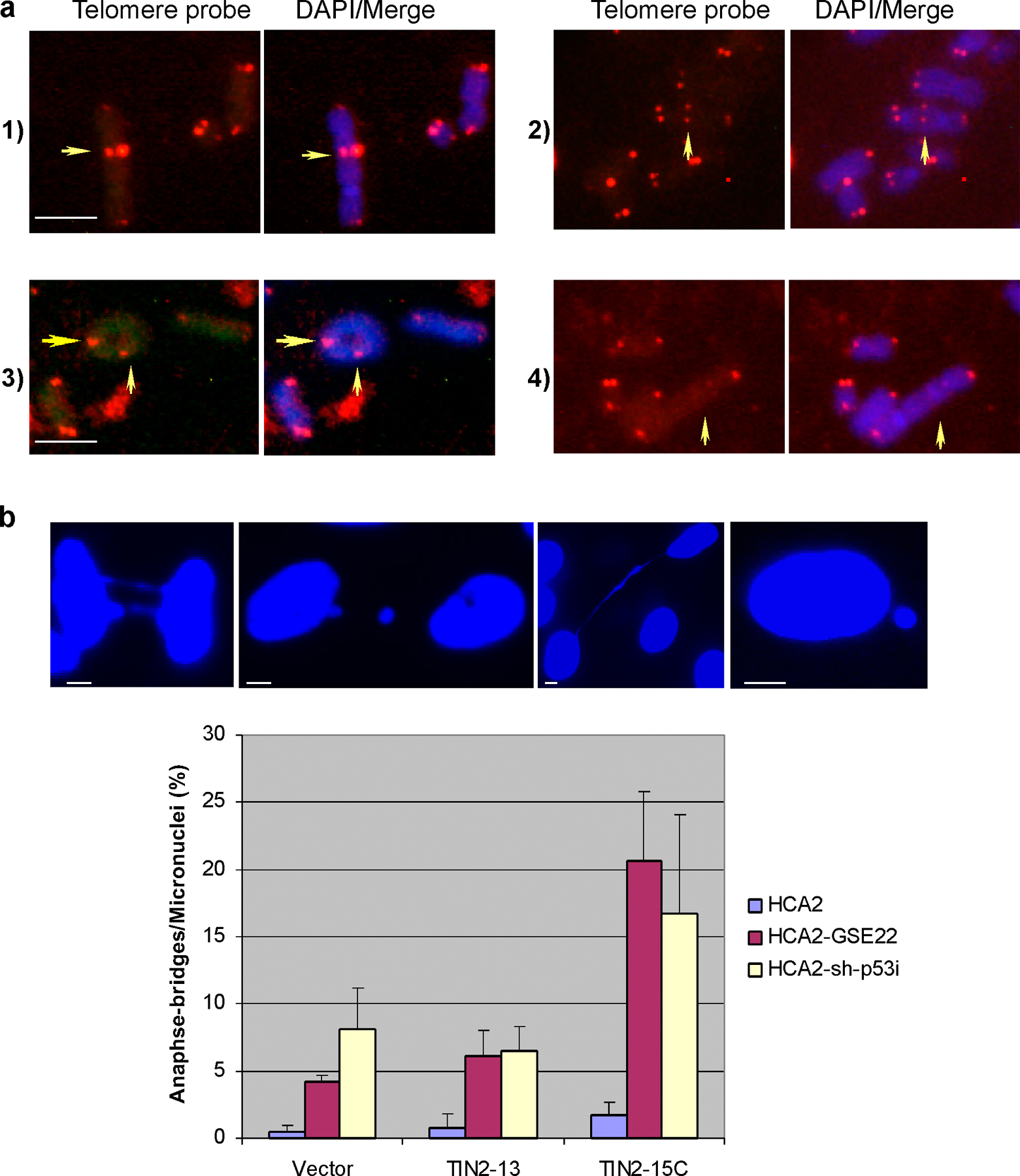
Although p53-deficient cancer cells underwent rapid cell death in response to TIN2-15C, p53 inactivation and TIN2-15C expression did not cause rapid cell death in presenescent or senescent normal human fibroblasts (in contrast to the effect of TIN2 depletion). Thus, additional events, such as cell cycle progression, chromosome fusion, and anaphase bridge formation might be necessary for the death of cells with lesions that inactivate p53 and complex B. To test this idea, we expressed GFP (control), TIN2-13, or TIN2-15C in HCA2 cells in which p53 was inactivated by GSE-22. Cells expressing TIN2-15C had a significantly more chromosome fusions (0.85 per metaphase) and anaphase bridges or micronuclei (∼21%) compared with cells expressing TIN2-13 (0.19 per metaphase and ∼6% anaphase bridges or micronuclei; Table I and Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200710028/DC1). We observed no chromosome fusions and ∼4% anaphase bridges or micronuclei in cells expressing GFP. Fluorescence in situ hybridization with a telomeric probe showed that most of the fused chromosomes in TIN2-15C–expressing cells possessed a telomeric signal (0.68 per metaphase), which is indicative of telomeric uncapping and subsequent fusion. These fusions are similar in type and level to those reported for cells expressing DN-TRF2 (van Steensel et al., 1998; Hockemeyer et al., 2005; Yang et al., 2005), which is consistent with findings that TIN2 and TRF2 interact and that TIN2-15C reduces telomeric TRF2 (Kim et al., 2004). A small fraction of fusions lacked telomeric signal (0.16 per metaphase), which is more common upon loss of POT1 function (Wu et al., 2006). These results suggest that TIN2-15C may cause the death of p53-deficient cells indirectly by driving chromosome fusions and mitotic catastrophe.
Table I. Telomeric fusions caused by expression of TIN2 mutants.
| Lentiviral vector | No. metaphases analyzed | No. chromosome fusions
|
Fusions per metaphase | Fusions per chromosome | |
|---|---|---|---|---|---|
| +Tel | –Tel | ||||
| GFP | 53 | 0 | 0 | 0 | 0 |
| TIN2-13 | 53 | 7 | 3 | 0.18 | 0.0034 |
| TIN2-15C | 61 | 42 | 10 | 0.85 | 0.0175 |
Presenescent HCA2 cells were infected with a retroviruses expressing GSE-22, selected, and infected with lentiviruses expressing GFP, TIN2-13, or TIN2-15C. The cells were treated with colcemid, and telomeres on metaphase chromosomes were identified by PNA-FISH as described in Materials and methods. Chromosome fusions were scored by fluorescence in situ hybridization for the presence (+Tel) or absence (−Tel) of telomeric DNA at the site of fusion.
Telomerase does not rescue TIN2-15C lethality
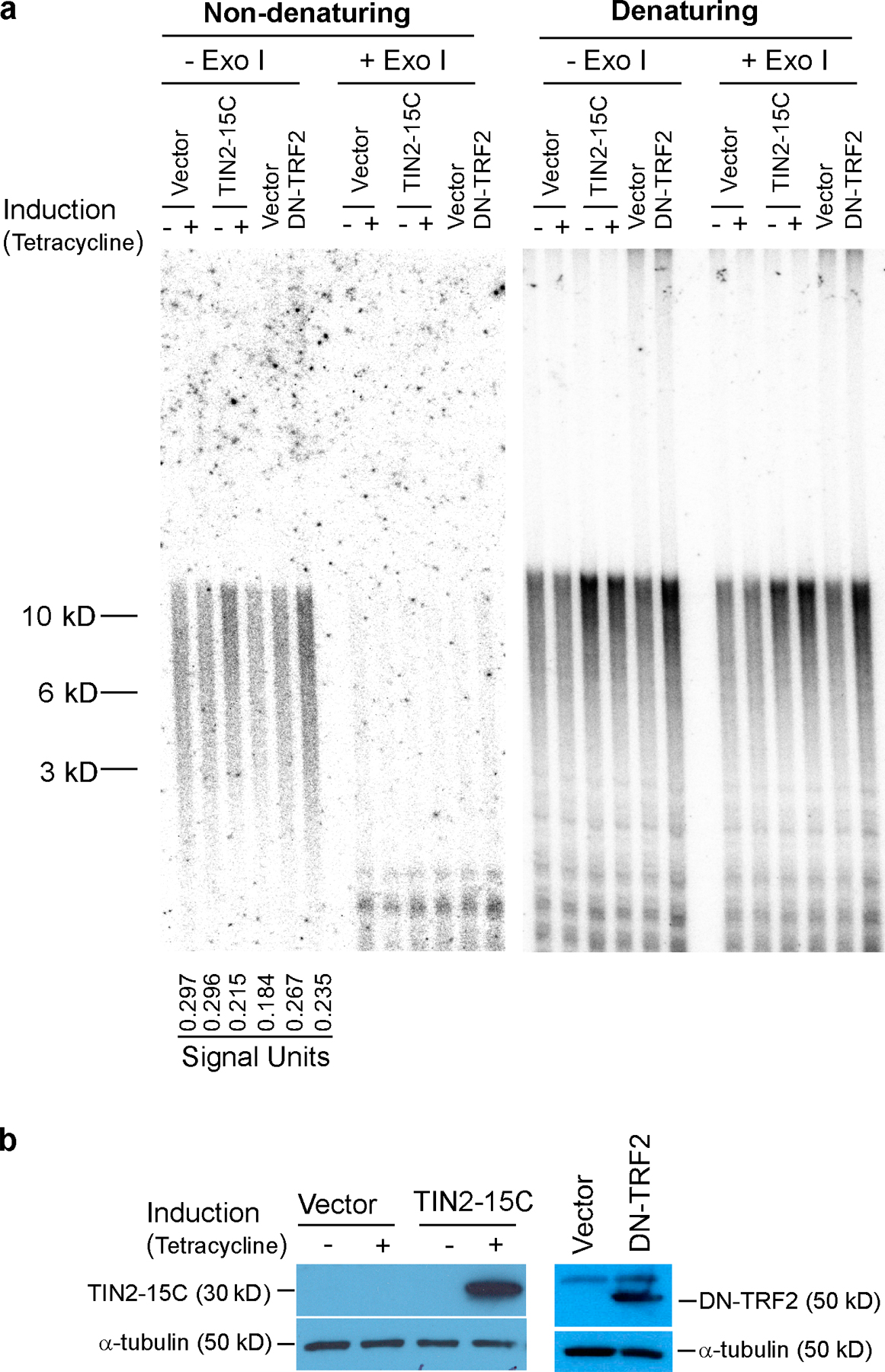
Telomerase can rescue senescent cells in which p53 has been inactivated from crisis caused by dysfunctional telomeres. To determine whether telomerase can similarly rescue p53-deficient senescent cells from the dysfunctional telomeres caused by TIN2-15C, we coexpressed the telomerase catalytic subunit (hTERT) and TIN2-15C in senescent HCA2 cells in which p53 was inactivated by GSE-22. Telomerase prevented the crisis that limits the proliferative capacity of these cells to ∼20 population doublings (Beausejour et al., 2003). However, coexpression of telomerase and TIN2-15C did not rescue the cells from loss of proliferative potential (Fig. 6, a and b). These results suggest that TIN2-15C causes cell death by disrupting telomeric structure and capping rather than by shortening telomere length.
Figure 6.

Effects of telomerase and a proposed model. (a) Telomerase does not rescue cell death by TIN2-15C. 5 × 104 senescent cells were infected with lentiviruses expressing GFP (control), the telomerase catalytic subunit hTERT, TIN2-15C, or both TIN2-15C and hTERT. The cells were then infected with lenti-GSE-22. After 2 d, the cells were subcultured and plated for colony formation. Colonies were fixed and stained 61 d later. (b) Colonies were counted 5, 11, and 61 d after plating. Colonies were scored in two or three independent experiments for each transfection. Error bars represent the standard deviation. (c) Proposed model for TIN2 complexes at telomeres. Complex A and B cooperate, albeit at different positions on telomeres, to form t-loops or other terminal structures. Complex B may localize preferentially or uniquely near t-loop junctions, whereas A complexes may modulate the tertiary structure of telomeres and promote B complex stability. TIN2-13 disrupts complex A, thereby reducing the tertiary telomeric structure and destabilizing B complexes, resulting in partial or mild disruption of t-loops and telomere uncapping. TIN2-15C directly disrupts B complexes, resulting in severe disruption of t-loops and telomere uncapping. Cells expressing wild-type p53 and TIN2-13 or TIN2-15 undergo growth arrest, whereas cells lacking functional p53 undergo cell death. In both cases, the consequences of TIN2-15C expression are more severe than that of expressing TIN-13.
Discussion
TIN2 telomeric complexes
Six telomere-associated proteins (TRF1–TIN2–TPP1–POT1–TRF2–hRAP1) have been isolated as a single soluble complex (Liu et al., 2004a; Ye et al., 2004a). TIN2 occupies a unique position in this complex because it interacts directly with TRF1 and TRF2 and indirectly (through TPP1) with POT1. In these studies, TRF2 complexes contained TIN2 and POT1 but not TRF1, which suggests that TRF1 is not required for the TIN2–TRF2/hRap1–TPP1/POT1 interaction (Houghtaling et al., 2004). TRF1 and TRF2 bind noncooperatively along telomeric repeats and have high off-rates in vitro (Bianchi et al., 1999). These results suggest that TIN2 may form dynamic TRF1 or TRF2 complexes at telomeres. We identified three major soluble TIN2 complexes (TRF1–TIN2, complex A; TIN2–TRF2/hRap1–TPP1/POT1, complex B; and TRF1–TIN2–TRF2/hRap1–TPP1/POT1, complex C) from nuclear extracts containing endogenously expressed proteins, which supports the idea that TRF1 and TRF2 complexes are separable.
Previously reports (Liu et al., 2004a; Ye et al., 2004a) did not detect complex A, which suggests that our nuclear extraction protocol, which used higher salt concentrations, was more effective at dissociating TRF1–TIN2 complexes from telomeres or the nuclear matrix. TRF1 cross-reactivity was detected as multiple bands in low molecular weight fractions, which is likely due to proteolysis after release from telomeres (Chang et al., 2003) or the nuclear matrix (Okabe et al., 2004). In addition, we did not detect complex C as a major complex in cells that overexpressed TRF1 and TIN2 and were extracted by physiological salt. Complex C formation may require telomeric DNA, t-loop formation, and/or posttranslational modifications to TRF1 or matrix-associated TRF1.
We used two TIN2 mutants with distinct abilities to disrupt the major TIN2 complexes (A and B). TIN2-13 affected the TRF1-TIN2 complex A, whereas TIN2-15C affected TRF2 complexes. If the major telomeric complex was a single entity (complex C), TIN2-13 and TIN2-15C should disrupt this complex similarly. However, our results are more consistent with the existence of two separable TIN2 complexes, which may have different functions in cells.
TIN2 complexes may function differently
In normal cells, telomeres shorten with each division, eventually causing senescence. TIN2-15C, which disrupts complex B, removes TRF2 from telomeres and induces DNA damage foci but TRF1 remains at telomeres (Kim et al., 2004). DN-TRF2 also removes TRF2 and induces DNA damage foci containing TRF1 (Takai et al., 2003). Senescent cells with short telomeres and telomeric DNA damage foci frequently lack TRF2 but not TRF1 (Herbig et al., 2004). Thus, short telomeres may preferentially attract A complexes, and senescent cells may be unable form enough complex B (TIN2–TRF2/hRap1–TPP1/POT1) because TIN2 is bound primarily in A (TRF1–TIN2) complexes. Alternatively, complexes A and B may have different preferred locations on the telomere or t-loop and cooperate in telomere capping.
TRF1–TIN2 complexes stimulate interactions between telomeric DNA tracts in vitro, which suggests that this complex modulates a tertiary telomeric structure (Kim et al., 2003). TIN2-13, which disrupts this complex (complex A), may decrease the complexity of the telomeric structure. However, deletion of TRF2 or POT1 causes telomere uncapping and chromosome fusion, which suggests that complex B is essential for telomere end protection (Hockemeyer et al., 2006; Wu et al., 2006). Consistent with this view, we found that TIN2-15C, which disrupts complex B, uncapped telomeres and caused telomeric fusions and anaphase bridges. Thus, we favor the hypothesis that TRF1–TIN2 (A) complexes and TIN2–TRF2/hRap1–TPP1/POT1 (B) complexes have different locations at telomeres and cooperate to form the telomeric cap (Fig. 6 c). B complexes are more important for ensuring a proper terminal or t-loop structure, whereas A complexes modulate the telomeric tertiary structure and enhance the stability and function of complex B. Although TIN2-13 induces some telomere uncapping and telomeric fusion, the effects of TIN2-13 are less pronounced than those of TIN2-15C, which suggests that complex A supports the functions of complex B.
Alternatively, complexes A and B may cooperate to form the six-protein complex C (also termed shelterin; de Lange, 2005), and this complex may be the essential entity for telomere capping and cell survival. TIN2-13 may be less efficient than TIN2-15C at disrupting complex C. TPP1 may be an important regulator of complex C formation because loss of TPP1 can reduce the TRF1–TRF2–Rap1 interaction (O'Connor et al., 2006).
Telomere-dependent cell survival in cells with or without p53 function
Late generation mice lacking both telomerase and p53 have a higher incidence of cancer relative to animals lacking only telomerase (Chin et al., 1999), implicating p53 as a key regulator of the response to telomere dysfunction. However, mice deficient in both p53 and telomerase lose sterility after only a few generations, which indicates a second p53-independent block to cell viability (Chin et al., 1999). Expression of dominant-negative telomerase proteins (Hahn et al., 1999) or mutant telomerase RNAs (Guiducci et al., 2001) induces death in cells with either wild-type or mutant p53, which supports the idea that the loss of viability caused by telomere dysfunction is not dependent on a functional p53 response. In contrast, cell death or growth inhibition caused by expression of DN-TRF2 is p53 dependent (Karlseder et al., 1999; Smogorzewska and De Lange, 2002).
Our results indicate that complex B disruption by TIN2-15C induces telomere dysfunction and cell cycle arrest in p53-positive cells. However, p53-deficient cells that express TIN2-15C continue to divide, developing chromosome-end fusions and anaphase bridges. These abnormalities result in cell death, which does not require p53 function. Thus, our findings support the idea that complex B disruption by TIN2-15C causes cell death that is independent of p53 due to severe genomic damage and mitotic catastrophe (Fig. 6 c). Our results also suggest that DN-TRF2 and TIN2-15C have different effects on the integrity of complex B.
In the absence of p53 activity, senescent cells that express little or no p16 can resume growth, although their proliferation is eventually limited by severe telomere shortening, crisis, and cell death (Beausejour et al., 2003). Expression of telomerase eliminated this growth limitation. Expression of TIN2-13 and DN-TRF2 only minimally affected this growth resumption, whereas TIN2-15C expression accelerated this growth limitation. Recent findings (Hockemeyer et al., 2005, 2006; Wu et al., 2006) suggest an explanation for why the effects of TIN2-15C differ from those of DN-TRF2. Although POT1 deficiency causes a telomere uncapping phenotype similar to that caused by TRF2 loss, POT1 and TRF2 likely have distinct functions in protecting telomeres, regulating nucleolytic processing, and controlling telomeric recombination. Thus, TIN2-15C and DN-TRF2 likely inactivate TRF2 and POT1 differently in complex B. Because DN-TRF2 induces p53-dependent apoptosis (Karlseder et al., 1999), whereas TIN2-15C caused cell death in p53-deficient cells, our findings offer an explanation for why ablation of TIN2 in cells (Fig. 1) and in the mouse germ line (Chiang et al., 2004) causes cell lethality or senescence regardless of p53 status.
It remains to be understood how telomere dysfunction causes cell death in p53-deficient cells, how dysfunctional telomeres are sensed as DNA damage, and the nature of the p53-dependent and -independent sensing mechanisms. Despite these gaps in our knowledge, telomerase inactivation has been proposed as an anticancer strategy because most cancer cells maintain telomere length by expressing this enzyme. Likewise, disruption of TIN2 complexes may provide a strategy for preferential killing of cancer cells, most of which lack p53 function. Thus, TIN2-15C–sensitive complexes may provide a sensitive target for selective elimination of p53-deficient cancer cells.
Materials and methods
Cell culture, senescence, and apoptosis characterization
We cultured all cells and measured senescence-associated β-galactosidase as described previously (Dimri et al., 1995). We determined cell death by collapse of the mitochondrial membrane potential (Davalos and Campisi, 2003) or by a cumulative assay in which cells are incubated with a caspase inhibitor for 3 d and scored for cytosolic cytochrome c (Goldstein et al., 2005).
Separation of native TIN2 complexes
Nuclei were isolated from HeLa-S3 cell suspension cultures (20 liters, 106 cells per ml; National Cell Culture Center) and extracted with 0.5 M KCl by a modified nuclear extraction method (Dignam et al., 1983). Extracts were dialyzed into S-300 chromatography buffer (50 mM Tris, pH 7.5; 150 mM KCl; 0.2 mM EDTA; 20% glycerol; 0.025% NP-40; 0.5 mM dithiothreitol; 1 μg/ml aprotinin, pepstatin A, and leupeptin; and 0.5 mM PMSF) and cleared by centrifugation at 15,000 rpm for 30 min at 4°C. The dialyzed sample (2.5 ml) was loaded onto a column (1.6/60 cm, 125 ml; Sephacryl S-300-HR; GE Healthcare) equilibrated with S-300 buffer. Fractions (1.4 ml) were collected and stored at 4°C. The S-300 column was calibrated using molecular mass standards (blue dextran, thyroglobulin, ferretin, aldolase, and conalbumin; GE Healthcare) according to the manufacture's instructions.
We expressed Flag-tagged TIN2 and HA-tagged TRF1 in HT1080 cells using the retroviral vector pLXSN as described previously (Kim et al., 1999). V5-tagged POT1 (a gift from P. Baumann, Stowers Institute of Medical Research, Kansas City, MO; Baumann et al., 2002) and myc-TIN2 mutants (TIN2-13 or TIN2-15C) were expressed using the pIRE2-EGFP vector (BD Biosciences) and transient transfection using Fugene 6 (Roche). 6 × 106 cells were washed with PBS, and 1 ml RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 10% glycerol, and protease inhibitor cocktail; Roche) was added to each plate. After incubation on ice for 30 min, cells were collected by scraping and centrifugation at 4°C and the supernatant (cell lysate) was recovered.
Immunoprecipitation and western analyses
We incubated cell lysates (200–300 μg protein) with 2 μg HA antibody (Roche), 10 μg Flag M2 antibody (Sigma-Aldrich), or 2 μg V5 antibody (Invitrogen) for 2 h at 4°C and added 50 μl of a 50% protein G–Sepharose slurry (Invitrogen) for 2 h at 4°C. We washed the immune complexes with RIPA buffer and analyzed proteins by Western blotting as described previously (Kim et al., 1999; Davalos and Campisi, 2003). Primary antibodies were monoclonal anti-TRF2 (IMG-124A; Imgenex), polyclonal anti-hRap1 (IMG-289; Imgenex), polyclonal anti-TRF1 (H-242 [Santa Cruz Biotechnology, Inc.] and ab1423 [Abcam]), polyclonal anti-POT1 (H200; Santa Cruz Biotechnology, Inc.), polyclonal anti-HA or -TRF2 or monoclonal anti-HA (Santa Cruz Biotechnology, Inc.), monoclonal anti-V5 (Invitrogen), polyclonal anti-TIN2 (Kim et al., 1999), and polyclonal TPP1 (a gift from Z. Songyang, Baylor College of Medicine, Houston, TX; O'Connor et al., 2006). The protein band intensities were analyzed by the Multi-Gauge program (Fujifilm).
Immunostaining
We immunostained cells as described previously (Kim et al., 1999). In brief, we cultured cells in chamber slides and cells were fixed with 4% formalin, permeabilized with 0.5% Triton X-100, and stained with anti-TRF2, anti-TIN2 (Kim et al., 1999), polyclonal anti-53BP1 (Abcam), monoclonal anti-p21 (BD Biosciences), monoclonal anti-p53 (Oncogene), or control (10% goat) serum (Vector Laboratories). After washing, we stained cells with secondary antibodies conjugated to Texas red or FITC (Invitrogen) and counterstained the nuclei with DAPI. Images of cells were acquired on a microscope (BX60; Olympus) using either a 100× UPlanFL 1.3 NA (Olympus) lens with oil or a 40× UPlanFL 0.5 NA (Olympus) lens without oil. Images were acquired with a charge-coupled device camera (Diagnostic Instruments, Inc.) and captured into SPOT imaging software (Diagnostic Instruments, Inc). All the modifications were applied to the whole image using Canvas 8 (Deneba) or Photoshop CS (Adobe).
Chromosome analyses
Telomeres were visualized by in situ hybridization (FISH) on metaphase spreads using a telomeric protein nucleic acid (PNA) probe as described previously (Hultdin et al., 1998). Cells treated with 0.1 μg/ml colcemid for 4 h were trypsinized and collected at 1,000 g (5 min). After hypotonic swelling in 30 mM sodium-citrate for 20 min at 37°C, the cells were fixed in methanol/acetic acid (3:1). FISH using a Cy3-labeled (CCCTAA)3 PNA probe (Applied Biosystem) and scoring for telomeric fusions were performed as described previously (Bailey et al., 1999).
3′-G–rich strand overhangs were measured as described previously (Kim et al., 1999). 4 μg of genomic DNA was hybridized with telomeric C-stranded probes (CCCTAA)5 in nondenaturing buffer to detect the 3′ overhang and in denaturing buffer to detect total telomeric DNA. DNA signals were quantified with the Multi-Gauge program (FLA-7000).
shRNA and expression vectors
Where indicated, cells were infected with a retrovirus expressing GSE-22 (Gudkov et al., 1993) or a lentivirus expressing sh-p53 (Nair et al., 2005), selected, and then transfected with expression vectors or infected with lentiviruses as described previously (Beausejour et al., 2003; Kim et al., 2004). To ablate TIN2 expression, we synthesized double-stranded DNAs to target the TIN2 mRNA (T2i-1, CAGGTGAAGCAGCTGTCAG; T2i-2, GGTCATATCTAATCCTGAG; and T2i-3, GTGGTGGTGGAGCTGATC) or SATB1, a nuclear protein that is not expressed in fibroblasts or HT1080 cells (AACAGCTACTATTGCCACT). We cloned the DNA into the pSuper vector (Brummelkamp et al., 2002) and transiently transfected packaging cells using FuGene6 (Roche). We cloned TIN2 mutants into the bicistronic pIRES2-EGFP (Clontech Laboratories, Inc.) or a pPRL-Sin18-lenti vector (Dull et al., 1998). Lentiviruses were used at equivalent titers sufficient to infect ∼80–90% of cells.
Colony formation assay
Senescent cells (<2% labeling index) were plated with 5 × 104 cells in 6-well plates and infected with lentiviruses expressing GFP, TIN2-15C, TIN2-13, hTERT, or GSE-22 as described previously (Beausejour et al., 2003). The cells were washed with PBS twice, fixed, and stained with 0.1% crystal violet in 10% ethanol for 5 min at room temperature, washed with PBS, and dried.
Online supplemental material
Fig. S1 shows that TIN2 reduction decreases TRF1 and TRF2 foci in immunostaining. Fig. S2 presents additional fractionation of TRF1–TIN2 complexes from HeLa cell nuclear lysate on a Superdex S-200 HR column and shows that nuclear extraction efficiently liberates telomeric proteins in our extraction protocol. Fig. S3 presents telomeric G-strand overhangs and telomere length analyses in cells expressing TIN2-15C and DN-TRF2. Fig. S4 shows telomeric damage responses that are costained for telomeric DNA in cells expressing TIN2-13 or TIN2-15C. Fig. S5 shows telomeric fusions and anaphase bridges in cells expressing TIN2-15C. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710028/DC1.
Supplemental Material
Acknowledgments
We thank Zhou Songyang for TPP1 antibodies, Peter Baumann for the POT1-V5 cDNA, and Yoichi Shinkai for critical reading of the manuscript.
We acknowledge support from the National Institutes of Health (grants AG011658-12 and AG024399-02 to J. Campisi and CA107798A to S.-h. Kim), the Department of Energy (grant DE-AC03-76SF00098), and the California Breast Cancer Research Program.
S.-h. Kim's present address is Vattikuti Urology Institute, Henry Ford Health System, Detroit, MI 48202.
C. Beausejour's present address is Département de Pharmacologie Centre de Recherche, Centre Hospitalier Universitaire Sainte-Justine, Montréal, Quebec H3T 1C5, Canada.
P. Kaminker's present address is Human Genome Sciences, Rockville, MD 20850.
Abbreviations used in this paper: DN-TRF2, dominant-negative TRF2; POT1, protector of telomeres 1; PNA, protein nucleic acid; shRNA, short hairpin RNA; TIN2, TRF1-interacting protein 2; TRF, telomeric repeat binding factor.
References
- Bailey, S.M., J. Meyne, D.J. Chen, A. Kurimasa, G.C. Li, B.E. Lehnert, and E.H. Goodwin. 1999. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl. Acad. Sci. USA. 96:14899–14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, P., and T.R. Cech. 2001. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 292:1171–1175. [DOI] [PubMed] [Google Scholar]
- Baumann, P., E. Podell, and T.R. Cech. 2002. Human Pot1 (protection of telomeres) protein: cytolocalization, gene structure, and alternative splicing. Mol. Cell. Biol. 22:8079–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausejour, C.M., A. Krtolica, F. Galimi, M. Narita, S.W. Lowe, P. Yaswen, and J. Campisi. 2003. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 22:4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, A., R.M. Stansel, L. Fairall, J.D. Griffith, D. Rhodes, and T. de Lange. 1999. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 18:5735–5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn, E.H. 2000. Telomere states and cell fates. Nature. 408:53–56. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- Celli, G.B., and T. de Lange. 2005. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 7:712–718. [DOI] [PubMed] [Google Scholar]
- Chang, W., J.N. Dynek, and S. Smith. 2003. TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev. 17:1328–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, Y.J., S.H. Kim, L. Tessarollo, J. Campisi, and R.J. Hodes. 2004. Telomere-associated protein TIN2 is essential for early embryonic development through a telomerase-independent pathway. Mol. Cell. Biol. 24:6631–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, L., S.E. Artandi, Q. Shen, A. Tam, S.L. Lee, G.J. Gottlieb, C.W. Greider, and R.A. DePinho. 1999. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 97:527–538. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna, F., P.M. Reaper, L. Clay-Farrace, H. Fiegler, P. Carr, T. Von Zglinicki, G. Saretzki, N.P. Carter, and S.P. Jackson. 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 426:194–198. [DOI] [PubMed] [Google Scholar]
- Davalos, A.R., and J. Campisi. 2003. Bloom syndrome cells undergo p53-dependent apoptosis and delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks. J. Cell Biol. 162:1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange, T. 2005. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19:2100–2110. [DOI] [PubMed] [Google Scholar]
- Dignam, J.D., R.M. Lebovitz, and R.G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri, G.P., X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E.E. Medrano, M. Linskens, I. Rubelj, O. Pereira-Smith, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull, T., R. Zufferey, M. Kelly, R.J. Mandel, M. Nguyen, D. Trono, and L. Naldini. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, J.C., F. Rodier, J.C. Garbe, M.R. Stampfer, and J. Campisi. 2005. Caspase-independent cytochrome c release is a sensitive measure of low-level apoptosis in cell culture models. Aging Cell. 4:217–222. [DOI] [PubMed] [Google Scholar]
- Griffith, J.D., L. Comeau, S. Rosenfield, R.M. Stansel, A. Bianchi, H. Moss, and T. de Lange. 1999. Mammalian telomeres end in a large duplex loop. Cell. 97:503–514 (see comments). [DOI] [PubMed] [Google Scholar]
- Gudkov, A.V., C.R. Zelnick, A.R. Kazarov, R. Thimmapaya, D.P. Suttle, W.T. Beck, and I.B. Roninson. 1993. Isolation of genetic suppressor elements, inducing resistance to topoisomerase II-interactive cytotoxic drugs, from human topoisomerase II cDNA. Proc. Natl. Acad. Sci. USA. 90:3231–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiducci, C., M.A. Cerone, and S. Bacchetti. 2001. Expression of mutant telomerase in immortal telomerase-negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene. 20:714–725. [DOI] [PubMed] [Google Scholar]
- Hahn, W.C., S.A. Stewart, M.W. Brooks, S.G. York, E. Eaton, A. Kurachi, R.L. Beijersbergen, J.H. Knoll, M. Meyerson, and R.A. Weinberg. 1999. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 5:1164–1170. [DOI] [PubMed] [Google Scholar]
- Herbig, U., W.A. Jobling, B.P. Chen, D.J. Chen, and J.M. Sedivy. 2004. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell. 14:501–513. [DOI] [PubMed] [Google Scholar]
- Hockemeyer, D., A.J. Sfeir, J.W. Shay, W.E. Wright, and T. de Lange. 2005. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 24:2667–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer, D., J.P. Daniels, H. Takai, and T. de Lange. 2006. Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. Cell. 126:63–77. [DOI] [PubMed] [Google Scholar]
- Houghtaling, B.R., L. Cuttonaro, W. Chang, and S. Smith. 2004. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 14:1621–1631. [DOI] [PubMed] [Google Scholar]
- Hultdin, M., E. Gronlund, K. Norrback, E. Eriksson-Lindstrom, T. Just, and G. Roos. 1998. Telomere analysis by fluorescence in situ hybridization and flow cytometry. Nucleic Acids Res. 26:3651–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano, T., M. Tachibana, M. Reth, and Y. Shinkai. 2004. Importance of TRF1 for functional telomere structure. J. Biol. Chem. 279:1442–1448. [DOI] [PubMed] [Google Scholar]
- Karlseder, J., D. Broccoli, Y. Dai, S. Hardy, and T. de Lange. 1999. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 283:1321–1325. [DOI] [PubMed] [Google Scholar]
- Kim, S.H., P. Kaminker, and J. Campisi. 1999. TIN2, a new regulator of telomere length in human cells. Nat. Genet. 23:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.H., S. Han, Y.H. You, D.J. Chen, and J. Campisi. 2003. The human telomere-associated protein TIN2 stimulates interactions between telomeric DNA tracts in vitro. EMBO Rep. 4:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.H., C. Beausejour, A.R. Davalos, P. Kaminker, S.J. Heo, and J. Campisi. 2004. TIN2 mediates functions of TRF2 at human telomeres. J. Biol. Chem. 279:43799–43804. [DOI] [PubMed] [Google Scholar]
- Li, S., J.E. Rosenberg, A.A. Donjacour, I.L. Botchkina, Y.K. Hom, G.R. Cunha, and E.H. Blackburn. 2004. Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res. 64:4833–4840. [DOI] [PubMed] [Google Scholar]
- Liu, D., M.S. O'Connor, J. Qin, and Z. Songyang. 2004. a. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J. Biol. Chem. 279:51338–51342. [DOI] [PubMed] [Google Scholar]
- Liu, D., A. Safari, M.S. O'Connor, D.W. Chan, A. Laegeler, J. Qin, and Z. Songyang. 2004. b. PTOP interacts with POT1 and regulates its localization to telomeres. Nat. Cell Biol. 6:673–680. [DOI] [PubMed] [Google Scholar]
- Loayza, D., and T. De Lange. 2003. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 423:1013–1018. [DOI] [PubMed] [Google Scholar]
- Nair, A.R., M. Schliekelman, M.B. Thomas, J. Wakefield, S. Jurgensen, and R. Ramabhadran. 2005. Inhibition of p53 by lentiviral mediated shRNA abrogates G1 arrest and apoptosis in retinal pigmented epithelial cell line. Cell Cycle. 4:697–703. [DOI] [PubMed] [Google Scholar]
- O'Connor, M.S., A. Safari, H. Xin, D. Liu, and Z. Songyang. 2006. A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc. Natl. Acad. Sci. USA. 103:11874–11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe, J., A. Eguchi, R. Wadhwa, R. Rakwal, R. Tsukinoki, T. Hayakawa, and M. Nakanishi. 2004. Limited capacity of the nuclear matrix to bind telomere repeat binding factor TRF1 may restrict the proliferation of mortal human fibroblasts. Hum. Mol. Genet. 13:285–293. [DOI] [PubMed] [Google Scholar]
- Rodier, F., S.H. Kim, T. Nijjar, P. Yaswen, and J. Campisi. 2005. Cancer and aging: the importance of telomeres in genome maintenance. Int. J. Biochem. Cell Biol. 37:977–990. [DOI] [PubMed] [Google Scholar]
- Smogorzewska, A., and T. De Lange. 2002. Different telomere damage signaling pathways in human and mouse cells. EMBO J. 21:4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska, A., and T. de Lange. 2004. Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 73:177–208. [DOI] [PubMed] [Google Scholar]
- Takai, H., A. Smogorzewska, and T. de Lange. 2003. DNA damage foci at dysfunctional telomeres. Curr. Biol. 13:1549–1556. [DOI] [PubMed] [Google Scholar]
- van Steensel, B., and T. de Lange. 1997. Control of telomere length by the human telomeric protein TRF1. Nature. 385:740–743. [DOI] [PubMed] [Google Scholar]
- van Steensel, B., A. Smogorzewska, and T. de Lange. 1998. TRF2 protects human telomeres from end-to-end fusions. Cell. 92:401–413. [DOI] [PubMed] [Google Scholar]
- Wu, L., A.S. Multani, H. He, W. Cosme-Blanco, Y. Deng, J.M. Deng, O. Bachilo, S. Pathak, H. Tahara, S.M. Bailey, et al. 2006. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell. 126:49–62. [DOI] [PubMed] [Google Scholar]
- Xin, H., D. Liu, M. Wan, A. Safari, H. Kim, W. Sun, M.S. O'Connor, and Z. Songyang. 2007. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature. 445:559–562. [DOI] [PubMed] [Google Scholar]
- Yang, Q., Y.L. Zheng, and C.C. Harris. 2005. POT1 and TRF2 cooperate to maintain telomeric integrity. Mol. Cell. Biol. 25:1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J.Z., J.R. Donigian, M. van Overbeek, D. Loayza, Y. Luo, A.N. Krutchinsky, B.T. Chait, and T. de Lange. 2004. a. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J. Biol. Chem. 279:47264–47271. [DOI] [PubMed] [Google Scholar]
- Ye, J.Z., D. Hockemeyer, A.N. Krutchinsky, D. Loayza, S.M. Hooper, B.T. Chait, and T. de Lange. 2004. b. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 18:1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, Y., A. Sfeir, S.M. Gryaznov, J.W. Shay, and W.E. Wright. 2004. Does a sentinel or a subset of short telomeres determine replicative senescence? Mol. Biol. Cell. 15:3709–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}