

Abstract
A sequential reaction methodology is employed for the complete derivatization of protein thiols, amines, and acids in high purity under denaturing conditions. Following standard thiol alkylation, protein amines are modified via reductive methylation with formaldehyde and pyridine-borane. Protein acids are subsequently amidated under buffered conditions in DMSO using the coupling reagent (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate. The generality of the approach is demonstrated with four proteins and with several amines yielding near-quantitative transformations as characterized by high-resolution Fourier transform mass spectrometry. The developed approach has numerous implications for protein characterization and general protein chemistry. Applications in mass spectrometry (MS) based proteomics of intact proteins (top-down MS) are explored, including the addition of stable isotopes for relative quantitation and protein identification through functional group counting. The methodology can be used for altering the physical and chemical properties of proteins, as demonstrated with amidation to modify protein isoelectric point and through derivatization with quaternary amines. Additionally, the chemistry has applications in the semisynthesis of mono-disperse polymers based on protein scaffolds. We prepare proteins modified with azides and alkynes to enable further functionalization via copper(I)-catalyzed 1,3-dipolar Huisgen cycloaddition (“click”) chemistry.
The reduction and alkylation of protein thiols is common practice for mass spectrometry (MS) analysis of both whole proteins and peptides. Complete chemical modification of other functional groups, such as amines and acids, has been limited to peptides.1,2 There is an extensive prior history in the chemical modification of proteins at several functional groups.3 However, these modifications typically do not produce homogeneous products and are therefore not useful for several applications, particularly for the analytical characterization of proteins. The completion of several transformations on a highly functionalized scaffold in high purity represents a unique synthetic challenge. Side reactions and incomplete modifications are quickly compounded on the whole protein scale and can create significant, artifactual heterogeneity. For a modification that does not proceed fully at n sites in a protein, there are 2n − 1 possible reaction products.4 For example, variable amine labeling for a protein with 10 amines can produce 1023 possible products. Additional complications arise from side reactions at nontargeted sites, which are difficult to predict.
While the global chemical modification of functional groups other than thiols has been limited to peptides for MS proteomic analysis, full modification of both protein amines and acids has been investigated for other purposes. Full dimethylation of amines by reductive alkylation has been accomplished for probing protein structure function relationships, incorporation of radioisotopes, or altering the crystallization properties of proteins.5–7 Complete acylation of protein amines has been used as an approach for the synthesis of monodisperse polymers based on protein scaffolds.8 Global modification of protein acids to amides using water-soluble carbodiimides has been used for determining the number of carboxyl groups within a protein using hydrolytic amino acid analysis.9,10 These amine and acid modifications have been accomplished on several proteins in reasonable purity, as assessed either by hydrolytic amino acid analysis or by capillary electrophoresis. To our knowledge, characterization of fully amine or acid-labeled, intact proteins by MS has not been investigated thoroughly.
In proteomic applications, the global chemical modification of protein thiol, amine, or acid functional groups has been generally implemented either for improving peptides or proteins as MS analytes or for incorporating labels for relative quantitation.2 The reduction and alkylation of protein thiols greatly facilitates MS analysis of both peptides and proteins. Removal of disulfide cross-links facilitates MS/MS sequencing and prevents the formation of random dimers upon thiol oxidation. In whole proteins, the removal of these cross-links enables the unfolding of protein structure. Electrospray ionization (ESI)-MS of unfolded proteins produces greatly increased signal intensity (up to 2 orders of magnitude) compared to folded proteins and also generates more highly charged ions at lower m/z ratios, which are generally considered more favorable for MS/MS fragmentation.11,12 Several additional chemical modifications applied to amine and acid functional groups have been utilized to improve peptide MS analysis. Guanidination of amine groups has been employed for the improvement of signal intensity in MALDI-MS analysis.13 Amine acylation with hydrophobic groups14 or hydrophobic groups with fixed charges15 has shown dramatic improvements in the signal intensity of peptides. Amine dimethylation via reductive alkylation has been used to improve MS/MS fragmentation of peptides for de novo sequencing by enhancement of a1 ions in collisonally activated dissociation (CAD).16 The modification of peptide acids by esterification or amidation has been shown to improve MS/MS fragmentation spectra by limiting preferential cleavage at these residues.17,18 Additionally, esterified peptides have also demonstrated decreased tendencies to form adducts with alkali metals.19
These chemical modifications have also been utilized in postgrowth covalent labeling strategies with peptides for the incorporation of stable isotope labels to determine relative differences in protein abundance. These approaches were popularized by the isotope-coded affinity tag for labeling protein thiols developed by Aebersold and co-workers.20 Briefly, the method involves comparison of two or more samples that are labeled with different forms of a labeling reagent containing the same molecular formula yet differing in mass due to substitution of stable isotopes (typically 2H for 1H, 15N for 14N, or 13C for 12C).1 After mixing the samples, analysis by liquid chromatography–mass spectrometry (LC–MS) indicates relative protein ratios by comparison of the ion current observed for each of the species in the pair. Several methods have been developed for stable isotope coding by labeling peptide amines.21 In addition to the acyl, alkyl, and guanidium groups mentioned above, amines have been modified by 2,4-dinitro-1-fluorobenzene22 and isocyanates23 for relative quantitation. Isotope coding through carboxyl groups on peptides has been achieved in several studies through esterification or by 18O incorporation during proteolysis.
The “top-down” approach to proteomics is focused largely on the direct characterization of intact proteins utilizing ESI and high-performance Fourier transform mass spectrometry (FTMS).24 This approach is in contrast to more typical “bottom-up” proteomic methods wherein proteins are characterized based on the analysis of their peptides obtained from proteolytic digests. While peptide analysis has been enormously useful and successful, the development of better methods to facilitate top-down proteomics is a highly desirable goal due to several inherent advantages.25 MS analysis of intact proteins can provide 100% sequence coverage, while bottom-up MS typically achieves only 40–90%.26 Precise measurement of an intact molecular weight contributes significantly to complete characterization of a protein. Top-down analysis avoids increases in sample complexity generated by digestion. In addition, top-down methods are beneficial for analysis of proteins with diverse post-translational modifications (PTMs), as increases in sequence coverage aid in identifying and localizing modifications.27 Top-down MS is particularly useful when proteins harbor multiple PTMs, as their combinations can be determined at the intact protein level. Recent work on histones28 exemplifies several of the advantages of the top-down approach, but many proteins are not as well behaved during chromatography and ESI-MS.
Here, we describe a sequential approach for the modification of three functional groups on intact proteins in denatured form (Scheme 1). Reduction and alkylation of thiols, reductive methylation of amines, and amidation of acids are carried out in excellent purity on whole proteins, as characterized by ESI-FTMS. The chemistry developed herein has broad implications for the characterization of proteins by mass spectrometry and for general applications in protein chemistry.
Scheme 1.

Protein Modification Strategy
EXPERIMENTAL SECTION
Materials
Synthetic reagents were obtained from Aldrich Chemical (Milwaukee, WI) unless specified otherwise. D2 form-aldehyde was purchased from Cambridge Isotope Laboratories (Andover, MA). (7-Azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP) was purchased from Applied Biosystems (Foster City, CA). 3-Azidopropylamine was prepared as previously described.29 (4-Aminobutyl)trimethylammonium dichloride was prepared as described below. D3 glycine methyl ester was prepared as previously described.30 Proton nuclear magnetic resonance (1H NMR) spectra were recorded at 300 MHz using a Bruker AC+ 300 spectrometer. High-resolution electrospray ionization mass spectra (HRESI-MS) for small molecules were obtained on a Micromass LCT. The four standard proteins (bovine ubiquitin, equine myoglobin (EC 309-705-0), bovine ribonuclease A (EC 3.1.27.5), and chicken lysozyme (EC 3.2.1.17)) were obtained from Sigma (St. Louis, MO). Prior to use, the heme from myoglobin was removed as previously described.31 Ribonuclease A (RNase A) was purified on a 5-mL HiTrap SPHP cation-exchange column (GE Healthcare) to remove additional protein contaminants on a AKTA FPLC system (GE Healthcare). Protein was applied to the column in 50 mM sodium acetate buffer pH 5.0 and eluted with a linear gradient of sodium chloride. Reversed-phase HPLC was conducted on a YMC C18 ODS-A 100 × 10 mm column on a Beckman Coulter System Gold HPLC.
General Protein Modification Procedures
(1)Thiol Alkylation
Proteins were dissolved in 60 mM NH4HCO3 buffer with 6 M guanidine-HCl and 20 mM DTT at 5 mg/mL protein concentration. Protein thiols were reduced at 37 °C for 1 h. Reactions were diluted to 2.5 mg/mL with 60 mM NH4HCO3, 6 M guanidine-HCl buffer and made to 25 mM iodoacetamide. Reactions were allowed to proceed at room temperature for 1.5 h in the dark.
(2) Amine Reductive Methylation
Thiol-alkylated protein or protein lacking thiols was dissolved in 300 mM triethanolamine buffer pH 7.5 with 6 mM guanidine-HCl at 1.25 mg/mL. Reactions were diluted to 1 mg/mL with methanol. Pyridine–BH3 (8 M in excess pyridine) was added to 30 mM. Last, formaldehyde was added to 20 mM. Reactions were then sonicated briefly in a water bath (~1 min) to suspend pyridine–BH3 and allowed to react at room temperature for 2 h.
(3) Amine Acylation
Amine acylation was performed with the difluorophenol ester of 4-(trimethylamino)-3-butyric acid (compound 1). Acylation was performed on ubiquitin (0.5 mg) dissolved in 300 mM HEPES buffer pH 8.0 with 6 M guanidine-HCl (0.25 mL). The active ester (9.6 mg, 25 μmol) was added and allowed to react overnight at room temperature. Protein was washed with 6 M guanidine-HCl in H2O (3 mL) in a 5K MWCO Amicon Ultra centrifugal filter device (Millipore) in 500-μL portions and then concentrated to 2 mg/mL. An equal volume of 200 mM LiOH in 6 M guanidine-HCl pH 12.5 was added to hydrolyze esters at 5 °C for 3 h.
(4) Acid Amidation
Thiol-alkylated (if thiols present), amine-methylated protein was dissolved in wet DMSO (~5% H2O) to an approximate concentration of 1.5 mg/mL. Proteins were heated slightly (~50 °C) and sonicated if necessary to dissolve. The solution was then made to 1 M glycine methyl ester HCl salt and 750 mM N-methylmorpholine (NMM). Last, PyAOP was added to 40 mM. Reactions were allowed to proceed at room temperature for 2 h and were then quenched with the addition of an equal volume of H2O. Reactions with other amines used slight modifications of the above conditions. Couplings with benzylamine and 3-azidopropylamine employed 1 M amine HCl and 500 mM NMM. Couplings with propargyl amine employed 500 mM amine HCl and 250 mM NMM. Couplings to (4-aminobutyl)trimethylammonium employed 500 mM amine diHCl and 350 mM NMM. Also reactions with the fixed charge amine used 60 mM PyAOP and required an additional 10% H2O to solubilize the amine salt.
(5) Reaction Purification
To reduce adverse effects from previous reaction components, proteins were purified and desalted by HPLC on a reversed-phase column between reaction steps. Similar results were obtained when protein was purified by ethanol or acetone precipitation, but recovery of protein was significantly lower. In the cases where reaction contaminants, such as tris-(pyrrolidino)phosphine oxide, coeluted with protein peaks on HPLC, proteins were first purified by washing with excess buffer in a 5K MWCO Amicon Ultra centrifugal filter device (Millipore, Billerica, MA) and subsequently desalted by HPLC. Proteins were eluted with a gradient of H2O with 0.05% trifluoroacetic acid (TFA) (as solvent A) and acetonitrile with 0.05% TFA (as solvent B) as follows: 15% B for 3 min, 15–95% B over 24 min, and 95% B for 3 min. Protein peaks were collected and then lyophilized. Care was taken to collect the entirety of the protein-containing peak. In the optimization of reaction conditions, we found protein side products such as protein with missed labeling events were not resolved from desired products using this gradient. Proteins modified with glycine methyl ester were handled carefully to minimize exposure to acidic, aqueous conditions to prevent ester hydrolysis.
Mass Spectrometric Analysis of Proteins
Lyophilized protein was dissolved in 49.5/49.5/1% water/methanol/formic acid. Mass spectra of proteins were obtained by direct injection on an 8.5-T custom quadrupole FTMS with an ESI Advion Nanomate source or for the acylated ubitiquitin sample on a 12-T LTQ FT Ultra Hybrid mass spectrometer (constructed in collaboration with Thermo Scientific). Fragment ion analysis was performed using the program ProSight PTM (https://prosightpt-m.scs.uiuc.edu) using single protein mode. Analysis of the yeast proteome database was performed with the UniProt Saccharomyces cerevisiae yeast protein database (http://www.pir.uniprot.org/), which was expanded to include the possible presence or absence of N-terminal methionine or acetylation of each protein.
Synthesis of (4-Aminobutyl)trimethylammonium dichloride
Boc-anhydride (4.13 g, 18.9 mmol) was added in dry CH2-Cl2 to N1,N1-dimethylbutane-1,4-diamine (2.0 g, 17.2 mmol) (Karl Industries, Aurora, OH) in CH2Cl2. The mixture was allowed to stir and reduce under N2 overnight. Resulting oil was dissolved in ethyl acetate and extracted against saturated NaHCO3, dried over MgSO4, and evaporated under reduced pressure. Without further purification, a portion of the resulting oil (1 g, 4.62 mmol) was dissolved in methanol (200 mL) and methyl iodide (1.4 mL, 23 mmol) was added dropwise. The reaction was allowed to continue overnight, and then solvent was removed under reduced pressure. The resulting thick oil was then triturated extensively in diethyl ether. Solid material was then dissolved in dry MeOH and added to a premixed solution of 20 mL of MeOH and 2 mL of acetyl chloride. This was allowed to stir and reduce under N2 overnight. Material was resuspended in MeOH and evaporated under reduced pressure to dryness. Solid was then triturated in acetonitrile to remove impurities. Yield (as the di Cl− salt): 87%. Spectra matched a previous report of the compound.32
Synthesis of [3-(2,6-Difluorophenoxycarbonyl)propyl]-trimethylammonium iodide
4-(Dimethylamino)-3-butyric acid HCl (0.5 g, 3.0 mmol) was added to difluorophenol (0.38 g, 3.0 mmol) and dicyclohexylcarbodiimide (0.68 g, 3.3 mmol) in DMF and allowed to react overnight under N2. The formed precipitate of dicyclohexylurea was filtered from the mixture and washed with dry acetonitrile. Solvent was removed from the collected filtrate, which was then resuspended in acetonitrile. KHCO3 (1.5 g, 16.5 mmol) and CH3I (0.94 mL, 16.5 mmol) were added and allowed to react overnight with stirring. Product was recovered by precipitation with ether and triturated with 2-propanol to remove impurities to give the final compound 1. Yield (as the I− salt) 60%. 1H NMR (ACN-d3, 300 MHz) δ 7.33 (m, 1H), 7.13 (m, 2H), 3.42 (m, 2H), 3.11 (s, 9H), 2.83 (t, 2H), 1.95 (m, 2H). MS (HRESI-MS) calculated for [C13H18F2NO2+] 258.1300, found 258.1310.
RESULTS AND DISCUSSION
Amine Modification
To preserve the charge properties important for MS detection in positive mode, efforts were initially focused on reductive methylation rather than acylation. Using modifications of a previously published procedure,33 the methylation of protein amines was accomplished using 20 mM formaldehyde and 30 mM pyridine-borane in 6 M guanidine-HCl, 20% MeOH, 300 mM triethanolamine buffer at pH 7.5, which produced fully modified proteins in excellent purity. The labeling of four proteins was demonstrated: ubiquitin (8.6 kDa, 8 amines, 0 thiols), myoglobin (16.9 kDa, 20 amines, 0 thiols), thiol-alkylated RNaseA (14.1 kDa, 11 amines), and thiol-alkylated lysozyme (14.8 kDa, 7 amines). Figure 1 shows ESI-FTMS spectra of modified ubiquitin and myoglobin showing a minimal reduction in purity. It was found that reactions with the more commonly used reducing agents NaBH3CN or NaBH4 produced protein products of poor purity. Various side reactions from reductive alkylations with NaBH3CN are known34 and have recently been well characterized in the modification of several peptides.35
Figure 1.
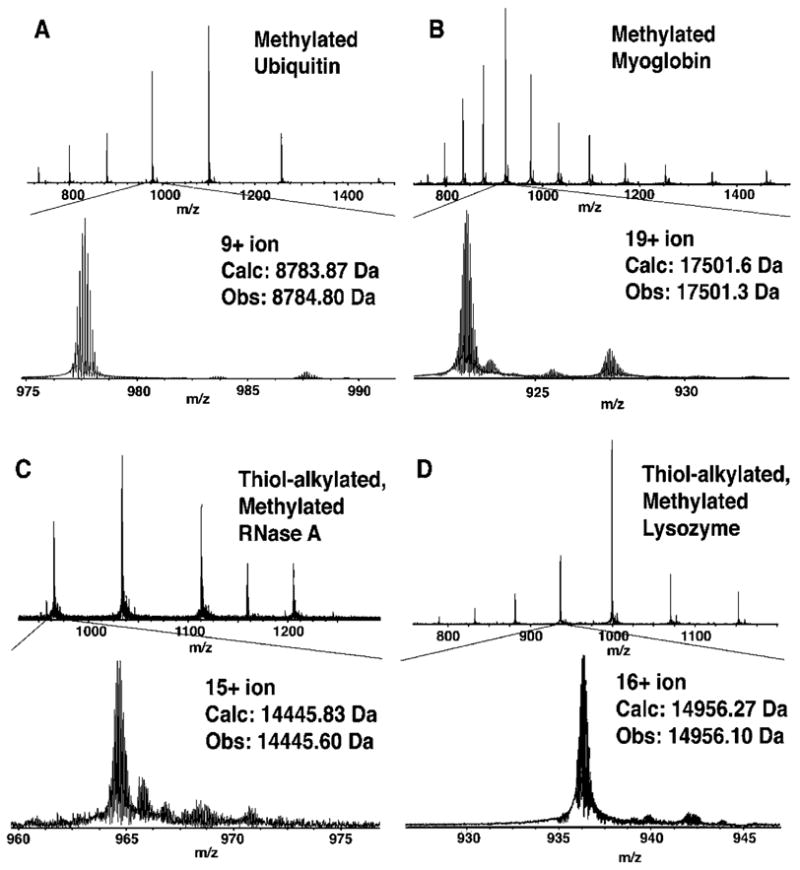
ESI-FTMS spectra of amine methylated proteins (A) ubiquitin, (B) myoglobin, (C) RNase A, and (D) lysozyme.
Additional chemistry that has been applied to peptides was briefly explored to enable alternative approaches to the modification of protein amines with preservation of positive charge. Amine guanidination with O-methylisourea has been used extensively in peptide modification.13 McLuckey and co-workers prepared a fully guanidinated ubiquitin for studying the effects of protein mobility on protein fragmentation.36 However, their modification was incomplete, and fully modified protein was obtained only after HPLC purification from the mixture. Similarly, we were unable to develop conditions for complete conversion of proteins using O-methylisourea.
As another alternative, the modification of amines with acyl groups bearing fixed charge amines was investigated, which has been successfully applied in the modification of peptides.15,37 An activated ester containing a quaternary ammonium (compound 1, Figure 2A) was prepared for the acylation of protein amines. Complete amine acylation of ubiquitin was accomplished with 100 mM 1 in 300 mM HEPES buffer with 6 M guanidine-HCl at pH 8.0. Removal of O-acylations by treatment with 100 mM LiOH at pH 12.5 was required to yield pure product. Figure 2B shows the ESI-MS spectrum of fully acylated ubiquitin. Unfortunately, these modified protein products were found to be poor substrates for subsequent acid modification reactions. Also, the highly basic hydrolysis conditions required may interfere with some post-translational modifications, particularly phosphorylations and O-linked sugars, which can eliminate to form reactive electrophiles.38,39 It was anticipated that the use of the mildly activating difluorophenol ester might enable selective N-acylation. Reducing the reaction pH to 7.5 or using fewer equivalents of acylating reagent was also unable to prevent acylation of protein alcohols. Therefore, the application of this chemistry was not explored further.
Figure 2.
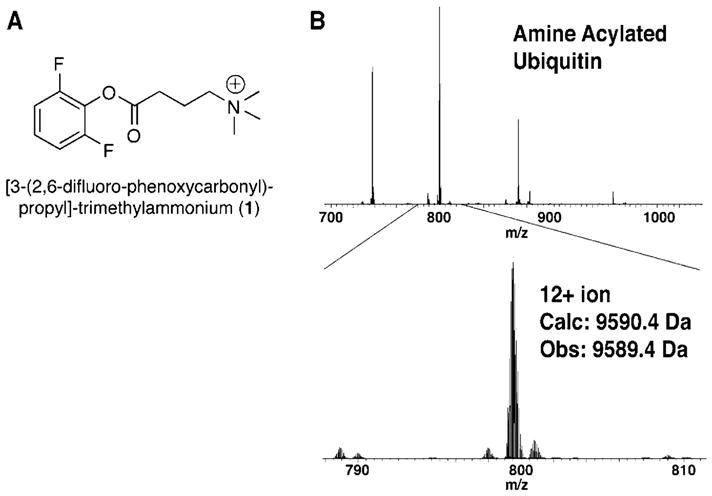
(A) Active ester of 4-(trimethylamino)-3-butyric acid prepared for acylation of protein amines. (B) ESI-FTMS of ubiquitin acylated with compound 1.
Acid Modification
In our hands, previously published procedures for protein acid amidation in water with carbodiimides using native or amine-methylated ubiquitin did not yield products with suitable purity for MS analysis.9,10 Our approach for high-purity protein acid amidation relies on prior thiol and amine alkyation with subsequent amidation reactions performed in DMSO. Labeling conditions employed the coupling agent PyAOP40 at 40 mM in wet DMSO (~5% H2O) with 1 M amine HCl and 750 mM N-methylmorpholine. These weakly basic, buffered conditions and the use of PyAOP as a coupling reagent were found to be critical for clean reaction conversions. The use of more strongly basic conditions resulted in incomplete modification and losses of ~18 Da in mass, presumably due to intramolecular couplings to protein alcohols forming esters. The active 1-hydroxy-7-azabenzotriazole (HOAt) esters formed in PyAOP couplings have been shown to promote sterically difficult couplings and coupling under weakly basic conditions.41,42 This promotion in reactivity is thought to occur through neighboring group coordination or deprotonation of the incoming amine by the pyridine nitrogen of HOAt during aminolysis of the activated ester.
The developed chemistry using glycine methyl ester was applied to four modified proteins: methylated ubiquitin (8.8 kDa, 12 acids), methylated myoglobin (17.5 kDa, 22 acids), thiol-alkylated, methylated RNaseA (14.5 kDa, 11 acids), and thiol-alkylated, methylated lysozyme (15.0 kDa, 10 acids). The reduction in purity of these highly modified proteins was generally minimal, as determined by ESI-FTMS (Figure 3). To further demonstrate the generality of labeling, thiol-alkylated, methylated lysozyme was amidated with four additional amines: (4-aminobutyl)trimethylammonium, benzylamine, propargylamine, and 3-azidopropylamine (Figure 4), using slight modifications of the procedure used for glycine methyl ester. The concentration of protonated amine and the relative ratio to the N-methylmorpholine base required empirical optimization for each amine to ensure complete conversions in high purity. We found that amidation with propargyl amine most consistently produced clean modifications (Figure S3).
Figure 3.
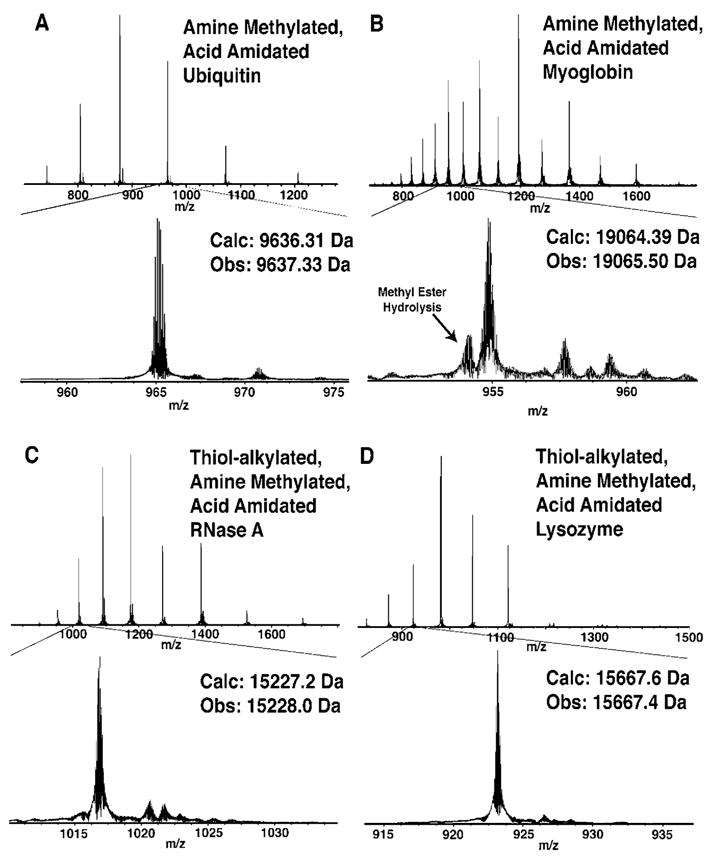
ESI-FTMS spectra of thiol alkylated (with iodoacetamide, if thiol present), amine methylated, acid amidated (with glycine methyl ester) proteins (A) ubiquitin, (B) myoglobin, (C) RNase A, and (D) lysozyme.
Figure 4.
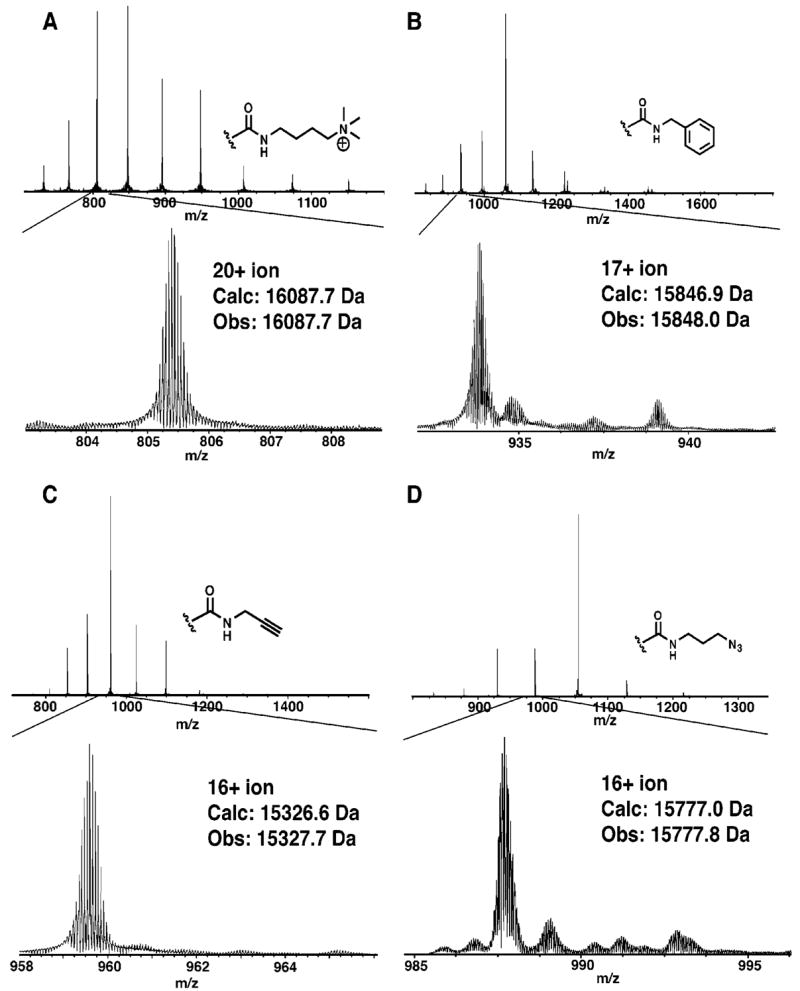
ESI-FTMS spectra of thiol alkylated (iodoacetamide), amine methylated, acid amidated lysozyme. The acid amidation used four separate amines, as indicated (A–D).
These acid labeling conditions should be generally applicable toward the modification of any protein. While there are few reports of protein solubility in nonaqueous solvents, DMSO is known to denature proteins,43 and proteins that have been assessed have shown excellent solubility (>0.5 mg/mL) in pure DMSO.44,45 Additionally, these conditions should be tolerant to post-translational modifications, such as phorphorylation.
Esterification was additionally considered for the modification of carboxyl groups on intact proteins, as esterification of peptides can be accomplished efficiently in anhydrous, acidic methanol or ethanol.1 However, these procedures were not applicable to intact proteins, primarily due to solubility. While some small proteins show good solubility in methanol, generally proteins are quite poorly soluble, even if they are previously denatured, reduced, and thiol alkylated.45 Thus, efforts were focused on acid amidation.
Potential Applications in MS Analysis
Modifying protein functional groups offers several possibilities for improving whole proteins as MS analytes in complex mixtures. Proteins with few basic residues, poor solubility, or very stable tertiary structures are difficult targets for MS.46 The extreme variability of protein physical properties makes chromatography prior to MS challenging and often results in large losses of certain proteins.47 Significant improvements in chromatography of intact proteins will be required to address the large range of protein abundances in cells (~106 in Escherichia coli, ~109 in humans), which exceeds the dynamic range of mass spectrometers (typically ~103–104).1 Substantial modification of protein isoelectric point (via acid amidation) or the addition of certain functionalities (such as fixed charges or additional basic groups) may improve the solubility and chromatography of proteins. Also, amidation with fixed charge or additional basic moieties offers the possibility of increasing the charging of proteins (Figures 4A and S4, Supporting Information), which can have benefits in facilitating MS analysis.48 Importantly, we found that neither amine methylation nor acid amidation interferes with common MS/MS fragmentation techniques (Figures S7–S9, Supporting Information). Modified proteins fragmented by CAD, electron capture dissociation, and infrared multiphoton dissociation produced fragmentation spectra typical for these methods.
This labeling methodology can be used to incorporate stable isotopes for quantitative proteomics studies with intact proteins (Figure 5) and should also be applicable to peptides and metabolites. Postgrowth labeling techniques that incorporate stable isotopes can be limited by cost, reaction selectivity, and efficiency.1 The reactions outlined here provide an efficient means for addition of stable isotopes to common functional groups (amines and acids). Deuterated and 13C formaldehyde are readily available and economical for amine methylation. The generality of the acid modification reaction could allow for several commercially available amines to be employed for isotope coding. Differential labeling of whole proteins at common functional groups is advantageous in that labeling reagents with small mass differences can be employed and the isotopic peaks of protein pairs can still be resolved. Labeling at rare functional groups such as thiols not only excludes the analysis of proteins without thiols but also requires the potentially expensive synthesis of labeling reagents with large mass differences. In the previous work of Du et al. in comparing expression ratios of intact proteins by postgrowth labeling, a fairly large mass difference was achieved conveniently through differential alkylation of protein thiols with either acrylamide or iodoacetamide (14-Da difference/residue).49 However, the different labels produced significant differences in retention time on reversed-phase chromatography, indicating that stable isotope labels would be required.
Figure 5.
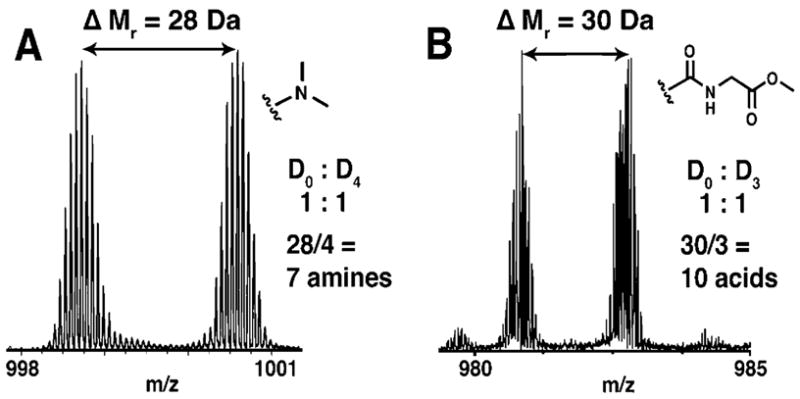
Relative quantitation of two protein samples and determination of protein amines and acids. (A) Mass spectrum of 15+ ions of 1:1 mixture of thiol-alkylated lysozyme methylated with either D0 or D2 formaldehyde. (B) Mass spectrum of 16+ ions of 1:1 mixure of thiol-alkyated, methylated lysozyme amidated with either D0 or D3 glycine methyl ester.
Mixed isotope reagents can also be employed to determine the number of functional groups present in a protein. This information, together with intact mass measurement, can be used as a search criterion to uniquely identify a protein from protein databases or genomic data. Figure 5 demonstrates use of this chemistry for the counting of protein amines and acids via isotopic labeling. This approach for protein identification was first investigated by Smith and co-workers with the incorporation of isotopically labeled leucine into proteins in auxotrophic E. coli.50 Similarly, gas-phase ion chemistry has been used to determine the number of acidic or basic sites in peptides and proteins through adduct formation with sodium or iodine ions, respectively.51,52 Recently, Du et al. explored this approach with post-growth labeling of protein thiols.49 The additional counting of amines and acids by postgrowth labeling could significantly improve this approach by lowering the required mass accuracy and increasing the confidence of protein identification. An analysis of the yeast proteome database determined that >95% confidence can be obtained in protein identification using intact mass and the number of thiols, amines, and acids as search constraints for proteins <50 kDa at 200 ppm mass accuracy (Figure 6B). This is a significant improvement over the counting of thiol groups alone (Figure 6A). The improved certainty could make functional group counting a viable approach for proteomic experiments and indicates that the approach may be applicable with proteomes more complex than yeast. The low mass accuracy required could enable this approach to be performed with widely available, low-resolution mass spectrometers. Additional plots with other functional group combinations are available in the Supporting Information (Figures S5 and S6). The main benefit of the technique would potentially be the increases in throughput attainable over MS/MS fragmentation approaches for protein identification. The time required to conduct MS/MS fragmentation of intact proteins poses limitations for their on-line analysis. Protein identification through functional group counting would require only intact mass determination after labeling. On-line capillary electrophoresis-MS and LC–MS have been used on-line successfully in the detection of intact masses.25
Figure 6.
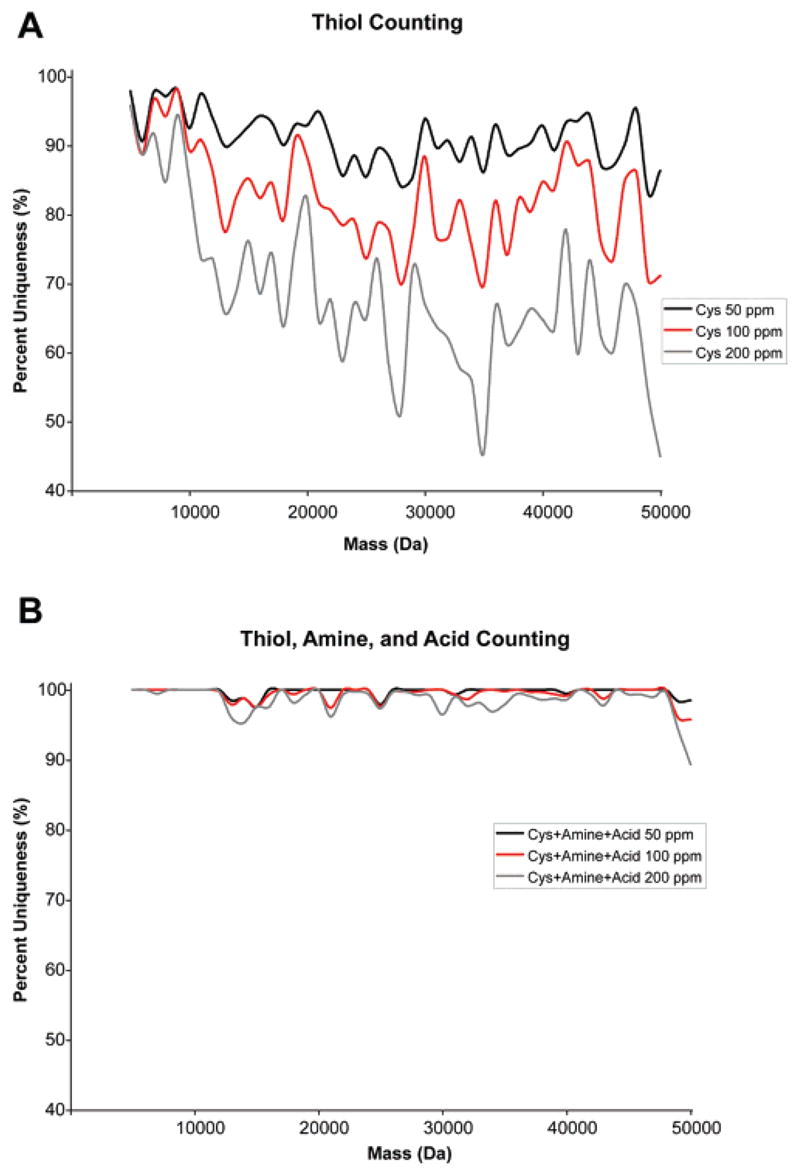
Percentage of unique proteins in the yeast proteome as a function of accuracy of the mass determination and the number of thiol (A) or thiol, amine, and acid (B) functional groups. Percent uniqueness was calculated from the number of proteins with unique numbers of functional groups within a certain mass accuracy (50, 100, or 200 ppm) divided by the total number of proteins within a 1000 Da mass window from 5 to 50 kDa in the yeast proteome.
Monodisperse Polymers
Additionally, this chemistry could be useful in the preparation of monodisperse polymers from protein scaffolds. As monodisperse polymers are difficult to prepare by conventional synthetic methods, semisynthetic strategies starting with a monodisperse scaffold, such as a protein, have been applied to synthesize these polymers. Derivatized protein polymers have been used as biological tools (e.g., polyvalent ligand displays53) and as building blocks for novel materials.54 We have prepared proteins modified with alkynes or azides (Figure 4C and D), which represent potentially useful scaffolds for additional functionalization through the bioorthogonal copper(I)-catalyzed 1,3-dipolar Huisgen cycloaddition or “click” chemistry.55,56 Our modification approach allows for proteins to retain their positive charges, which may be advantageous for some applications. For example, positively charged protein polymers may show utility for use inside cells since many highly positively charged proteins and peptides have demonstrated the ability to enter cells.57,58
CONCLUSIONS
Thiol reduction and alkylation so greatly improves proteins as MS analytes that this modification is likely to remain part of MS characterization for some time to come. Additional functional group modifications offer additional, significant benefits for analysis and would greatly aid MS top-down proteomics. Until now, no one has achieved the clean reaction conversions necessary to minimize added complexity. We have developed efficient reaction conditions for the reductive methylation of protein amines and the amidation of protein acids. These conditions were employed for the sequential modification of thiols, amines, and acids on denatured, intact proteins. These modifications may facilitate the purification and analysis of whole proteins and should find application in proteomics and protein-based polymer synthesis. Further investigation will develop the approach for larger proteins and proteomic mixtures.
Acknowledgments
The authors thank Thomas Rutkoski for assistance with RNase A purification. We thank Dr. Brian Frey and Dr. Marissa Rosen for insightful discussions. C.J.K. was supported by the NIH Biotechnology Training Grant Fellowship. This work was supported by the NIH (grants GM 065406 to P.J.B. and GM 067193 to N.L.K.).
Footnotes
SUPPORTING INFORMATION AVAILABLE Additional MS spectra, MS/MS spectra, representative HPLC traces, and additional plots of protein certainty determination with functional group counting of the yeast proteome. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Julka S, Regnier F. J Proteome Res. 2004;3:350–363. doi: 10.1021/pr0340734. [DOI] [PubMed] [Google Scholar]
- 2.Leitner A, Lindner W. Proteomics. 2006;6:5418–5434. doi: 10.1002/pmic.200600255. [DOI] [PubMed] [Google Scholar]
- 3.Lundblad RL. Chemical Reagents for Protein Modification. 3. CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- 4.Zhao JY, Waldron KC, Miller J, Zhang JZ, Harke H, Dovicki NJ. J Chromatogr. 1992;608:239–242. doi: 10.1016/0021-9673(92)87129-v. [DOI] [PubMed] [Google Scholar]
- 5.Means GE, Feeney RE. Anal Biochem. 1995;224:1–16. doi: 10.1006/abio.1995.1001. [DOI] [PubMed] [Google Scholar]
- 6.Jentoft N, Dearborn DG. Methods Enzymol. 1983;91:570–579. doi: 10.1016/s0076-6879(83)91052-2. [DOI] [PubMed] [Google Scholar]
- 7.Rayment I. Methods Enzymol. 1997;276:171–179. [PubMed] [Google Scholar]
- 8.Yang J, Gitlin I, Krishnamurthy VM, Vazquez JA, Costello CE, Whitesides GM. J Am Chem Soc. 2003;125:12392–12393. doi: 10.1021/ja035978l. [DOI] [PubMed] [Google Scholar]
- 9.Hoare DG, Koshland DE., Jr J Biol Chem. 1967;242:2447–2453. [PubMed] [Google Scholar]
- 10.Carraway KL, Koshland DE., Jr Methods Enzymol. 1972;25:616–623. doi: 10.1016/S0076-6879(72)25060-1. [DOI] [PubMed] [Google Scholar]
- 11.Kuprowski MC, Konermann L. Anal Chem. 2007;79:2499–2506. doi: 10.1021/ac0620056. [DOI] [PubMed] [Google Scholar]
- 12.Loo JA, Edmonds CG, Udseth HR, Smith RD. Anal Chem. 1990;62:693–698. doi: 10.1021/ac00206a009. [DOI] [PubMed] [Google Scholar]
- 13.Beardsley RL, Reilly JP. Anal Chem. 2002;74:1884–1890. doi: 10.1021/ac015613o. [DOI] [PubMed] [Google Scholar]
- 14.Frahm JL, Bori ID, Comins DL, Hawkridge AM, Muddiman DC. Anal Chem. 2007;79:3989–3995. doi: 10.1021/ac070558q. [DOI] [PubMed] [Google Scholar]
- 15.Mirzaei H, Regnier F. Anal Chem. 2006;78:4175–4183. doi: 10.1021/ac0602266. [DOI] [PubMed] [Google Scholar]
- 16.Hsu J, Huang S, Shiea J, Huang W, Chen S. J Proteome Res. 2005;4:101–108. doi: 10.1021/pr049837+. [DOI] [PubMed] [Google Scholar]
- 17.Ma M, Kutz-Naber KK, Li L. Anal Chem. 2007;79:673–681. doi: 10.1021/ac061536r. [DOI] [PubMed] [Google Scholar]
- 18.Sekiya S, Wada Y, Tanaka K. Anal Chem. 2004;76:5894–5902. doi: 10.1021/ac049374r. [DOI] [PubMed] [Google Scholar]
- 19.Poon C, Kaplan H, Mayer PM. Eur J Mass Spectrom. 2004;10:39–46. doi: 10.1255/ejms.582. [DOI] [PubMed] [Google Scholar]
- 20.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 21.Regnier FE, Julka S. Proteomics. 2006;6:3968–3979. doi: 10.1002/pmic.200500553. [DOI] [PubMed] [Google Scholar]
- 22.Chen X, Chen YH, Anderson VE. Anal Biochem. 1999;273:192–203. doi: 10.1006/abio.1999.4243. [DOI] [PubMed] [Google Scholar]
- 23.Mason DE, Liebler DC. J Proteome Res. 2003;2:265–272. doi: 10.1021/pr0255856. [DOI] [PubMed] [Google Scholar]
- 24.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. J Am Chem Soc. 1999;121:806–812. [Google Scholar]
- 25.Kelleher NL. Anal Chem. 2004;76:197A–203A. [PubMed] [Google Scholar]
- 26.Gygi SP, Aebersold R. Curr Opin Chem Biol. 2000;4:489–494. doi: 10.1016/s1367-5931(00)00121-6. [DOI] [PubMed] [Google Scholar]
- 27.Bogdanov B, Smith RD. Mass Spectrom Rev. 2004;2:168–200. doi: 10.1002/mas.20015. [DOI] [PubMed] [Google Scholar]
- 28.Boyne MTII, Pesavento JJ, Mizzen CA, Kelleher NL. J Proteome Res. 2006;5:248–253. doi: 10.1021/pr050269n. [DOI] [PubMed] [Google Scholar]
- 29.Lewis WG, Magallon FG, Fokin VV, Finn MG. J Am Chem Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 30.Jacobsen JR, Cochran AG, Stephans JC, King DS, Schultz PG. J Am Chem Soc. 1995;117:5453–5461. [Google Scholar]
- 31.Harrison SC, Blout ER. J Biol Chem. 1965;240:299–303. [PubMed] [Google Scholar]
- 32.Yudovin-Farber I, Yanay C, Azzam T, Linial M, Domb AJ. Bioconjugate Chem. 2005;16:1196–1203. doi: 10.1021/bc050066p. [DOI] [PubMed] [Google Scholar]
- 33.Wong WSD, Osuga DT, Feeney RE. Anal Biochem. 1984;139:58–67. doi: 10.1016/0003-2697(84)90388-9. [DOI] [PubMed] [Google Scholar]
- 34.Gidley MJ, Sanders JK. Biochem J. 1982;224:331–334. doi: 10.1042/bj2030331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu Q, Li L. Anal Chem. 2005;77:7783–7795. doi: 10.1021/ac051324e. [DOI] [PubMed] [Google Scholar]
- 36.Pitteri SJ, Reid GE, McLuckey SA. J Proteome Res. 2004;3:46–54. doi: 10.1021/pr034054u. [DOI] [PubMed] [Google Scholar]
- 37.Fricker LD. Endocrinology. 2007;148:4185–4190. doi: 10.1210/en.2007-0123. [DOI] [PubMed] [Google Scholar]
- 38.Tinett S, Feyereisen R, Robichon A. J Cell Biochem. 2007;100:875–882. doi: 10.1002/jcb.21070. [DOI] [PubMed] [Google Scholar]
- 39.Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mol Cell Proteomics. 2002;1:791–804. doi: 10.1074/mcp.m200048-mcp200. [DOI] [PubMed] [Google Scholar]
- 40.Albericio F, Cases M, Alsina J, Triolo SA, Carpino LA, Kates SA. Tetrahedron Lett. 1997;38:4853–4856. [Google Scholar]
- 41.Montalbetti CAGN, Falque V. Tetrahedron. 2005;61:10827–10852. [Google Scholar]
- 42.Shaginian A, Rosen MC, Binkowski BF, Belshaw PJ. Chem Eur J. 2004;10:4334–4340. doi: 10.1002/chem.200400267. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharjya S, Balaram P. Proteins. 1997;29:492–507. doi: 10.1002/(sici)1097-0134(199712)29:4<492::aid-prot9>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 44.Houen G. Acta Chem Scand. 1996;50:68–70. doi: 10.3891/acta.chem.scand.50-0788. [DOI] [PubMed] [Google Scholar]
- 45.Houen G, Svaerke C, Barkholt V. Acta Chem Scand. 1999;53:1122–1126. doi: 10.3891/acta.chem.scand.53-1122. [DOI] [PubMed] [Google Scholar]
- 46.Chait BT, Kent SBH. Science. 1992;257:1885–1894. doi: 10.1126/science.1411504. [DOI] [PubMed] [Google Scholar]
- 47.Chait BT. Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- 48.Iavarone AT, Jurchen JC, Williams ER. Anal Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du Y, Parks BA, Sohn S, Kwast KE, Kelleher NL. Anal Chem. 2006;78:686–694. doi: 10.1021/ac050993p. [DOI] [PubMed] [Google Scholar]
- 50.Veenstra TD, Martinovic S, Anderson GA, Pasa-Tolic L, Smith RD. J Am Soc Mass Spectrom. 2000;11:78–82. doi: 10.1016/S1044-0305(99)00120-8. [DOI] [PubMed] [Google Scholar]
- 51.Timofeev O, Zhu MM, Gross ML. Int J Mass Spectrom. 2004;231:113–117. [Google Scholar]
- 52.Stephenson JL, McLuckey SA. Anal Chem. 1997;69:281–285. doi: 10.1021/ac961119m. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Kiick KL. J Am Chem Soc. 2005;127:16392–16393. doi: 10.1021/ja055102+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woerdeman DL, Veraverbeke WS, Parnas RS, Johnson D, Delcour JA, Verpoest I, Plummer CJG. Biomacromolecules. 2004;5:1262–1269. doi: 10.1021/bm034530+. [DOI] [PubMed] [Google Scholar]
- 55.Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 56.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 57.Yang Y, Ma J, Song Z, Wu M. FEBS Lett. 2002;532:36–44. doi: 10.1016/s0014-5793(02)03624-4. [DOI] [PubMed] [Google Scholar]
- 58.Futami J, Kitazoe M, Maeda T, Nukui E, Sakagucki M, Kosaka J, Miyazaki M, Kosaka M, Tada H, Seno M, Sasaki J, Huh NH, Namba M, Yamada H. J Biosci Bioeng. 2005;99:95–103. doi: 10.1263/jbb.99.95. [DOI] [PubMed] [Google Scholar]