

Abstract
We show that localized expression of the integrin α3 protein is regulated at the level of RNA localization by the human homologue of Drosophila Muscleblind, MLP1/MBLL/MBNL2, a unique Cys3His zinc-finger protein. This is supported by the following observations: MLP1 knockdown abolishes localization of integrin α3 to the adhesion complexes; MLP1 is localized in adhesion plaques that contain phospho-focal adhesion kinase; this localization is microtubule-dependent; integrin α3 transcripts colocalize with MLP1 in distinct cytoplasmic loci; integrin α3 transcripts are physically associated with MLP1 in cells and MLP1 binds to a specific ACACCC motif in the integrin α3 3′ untranslated region (UTR) in vitro; and a green fluorescent protein (GFP) open reading frame–integrin α3 3′ UTR chimeric gene directs GFP protein localization to distinct cytoplasmic loci near the cell periphery, which is dependent on MLP1 and is mediated by the ACACCC motif but is independent of the integrin α3 signal peptide.
We have recently identified a gene that is differentially expressed in many tumours. The cognate protein was named MLP1 for Muscleblind-like protein 1 (GenBank accession #AF491866) because of its high degree of homology with the Drosophila Muscleblind (Mbl) protein. The Drosophila mbl gene is involved in regulatory processes that are associated with the terminal differentiation of photoreceptors and muscles1,2. The Mbl protein is characterized by novel Cys3His zinc-finger motifs, CX7CX6CX3H-CX7CX4CX3H. Highly conserved homologues of mbl are found from worms and insects to mammals. Three human Mbl-like proteins — named EXP/MBNL1, MBLL/MBNL2 and MBXL/CHCR/MBNL3 — have been partially characterized3–7. MLP1 is the same as MBLL/MBNL2 and, as the former name was first used in the GenBank database, we will continue to use it throughout this report for brevity. These three proteins display RNA-binding activity towards transcripts that contain expanded trinucleotide repeats in myotonic dystrophy muscle cells, presumably sequestering the target RNAs3–5. These observations, however, did not shed light on the normal cellular functions of this family of proteins, although a role in mRNA processing has been implicated7. The developmental functions of Drosophila Mbl do not include early proliferation or differentiation of the muscle precursors2. In mbl mutants, muscles detach from the epidermis, indicating a defect in integrin-mediated adhesion. In addition, Mbl protein is found associated with the Z-band that is rich in actin. Therefore, these mbl homologues may represent a unique group of regulatory genes that control the cellular functions that are related to cell–matrix interactions and cytoskeletal organization.
To identify possible MLP1 targets that are relevant to these functions, a limited cDNA array analysis for structure- and motility-related genes was conducted. We compared the expression profiles of H520 cells, a lung squamous carcinoma line that expresses low endogenous levels of MLP1 (Fig. 1 a, b), with or without re-expression of MLP1 (see Supplementary Information, Fig. S1). The analysis identified integrin α3 as a potential positive regulatory target for MLP1. The integrin α3/β1 dimer is a major pro-migratory adhesion molecule in many mesenchymal and metastatic cell types8–10. To verify the function of MLP1, affinity-purified anti-MLP1 antisera were used in western blot analyses against crude extracts from various cell lines and detected only one protein band, which had an expected MLP1 relative molecular mass of 40,000. Three cell lines are shown in Fig. 1a: lung carcinoma H520 and A549, and colorectal carcinoma HT29, which express low, moderate and high levels of MLP1, respectively. These three cell lines were used throughout this study. Increased expression of MLP1 in H520 cells resulted in a concurrent increase of the integrin α3 protein level (Fig. 1b) and elevated migratory capacity (Fig. 1c). Interestingly, reverse transcription polymerase chain reaction (RT-PCR) analysis of the integrin α3 transcripts showed that, in the presence of MLP1, the total level of transcripts was increased without a concurrent increase in the pre-spliced transcripts (Fig. 1d). Therefore, upregulation of integrin α3 by MLP1 is not at the transcriptional level but is post-transcriptional. Upregulation of the level of integrin α3 transcripts was specific, as there was no appreciable increase of the paxillin and focal adhesion kinase (FAK) transcripts (Fig. 1d). By contrast, the transcripts of β-actin, which are also a potential target of MLP1 upregulation (see Supplementary Information, Fig. S1), were increased by twofold (Fig. 1d).
Figure 1.
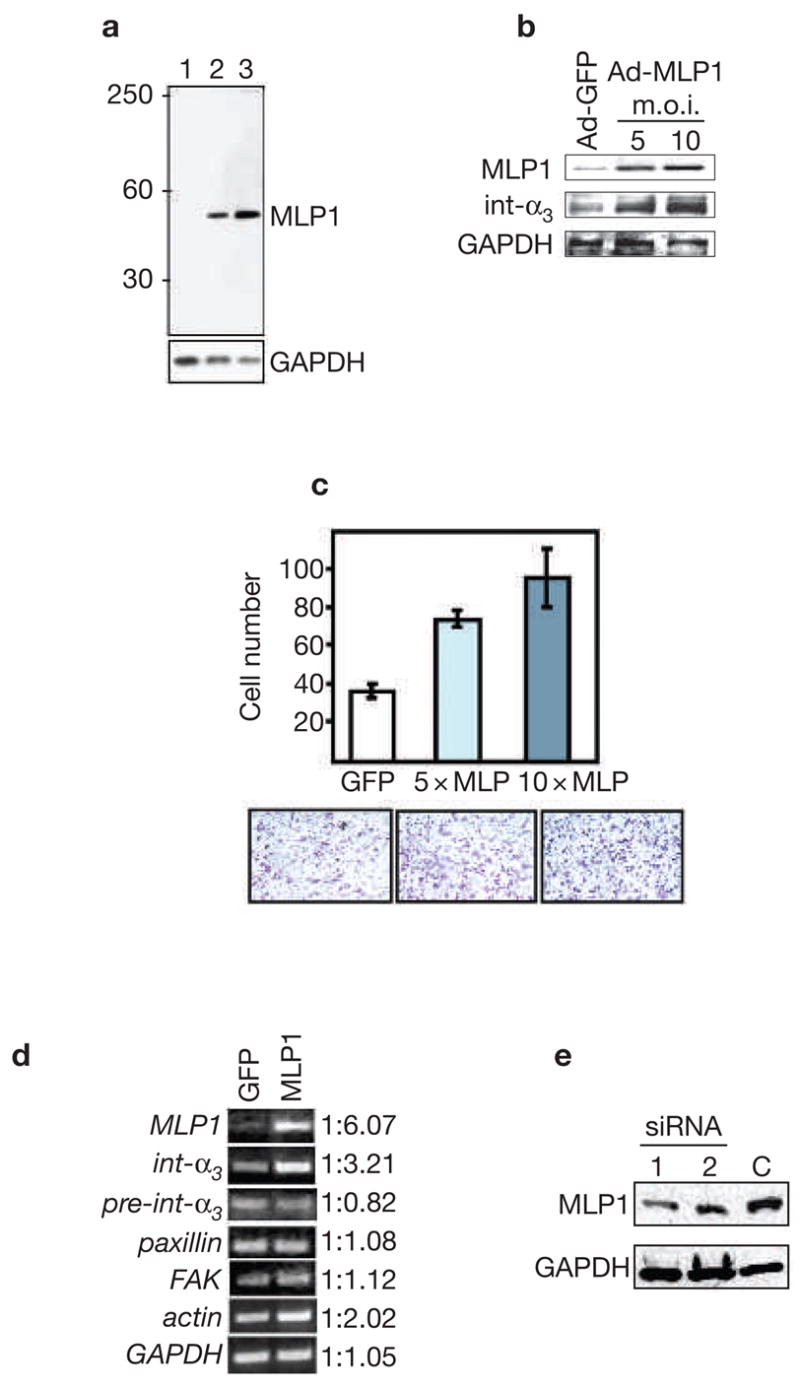
MLP1 regulates integrin α3 expression at the post-transcriptional level. (a) Protein extracts (10 μg/lane) from three cell lines were probed with rabbit anti-MLP1 antibody. The blot was stripped and re-probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the loading control. Lane 1: H520 cells; lane 2: A549 cells; lane 3: HT29 cells. A single band of expected size (relative molecular mass of approximately 40,000) was detected. MLP1 was not detectable in H520 cells with the amount of total protein extracts used. (b) H520 cells were infected with empty adenoviral vector (Ad-GFP; m.o.i. 10) or vector containing the MLP1 coding sequence (Ad-MLP1) at m.o.i. of 5 or 10. Total protein extracts (50 μg; higher amount needed for detecting integrins) were probed with the antibodies indicated on the left. GAPDH served as the loading control. Increased expression of MLP1 correlates with increased expression of integrin α3. (c) H520 cells were transfected with adenoviral vector expressing green fluorescent protein (GFP) alone (at 10× m.o.i.), or GFP plus MLP1 (at 5× and 10× m.o.i.), and plated onto the upper chamber of the transwell. Cells that migrated to the underside of the upper chamber were stained with crystal violet and the numbers compared. Lower panels show representative views of the migrated cells. (d) Reverse transcription polymerase chain reaction (RT-PCR) analysis using primers specific for MLP1 mRNA, integrin α3 exon (int-α3), integrin α3 intron–exon junction (pre-int-α3), or mRNAs of paxillin, focal adhesion kinase (FAK), actin and GAPDH. Total RNAs were isolated from H520 cells infected with adenoviral vector (GFP) or vector containing the MLP1 coding sequence at 5× m.o.i. The linear range of PCR amplification was empirically determined for each primer set (see Methods). At the mid-point of the linear curve, amplified samples were run on ethidium-bromide-stained agarose gel and quantified. Integrin α3 RNA level increases in step with increased expression of MLP1, whereas the pre-spliced integrin α3 RNA level remains unchanged. There is a moderate increase of actin in the presence of MLP1 and no change for paxillin, FAK or GAPDH. Note that without reverse transcriptase, the PCR reactions did not yield any amplified products (data not shown). (e) A549 cells were transfected with empty vector (C) or vector containing one of the two MLP1-specific small interfering RNA (siRNA)-encoding oligonucleotide duplexes.
We next examined whether the functional relationship between MLP1 and integrin α3 can be discerned in cells. MLP1-expressing A549 cells were transfected with vectors expressing either of two small-interfering RNAs (siRNAs) and the levels of MLP1 were examined. The transfection efficiency can be monitored by the expression of red fluorescent protein (RFP) that is also encoded by the vector. As shown in Fig. 1e, siRNA #1 achieved ~70% knockdown of MLP1 in the total cell extracts. As the transfection efficiency in this experiment was ~80% (data not shown), the efficiency of knockdown in the cells that received the siRNA vector was probably close to 90%. Interestingly, in cells transfected with the empty vectors, integrin α3 was localized to adhesion plaques mostly in the migration fronts that also contain the protein paxillin (Fig. 2a, vector), a known component of the focal complexes. By contrast, in cells transfected with MLP1-specific siRNA #1, integrin α3 was mainly localized in the cell body, not with adhesion plaques (Fig. 2a, siMLP1). Conversely, in H520 cells that express a low level of MLP1, re-expression of MLP1 resulted in re-localization of integrin α3 from within the cell body to plaques at the cell periphery that also contain phosphorylated FAK (pFAK), another component of the adhesion complexes (Fig. 2b, compare vector with MLP1). These results also indicate that the MLP1 function is selective, as paxillin and pFAK localization is not affected by MLP1.
Figure 2.
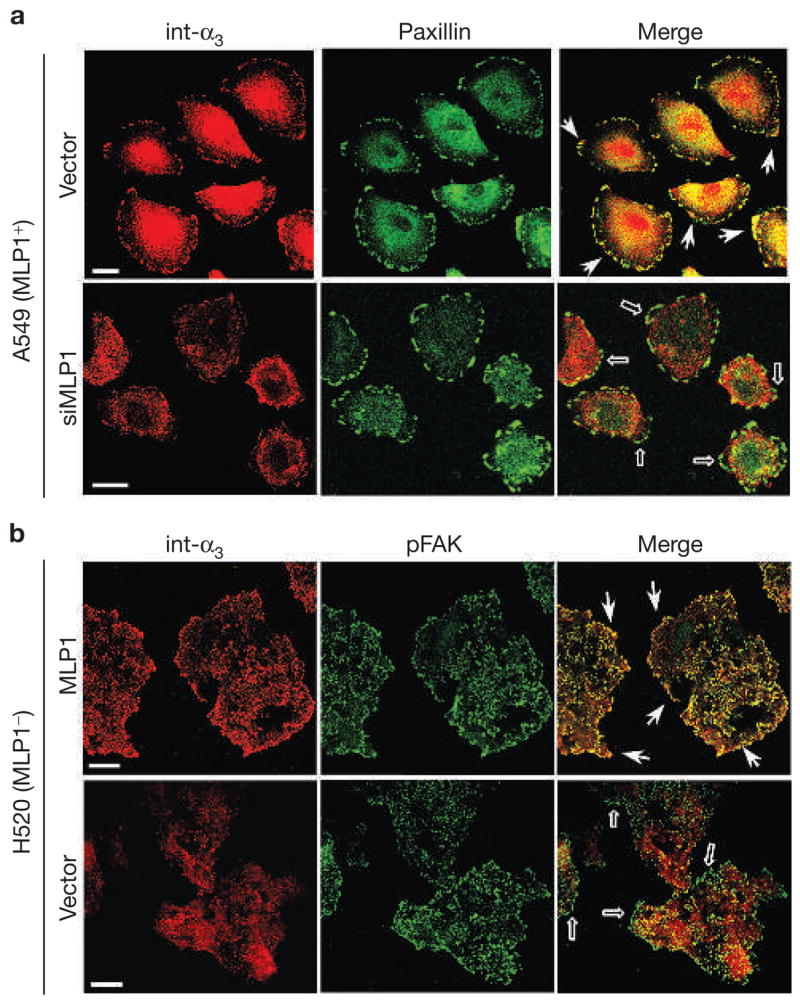
MLP1 is necessary for integrin α3 protein localization to adhesion plaques. (a) A549 cells transfected with empty vector or vector containing MLP1-specific oligonucleotide duplex #1 (siMLP1), as in Fig. 1e, were double-stained for paxillin (green) and integrin α3 (int-α3; red). In vector-transfected cells, integrin α3 colocalized prominently with paxillin in adhesion plaques at the migration front (examples indicated by arrows). In cells transfected with vector expressing MLP1-specific small interfering RNA (siRNA), integrin α3 no longer colocalized with paxillin in the cell periphery (empty arrows). (b) H520 cells were infected with empty adenovector or vector containing the MLP1 coding sequence at the m.o.i. of 10 (as in Fig. 1b), and double-stained for phospho-focal adhesion kinase (pFAK; green) and integrin α3 (red). Each stained area is a cluster of 5–10 cells but the peripheral pFAK appears mostly in cells at the leading edge. Without MLP1, integrin α3 is not colocalized with pFAK at the periphery (empty arrows). Expression of MLP1 results in re-localization of integrin α3 to pFAK-containing plaques at the periphery (arrows). Scale bars, 20 μm.
To gain insights into how MLP1 regulates integrin α3, we first examined the subcellular localization of MLP1. In A549 cells (Fig. 3a), the endogenous MLP1 protein was mainly expressed in or near large cytoplasmic plaques that also contain pFAK (Fig. 3a). Interestingly, localization of MLP1 to the pFAK-containing plaques was microtubule-dependent. For example, immediately after treatment with the microtubule-destabilizing drug nocodazole (0 h in Fig. 3b), MLP1 in HT29 cells was mostly localized in the nucleus. At 24 h after nocodazole-free recovery, MLP1 again remained mostly in the cytoplasm in association with pFAK. As the mammalian Muscleblind-related proteins have been shown to display RNA-binding activity3,5, we consider a possibility that MLP1 may be an integrin α3 RNA transporter from the nucleus to the cytoplasmic sites, where integrin α3 proteins are ultimately made and engaged in adhesion complexes. Without proper localization, the integrin α3 RNA is not efficiently translated, so there are reduced integrin α3 RNA and protein levels in cells expressing low levels of MLP1.
Figure 3.
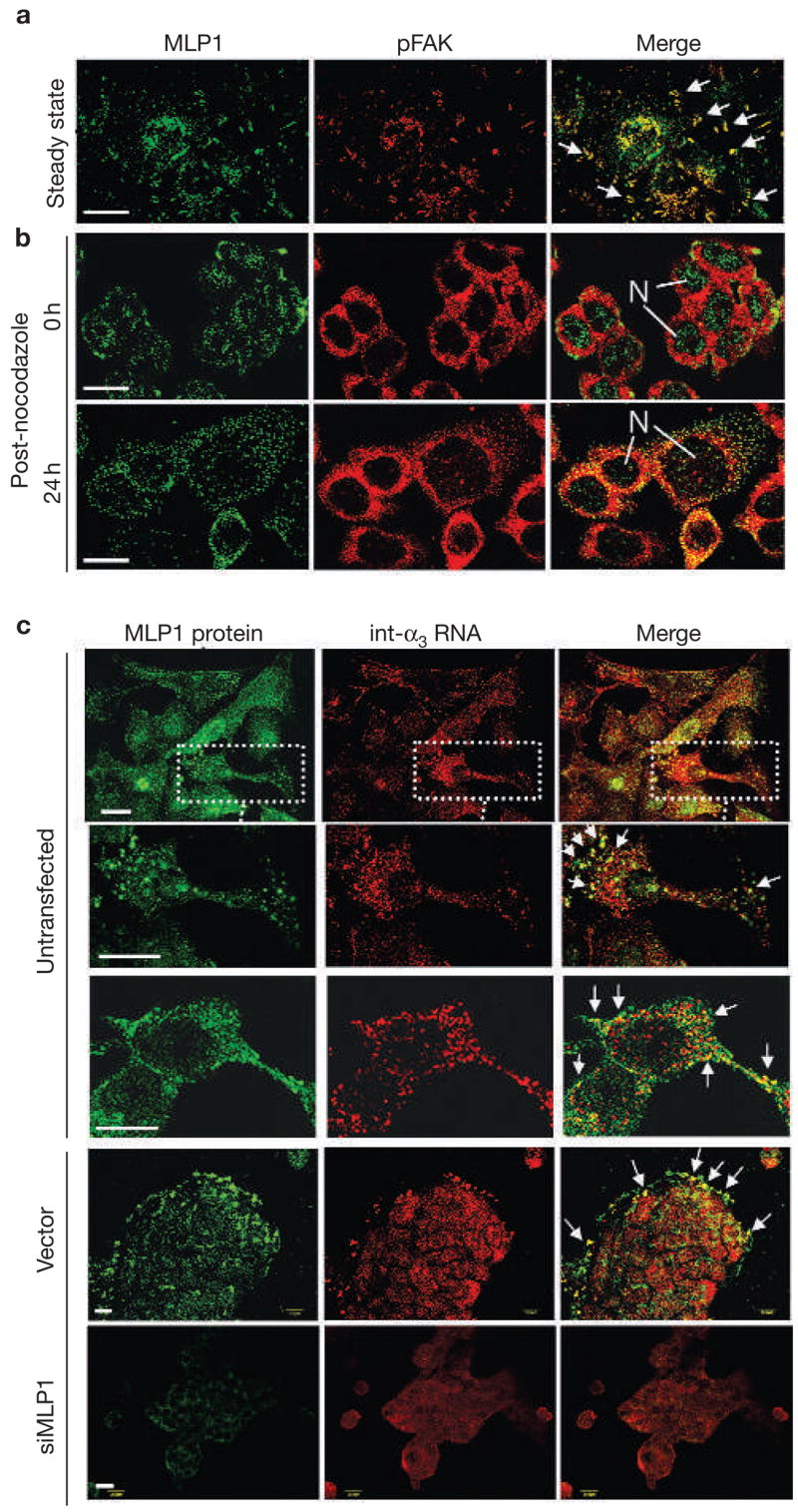
MLP1 regulates localization of integrin α3 RNA. (a) A549 cells were double-stained for MLP1 (green) and phospho-focal adhesion kinase (pFAK; red). Arrows mark examples of prominent adhesion plaques, where MLP1 and pFAK colocalize. (b) HT29 cells were treated with nocodazole and then returned to normal media. Cells were fixed and double-stained for MLP1 (green) and pFAK (red) at 0 and 24 h after recovery. Before recovery from nocodazole treatment (0 h), MLP1 was mainly localized in the nuclei (N). Twenty-four hours after recovery, MLP1 distribution returned to a punctated pattern with substantial colocalization with pFAK. (c) HT29 cells were double-stained for MLP1 protein and integrin α3 (int-α3) RNA. Untransfected cells and cells transfected with empty vector or vector expressing MLP1 small interfering RNA (siRNA) are indicated. In untransfected samples, a cell cluster and two enlarged single-cell views are shown to better visualize the large complexes that have been co-stained for integrin α3 RNA and MLP1 protein. In vector and siMLP1-transfected samples, clusters of cells are shown and the integrin α3 RNA–MLP1-containing complexes are mostly at the leading edges (arrows). Sense integrin α3 RNA probe showed no signals (data not shown). Scale bars, 20 μm.
This possibility was strengthened when RNA in situ hybridization-immunohistochemistry double-staining showed that integrin α3 transcripts were localized in a punctated pattern and that many of these large RNA-containing complexes colocalize with MLP1, particularly at or near the cell periphery (Fig. 3c, untransfected). Note that not all integrin α3 RNA punctates contain MLP1, and vice versa. This indicates that the association between these two molecules is transient, that not all integrin α3 mRNA molecules are transported by MLP1, or that a subset of MLP1-containing particles may colocalize with other mRNA species. These other potential MLP1-associated mRNA species will probably include those of β-actin. As is shown below, β-actin RNAs co-precipitate with the MLP1 protein (Fig. 4a). Importantly, the punctated integrin α3 RNA pattern can be dispersed when MLP1 is knocked down by siRNA (compare the vector with siMLP1 in Fig. 3c).
Figure 4.
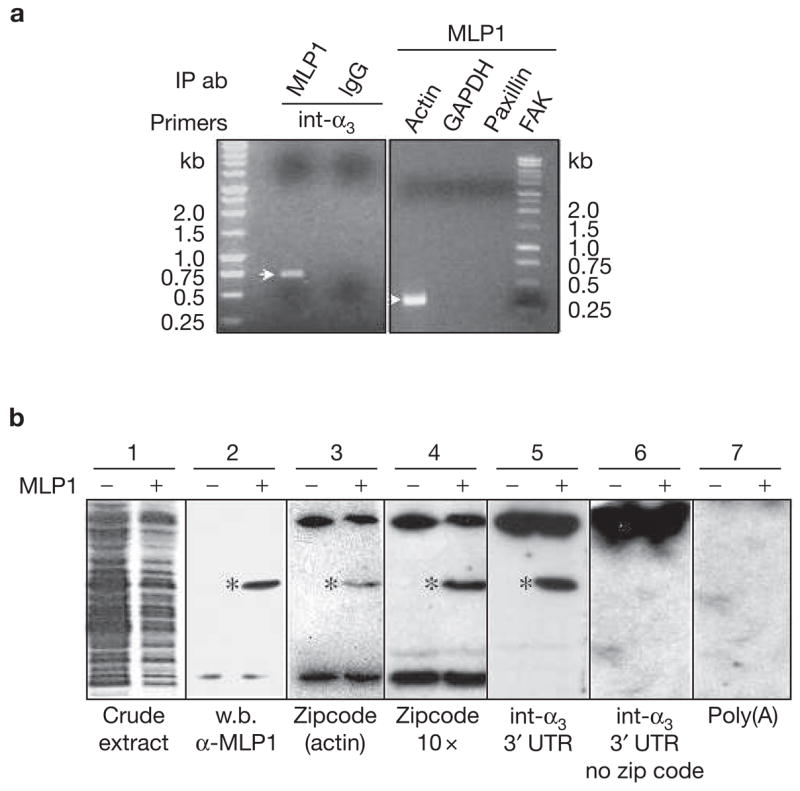
MLP1 interacts with integrin α3 3′ untranslated region (UTR) in vivo and in vitro. (a) H520 cells were transfected with 10× m.o.i. of adenovirus-containing MLP1 coding sequence and total RNAs were subjected to RNA pulldown and reverse transcription polymerase chain reaction (RT-PCR). Integrin α3 (int-α3) RNA was detected in the fraction that was pulled down by MLP1 antibody, yielding an expected fragment size (see Methods) on an ethidium-bromide-stained agarose gel. Pulldown by purified rabbit immunoglobulin G (IgG) served as a negative control. In addition, actin mRNA was present in the MLP1-containing complex, which was detected by RT-PCR yielding the expected 337-bp fragment, whereas paxillin, focal adhesion protein (FAK) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were not detectable. IP ab, immunoprecipitated antibodies (b) Extracts from BL21<DE3> bacteria transformed with (+) or without (−) plasmids containing MLP1 were run on SDS–PAGE and stained with Coomassie brilliant blue (blot 1) or western-blotted (w.b.) with anti-MLP1 antibody (blot 2). The MLP1 protein band is marked (*). Duplicate lanes were transferred to nitrocellulose membrane, renatured and hybridized with various 32P-labelled RNAs indicated at the bottom (blots 3–7). MLP1 (*) binds only to RNA molecules containing the zipcode (blots 3, 4 and 5). The bands at the top and bottom of the gels, present in both MLP1+ and MLP1− samples, were probably bacterial RNA-binding proteins.
We next assessed whether MLP1 is associated with integrin α3 transcripts in cells. The MLP1-containing complex was immunoprecipitated from extracts of H520 cells that were infected with MLP1-expressing adenovirus. Specific mRNA species were then detected by RT-PCR. As shown in Fig. 4a, MLP1 is indeed associated with integrin α3, as well as with actin RNAs, two species that were initially identified as upregulated targets of MLP1 (see Supplementary Information, Fig. S1). This interaction is specific, as the MLP1 complex does not contain paxillin, FAK or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA.
The inferred MLP1 function indicates that certain classes of mRNA are localized to specific subcellular loci, where the cognate proteins are needed. Actin mRNA has been shown to localize in such a manner in growth cones and cell protrusions11–13. The localization is dependent on a small sequence motif, ACACCC, termed a zipcode, within the 3′ UTR. Sequence analysis revealed that integrin α3 3′ UTR contains one copy of the zipcode. A ‘Northwestern’ assay was then devised to examine whether MLP1 binds to the zipcode in integrin α3 3′ UTR. BL21<DE3> bacteria were transformed with or without expression vectors carrying the full-length MLP1. Total bacterial proteins were separated by SDS–PAGE, transferred to nitrocellulose membrane and then re-natured in the blot. Note that these cells were not treated with isopropyl-beta-3-thiogalactopyranoside (IPTG), which induces exceedingly high levels of MLP1 expression and may cause non-specific binding. Moderate expression can be achieved by the background ‘leaky’ expression of MLP1. Low-level MLP1 was not readily discernable by Coomassie staining (Fig. 4b, blot 1), but could be specifically detected by western blotting (Fig. 4b, blot 2).
As a control, we showed that MLP1 binds to the 40-nucleotide zip-code-containing segment of the actin 3′ UTR (Fig. 4b, blot 3), as well as to an artificial RNA sequence consisting of 10 tandem repeats of the zipcode (Fig. 4b, blot 4). Towards the 1.2-kb integrin α3 3′ UTR RNA, MLP1 showed remarkable binding specificity. The interaction occured only in the presence of the 6-nucleotide ACACCC motif (compare blots 5 and 6 in Fig. 4b). Failure of MLP1 to bind to integrin α3 3′ UTR without the zipcode or to the poly(A) homopolymer (Fig. 4b, blot 7) indicated that MLP1 is not a promiscuous RNA-binding protein.
To assess the role of the integrin α3 3′ UTR in protein localization, the entire integrin α3 3′ UTR (nucleotides 3275–4484, accession number NM_005501), including the poly-adenylation signal, was placed immediately downstream of the green fluorescent protein (GFP) open reading frame (ORF) in the pEGFP–C1 vector. The original GFP cistron in the vector contains a ~180-nucleotide 3′ UTR that was derived from SV40 virus. Pools of stable transformants of HT29 cells carrying either of these plasmids were established. With the GFP–integrin-α3 chimeric cDNA, the GFP pattern of expression appeared punctated, which is markedly in contrast to the typical confluent expression pattern that is associated with the GFP–SV40 chimaera (Fig. 5a). As the two chimaeras contain the same GFP ORFs, we conclude that the difference in protein localization must be the result of differently localized transcripts. Significantly, the GFP punctates also colocalized with MLP1 (Fig. 5b).
Figure 5.
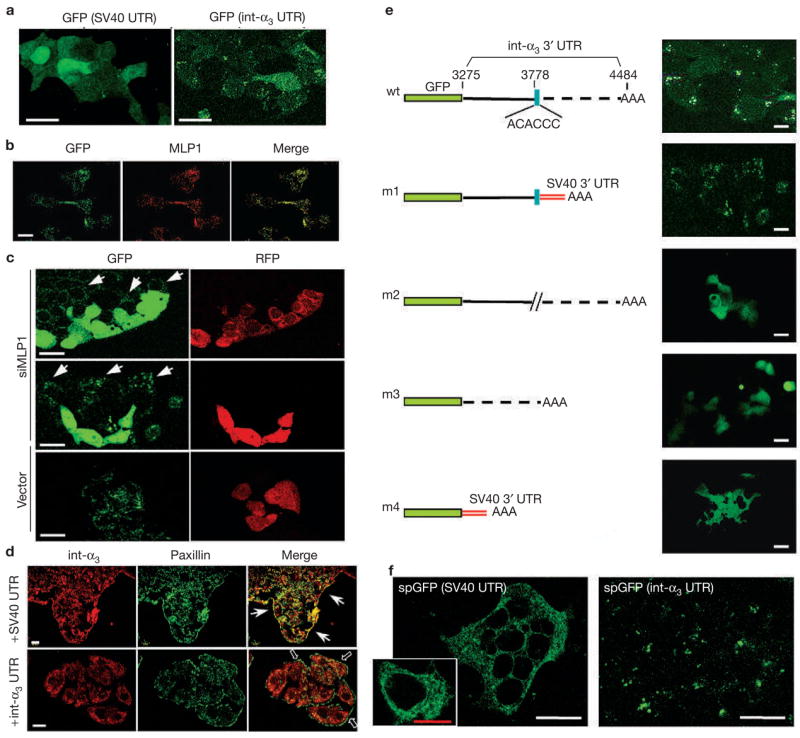
Localized GFP expression mediated by the zipcode in integrin α3 3′ UTR. (a) HT29 cells were transfected with vectors containing enhanced green fluorescent protein (eGFP) open reading frame upstream of the SV40 3′ untranslated region (UTR) or the integrin α3 (int-α3) 3′UTR, and observed for GFP expression. With SV40 3′ UTR, GFP showed a diffuse expression pattern throughout the cell body. With integrin α3 3′ UTR, GFP was localized in a punctated pattern. (b) The same HT29 cells that were transfected with GFP–integrin-α3 chimaeras were fixed and stained for GFP and MLP1. The two proteins colocalized in subcellular punctates. (c) HT29 cells expressing GFP–integrin-α3 chimaeras were transfected with vector containing red fluorescent protein (RFP) alone (vector) or RFP and MLP1 small interfering RNA (siRNA) #1 (siMLP1; see Fig. 1). Live cells were examined. In the presence of siMLP1 (co-expressing RFP) the GFP became diffuse, whereas in neighbouring untransfected cells (arrows) the GFP remained in a punctated pattern. Transfection by the vector alone did not have any effects on the punctated GFP expression. (d) Transfected HT29 cells (+SV40 UTR or +int-α3 UTR) were stained for the endogenous integrin α3 (red) and paxillin (green). Ectopic overexpression of integrin α3 3′ UTR resulted in dispersion of endogenous integrin α3 protein from the paxillin-containing plaques (empty arrows), whereas overexpression of SV40 3′ UTR had little effect. Arrows point to plaques containing integrin α3 and paxillin at the leading edges. (e) Various deletion constructs of the integrin α3 3′ UTR were tested for the essential cis-acting localization element. AAA depicts the polyadenylation signal. SV40 3′ UTR that was contained in the cloning vector provided the default polyadenylation signal for the m1 and m4 constructs. The zipcode (blue box) seemed to be the most critical cis element. wt, wild type. (f) Chimeric integrin α3 signal peptide (sp)-GFP–SV40 or spGFP–integrin-α3 3′ UTR were transfected into HT29 cells and the GFP patterns were observed. With SV40 3′ UTR, the expected membrane-targeted GFP (spGFP) appears associated with an intracellular network, presumably the ER. The inset shows an enlarged view of a single cell displaying the network-bound GFP. With the integrin α3 3′ UTR, the GFP remains in the same punctated pattern as without the signal peptide. Scale bars, 20 μm, except the red bar in the inset, which is 10 μm.
To establish the role of MLP1 in regulating GFP expression under control of the integrin α3 3′ UTR, HT29 cells stably expressing the GFP–integrin-α3 chimaeras were treated with the MLP1-specific siRNA as described above (Fig. 1). As shown in Fig. 5c, the GFP in siRNA/RFP-expressing cells became diffuse, whereas the GFP remained in the punctates near the cell periphery in the neighbouring ‘wild-type’ cells (arrows). Thus, integrin α3 3′ UTR-mediated GFP protein localization is regulated by MLP1. Furthermore, the exogenously overexpressed integrin α3 3′ UTR can disrupt the endogenous integrin α3 protein localization to the paxillin-containing plaques at the periphery, whereas the generic SV40 3′ UTR cannot (Fig. 5d), indicating that trans-acting factors such as MLP1 are involved in RNA-mediated protein localization.
The specific role of the zipcode motif was then assessed. More than half of the integrin α3 3′ UTR could be deleted from the 3′ end up to the zipcode motif, without affecting the punctated GFP distribution pattern (Fig. 5e, compare wt with m1). Note that in this context (m1 variant), the integrin α3 polyadenylation signal is removed and the chimaeras utilize the downstream SV40 3′ UTR that is present in the cloning vector. This indicates that the punctated GFP pattern is not a default state that can be overridden by the SV40 3′ UTR. This notion is further supported when the exact 6-nucleotide zipcode motif was removed and the GFP became diffuse (Fig. 5e, m2). In addition, the 3′ half of the 3′ UTR did not contain any localization signal (Fig. 5e, m3). Complete elimination of the integrin α3 3′ UTR resulted in confluent GFP expression, as expected (Fig. 5e, m4). The GFP reporter assay described above, although informative, may not completely reflect the endogenous integrin α3 expression, as GFP is not normally membrane-targeted. To address this potential problem, the native integrin α3 signal peptide coding sequence, together with the endogenous Kozak translation initiation signal (see Methods), was fused to the GFP coding sequence in the presence or absence of the integrin α3 3′ UTR. As shown in Fig. 5f, the membrane-targeted GFP (spGFP) that was directed by the SV40 3′ UTR still appeared throughout the cells, but was now associated with an intracellular network, presumably the endoplasmic reticulum (ER). When directed by the integrin α3 3′ UTR, the membrane-targeted GFP retained the same punctated pattern. Taken together, our results indicate that the integrin α3 3′ UTR-mediated protein localization is independent of, and probably precedes, the signal-peptide-directed membrane targeting.
mRNA localization is a major cellular mechanism that generates asymmetry in oocyte development and early embryogenesis14,15. However, some evidence indicates that mRNA trafficking may, in fact, play an equally important role in the normal maintenance of somatic cells16–18. In this report, we describe the first mammalian transmembrane protein that is regulated by RNA localization, which is dependent on the function of MLP1 and is mediated in cis by a 3′ UTR motif — the zipcode ACACCC — first identified in the β-actin mRNA19. In the case of actin mRNA, the localization in neurons and in fibroblasts can be mediated by a few unrelated zipcode-binding proteins13,19. It is possible that different proteins may regulate zipcode-containing messages independently or in different combinations in different cell types. Interestingly, we showed that MLP1 can bind to the actin zipcode and that actin may be similarly regulated by MLP1. We demonstrated that integrin α3 3′ UTR can localize GFP protein independently of the native integrin α3 translation initiation signal and the signal peptide. This further indicates that the RNA-directed protein localization mechanism may consist of distinct steps. We propose that the zipcode-containing RNAs are transported to distinct subcellular loci, some of which are near the cell periphery, where translation occurs. Cytosolic proteins are then engaged in the nearby structures (for example, adhesion plaques), where they are needed. Signal-peptide-containing proteins are synthesized in the ER at the cell periphery and translocated directly to the nearby membranous domains. The precise mechanism of the post-translational transport indicated here remains to be elucidated; however, new data do support some of the features of the model. A recent report demonstrated that translational machinery, such as ribosomes, along with mRNAs and hnRNPs, could be found in nascent adhesion structures during cell spreading20. Also, membrane localization of the yeast surface protein Ist2 is dependent on peripherally localized mRNA21. These findings are consistent with the suggestion that integrin α3 and other cytoskeletal components can be synthesized at or near the adhesion complexes. Also, it has been observed that the ER network is dynamic and continues to extend to the cell periphery in step with lamellipodia spreading22. As such, the emerging importance of RNA-directed protein localization may entail an underlying mechanism that allows for rapid expression of adhesion components at specific sites at which they are needed.
METHODS
Cell culture and transfection
H520 cells derived from human lung squamous cell carcinoma, A549 cells derived from lung carcinoma and HT29 cells derived from human colon adenocarcinoma were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in RPMI, DMEM and McCoy’s 5A, respectively, and all supplemented with 10% fetal calf serum (FCS). Transfection was carried out using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) in a six-well format.
For nocodazole treatment, HT29 cells were grown on coverslips until 80% confluency. Fresh prewarmed media supplemented with 0.05 μg ml−1 nocoda-zole (Sigma, St. Louis, MO) was used to replace the old media and plates were returned to the incubator for 4 h before nocodazole was rinsed off and fresh media was added.
MLP1 antibody
MLP1 antibody was generated as follows: the entire MLP1 ORF was cloned into the pCRT7/CT–TOPO vector (Invitrogen) that was fused to a His6-tag at the carboxyl terminus. BL21<DE3> bacteria-produced full-length MLP1 protein was purified using a TALON protein purification column (Clontech, Mountainview, CA). Purified MLP1 protein was used for injection into rabbits at the MUSC Antibody Production Facility (Medical University of South Carolina, Charleston, SC). The antibody was affinity-purified against MLP1 protein immobilized on CNBr-activated Sepharose-6MB (Amersham, Picataway, NJ). The specificity of the purified MLP1 antibody was confirmed by western blot analysis (Fig. 1a, d).
Western blot analysis
For protein analysis, near-confluent cells from three wells of a six-well plate were scraped, washed with phosphate-buffered saline (PBS) and resuspended in 150 μl RIPA containing a protease-inhibitor cocktail (Roche, Indianapolis, IN; one tablet dissolved in 1 ml H2O and used as 10 × stock). The cell suspension was sonicated with two 5-s pulses. Cleared lysate was obtained by centrifugation of the sonicate at 10,000 g at 4 °C in a microfuge. Protein concentration was estimated using a BCA protein assay kit (Pierce, Rockford, IL). Proteins (10–50 μg/lane of total protein) were separated on 10% SDS–PAGE gels before being electroblotted onto a nitrocellulose membrane (Invitrogen) and incubated with primary antibody.
Mouse monoclonal antibodies used were: GAPDH (1:6000; Research Diagnostics, Flanders, NJ); and integrin α3 (CD49c) (1:1000; Chemicon, Temecula, CA). Rabbit polyclonal antibodies used were: MLP1 (1:2000; see above). HRP-conjugated goat anti-mouse immunoglobulin G (IgG) (1:6000; Sigma) and goat anti-rabbit IgG (1:3000; Pierce) were used for western blotting and visualized using enhanced chemiluminescence reagents (Pierce).
MLP1 recombinant adenovirus
Recombinant adenovirus of MLP1 was produced using the AdEasy System (Qbioben, Irvine, CA), according to the manufacturer’s instructions. The Ad5CMV vector carried GFP for monitoring infection. Expression of MLP1 by the recombinant viruses was verified by western blotting (Fig. 1b). For infections of H520 cells, viruses were used at multiplicity of infection (m.o.i.) values of 5 or 10.
Migration assays
In transwell (Corning, Acton, MA) migration assays, both sides of the filters (pore size: 8.0 μm) were coated at 4 °C overnight with 0.25 μg ml−1 of laminin-5 (BD Biosciences) diluted in PBS. Excessive laminin was rinsed off with PBS and the filter was air-dried at room temperature. H520 cells (3 × 104 cells/filter) were seeded into the upper chamber in RPMI medium supplemented with 1% FCS, and allowed to migrate towards the lower chamber media with a high serum concentration (10%) for 12 h at 37 °C. Cells were then rinsed in PBS, fixed with methanol and stained using crystal violet. The upper side of the filter was wiped with a cotton swab to remove cells that had not migrated through the filter. Filters were viewed under bright-field optics and stained cells were counted in four fields (using a ×10 objective). Each experiment was performed in triplicate and the data were indicated from an average of three such independent experiments.
RT-PCR analyses
Total RNA was isolated from H520 cells infected with adeno-virus expressing MLP1 or GFP (×5 m.o.i) using the Trizol reagent (Invitrogen). RT-PCR was performed using the ProtoScript kit (New England Biolabs, Ipswitch, MA). Briefly, 1 μg of total RNA was reverse-transcribed using random primers and M-MuLV reverse transcriptase. One-tenth of the cDNA products were used for subsequent PCR reactions. The integrin α3 exon 32 primers used were: 5′ CAGATCATGCCCAAGTACCA and 3′ AAAGCAAGCCTTCTGGGTC, which generated an expected 741-bp product. The integrin α3 intron 31/exon 32 primers used were: 5′ CCTCTCTGTGATGACCTTGC and 3′ AAAGCAAGCCTTCTGGGTC, which generated an expected 1283-bp product. MLP1 cDNA primers used were: 5′ CAATTTATCACTCACCTTCA GACTTAC and 3′ TTGTGTCGATCATGGTGCTGTC, which generated an expected 754-bp product across exons 2–5. β-actin cDNA primers used were: 5′ AGCGGGAAATCGTGCGTGAC and 3′ ATCTCCTTCTGCATCCTGTCGG, which generated a 337-bp fragment across exons 3–4. GAPDH cDNA primers used were: 5′ ATGCCTCCTGCACCACCAACTG and 3′ TGCCAGTGAGCTTCCCGTTCAG, which generated a 246-bp fragment across exons 6–7. Paxillin cDNA primers used were: 5′ CTCCAAACGGCCTGTGTTCTTG and 3′ GGAGCCGTACACAGGTGATGAG, which generated a 217-bp fragment across exons 2–3. FAK cDNA primers used were: 5′ ATGCTTCAAGTGTGCTCTTG and 3′ TTAGGTCAGCCATATTCTCC, which generated a 263-bp fragment across exons 3–6. PCR cycling parameters were: denaturation at 94 °C for 1 min, annealing at 58 °C for 1 min and extension at 72 °C for 1 min. The cycle numbers were empirically determined for each primer set so that the product amplification was within the linear range: 31 cycles for the integrin α3 intron–exon primers, 30 cycles for the integrin α3 exon primers, 27 cycles for the MLP1 exon primers, and 26 cycles each for GAPDH, paxillin, FAK and actin primers.
Plasmid-based siRNA gene silencing
The duplex oligonucleotides encoding siRNAs (MLP1 siRNA #1: 1740GGTCAGTTTTGTGCATGAC1758; siRNA #2: 1417CAACACCGTAACCGTTTGT1435; numbers correspond to the nucleotide positions in AF491866) were initially cloned into pSuppressor-Neo (Imgenex, Sorrento Valley, CA) under the control of a U6 promoter. The entire U6-promoter–siRNA-cassette (BamHI-BglII fragment) was blunt-ended by T4 polymerase (New England Biolabs) and subcloned into the AseI site of pDsRed1–N1 (BD Biosciences) to create a bicistronic vector expressing both RFP and MLP1 siRNA, with the respective promoters being oriented in the opposite direction to minimize promoter interference.
Immunofluorescence
For indirect immunofluorescence, cells were grown on coverslips and fixed in PBS + 3.7% formaldehyde for 10 min, and washed twice with PBS. Cells were then permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature and stained with respective antibodies in PBS + 1% bovine serum albumin (BSA). Antibodies used were: rabbit anti-MLP1 (1:100); mouse anti-integrin α3 (1:100; Chemicon); mouse anti-pFAK-Y397 (1:100; BD Biosciences, San Jose, CA); mouse anti-GFP (1:100; Stressgen, San Diego, CA); and rabbit anti-paxillin (1:100; Cell Signaling, Beverly, MA). Secondary antibodies (Invitrogen) were highly cross-absorbed goat antibodies against rabbit IgG or mouse IgG, conjugated to either AlexaFluor 488 or 546 and used at 1:150 dilutions in PBS + 1% BSA.
Co-detection of integrin α3 RNA and MLP1 protein
The integrin α3 (NM_005501) 3′ UTR probe was generated as follows: the 3′ UTR (nucleotides 3275–4484) cDNA fragment was generated by PCR and cloned into the KpnI–HincII sites of pBlueScript II KS (Stratagene, La Jolla, CA). The integrin α3 3′ UTR DNA fragment was PCR-amplified using a T7/T3 primer pair. A digoxygenin-labelled antisense RNA probe was synthesized using the DIG labelling mix and a T7 RNA polymerase (Roche). Sense probe (synthesized from the T3 promoter) was used as a negative control. The probes were then purified on an RNase-free Sephadex G50 spin column (Roche), and ethanol precipitated. The resulting pellet was dried and resuspended in 95 μl H2O. To optimize the probe size, the intact 1,300-nucleotide-labelled RNA (containing the 1,200-nucleotide integrin α3 3′ UTR and ~100-nucleotide plasmid sequence) was partially hydrolysed by incubating the probe in 0.1 M Na-carbonate, pH 10.2, at 60 °C for 28 min, aiming to generate an average probe size of ~260 nucleotides based on the equation: time (min) = (Lo−Lf)/(K)(Lo)(Lf); Lo = starting length in kb, Lf = final length in kb, K(rate constant) = 0.11 kb−1min−1. The reaction was stopped by changing the hydrolysis solution to 0.5% acetic acid. RNA was then ethanol-precipitated, and the resulting pellet was dried and resuspended in 10 μl H2O. The labelled RNA content was measured against control, labelled actin RNA and reagents from the DIG labelling and detection kit (Roche).
Cells were grown directly onto coverslips, washed once in PBS and fixed for 10 min at room temperature in PBS containing 3.7% formaldehyde and 10% acetic acid. After two 5-min washes in PBS, cells were permeabilized by treatment with PBS containing 0.5% Triton X-100 for 10 min. Cells were prehybridized for 5 min at room temperature in 1× SSC/50% formamide. Cover slips were then incubated overnight at 50°C in hybridization mixture (1× SSC/40% formamide, 10% dextran sulphate, 0.4% BSA, 20 mM ribonucleotide–vanadyl complex, 10 μg ml−1 salmon testes DNA, 10 μg ml−1 tRNA, 10 mM Na-phosphate, pH 7, 0.75 ng ml−1 labelled probe). Following hybridization, coverslips were washed twice with 4× SSC/40% formamide at 37 °C for 10 min each, twice with 2× SSC/40% formamide at 37 °C for 10 min each, and three times with 2× SSC at room temperature for 10 min each. The coverslips were then blocked in TBS (25 mM Tris-HCl, pH 7.6/125 mM NaCl) containing 2% BSA and 2% FBS at 37 °C for 1 h and then incubated with rabbit anti-MLP1 antibodies (1:100) in TBS containing 1% BSA at 37 °C for 1 h, followed by two washes of 10 min each with TBS containing 1% BSA. Cover slips were then incubated at 37 °C for 1 h with Cy3-conjugated monoclonal antibody against digoxigenin (1:100; Roche) and goat anti-rabbit-AlexaFluor 488 (Invitrogen) in TBS containing 1% BSA.
RNA pulldown
MLP1-expressing adenovirus was used to infect H520 cells (8.8 × 106 cells in a 100-mm dish) at 10 m.o.i. After 12 h, cells were harvested, washed in cold PBS and homogenized by passing through a 21-gauge needle in 1 ml of ice-cold lysis buffer (10 mM HEPES, pH 7.4, 200 mM NaCl, 0.5% Triton X-100, 30 mM EDTA) in the presence of protease inhibitors (Roche) and 30 U/ml RNasin (Promega, Madison, WI). Cleared lysate was obtained by centrifugation at 10,000 g at 4°C in a microcentrifuge. Lysates were then preadsorbed for 1 h with 20 μl of lysis-buffer-equilibrated protein-A agarose (50% slurry; Biorad, Hercules, CA). The preadsorbed lysate was incubated with 15 μg of affinity-purified MLP1 polyclonal antibodies and 40 μl of lysis-buffer-equilibrated protein A agarose for 12 h at 4 °C, then rinsed three times with lysis buffer at 4 °C. 50 U/ml RNase-free DNase I (Roche) was added in the last rinse. The immunoprecipitate was resuspended in 100 μl H20 and boiled for 2 min. The immunoprecipitated RNA was isolated using Trizol reagent (Invitrogen), purified with the RNeasy Mini Kit (Qiagen, Valencia, CA) and eluted in 30 μl H20. The concentration and purity of the RNA were determined by measurement of the optical densities at 260 and 280 nm. First-strand cDNA was generated by reverse transcription that was performed on one-fifth of the co-immunoprecipitated RNA in a 20 μl volume using the ProtoScript kit (New England Biolabs) and random primers. The cDNA products were then used directly for the subsequent PCR amplification using ThermoPol (New England Biolabs) and 25 pmoles each of the integrin α3 3′ UTR primers3149CAGATCATGCCCAAGTACCA3168 and 3898AAAGCAAGC CTTCTGGGTC3880 (as in sequence NM_005501), as well as other gene-specific primers (paxillin, FAK, actin and GAPDH) described above.
RNA probes in binding assay
The 40-nucleotide native actin probe containing the zipcode (underlined) motif found in the human β-actin mRNA 3′ UTR was purchased from Qiagen: CUUAGUUGCGUUACACCCUUUCUUGACAAAA CCUAACUUG. The artificial zipcode 10× repeat probe was purchased from Qiagen. Poly(A) RNA was purchased from Pharmacia. The integrin α3 3′ UTR fragment was PCR-amplified using T3/T7 primers, as described above, and gel-purified. This T3/T7 promoter-containing DNA fragment was then used as a template for in vitro RNA synthesis using T3 RNA polymerase (Roche). The zipcode sequence (nucleotides 3778–3783) was precisely deleted from the integrin α3 3′ UTR by site-directed mutagenesis using the ExSite PCR mutagenesis kit (Stratagene). Specifically, the pBlueScript II KS plasmid containing the integrin α3 3′ UTR fragment was used as a template and primers (3784ATGCCAGAGA GGTGGGGATCC3804, 3777CAATTGGGGGCCCACCAGCCA3757) were used to generate the site-directed deletion of the zipcode motif.
All probes were treated with shrimp alkaline phosphatase (Roche) and 5′ end-labelled in the presence of γ-32P-ATP and T4 polynucleotide kinase (New England Biolabs). Labelled RNAs were purified through a RNase-free Sepadex G25 spun column (Roche). RNA probes were heated at 80°C for 3 min and chilled on ice for 5 min just before use. An aliquot (5,000 cpm) was run on polyacrylamide gel to verify the integrity.
In vitro RNA binding (Northwestern) assay
BL21<DE3> cells carrying pCRT7/CT–TOPO or a vector containing the MLP1 ORF, as described above, were grown to mid-log phase. Whole-cell lysates were run on 10% SDS–PAGE and electrob-lotted onto a nitrocellulose membrane. The transferred proteins were renatured as follows: the blot was first incubated in binding buffer (0.1 M HEPES, pH 7.9, 0.1 M MgCl2, 0.1 M KCl, 0.5 μM ZnSO4) containing 1 mM DTT and 6 M urea for 5 min at room temperature. The blot was then incubated, for 5 min each, through five serial twofold dilutions of urea in binding buffer containing 1 mM DTT. The blot was then placed in binding buffer/1 mM DTT without urea. Prehybridization was carried out in binding buffer (1 mM DTT, 5% BSA, 1 μg ml−1 yeast tRNA) for 1 h at room temperature. After addition of the 32P-labelled RNA probe (0.6 × 106 cpm ml−1 in binding buffer (1 mM DTT, 0.25% BSA, 1 μg ml−1 yeast tRNA), the membrane was hybridized overnight at 4°C. The membrane was then washed once for 10 min using chilled hybridization buffer and incubated at room temperature, then rinsed twice for 10 min in 2 × binding buffer containing 1 mM DTT, 0.25% BSA and 0.1% Triton X-100 at room temperature. The membranes were air-dried and autoradiographed.
GFP reporter constructs
The KpnI–HincII fragment of the integrin α3 3′ UTR, as described in RNA in situ, was subcloned into the KpnI–SmaI site of the pEGFP–C1 plasmid immediately downstream of the GFP coding sequence. Control vector or the integrin α3 3′ UTR-containing plasmid was transfected into HT29 cells. Pools of stable transformants were maintained. For membrane-targeted GFP constructs, the integrin α3-specific PCR primers —5′ CGCTTCCGCTGGCAGCCAT and 3′ CTGGCTGGCCACAGTCACT — were used to generate a fragment encompassing 17-nucleotides upstream of the start codon and the coding sequence of the first 125 amino acids. The fragment was cloned into the blunt-ended NheI site of the pEGFP–C1 plasmids described above, generating an in-frame fusion. All chimeric plasmids were sequence-verified before use.
Supplementary Material
Note: Supplementary Information is available on the Nature Cell Biology website.
Acknowledgments
The work was supported by a Department of Defense Phase V Research Grant to Y.A., a Department of Defense Phase VII Geo Center grant to V.D., a grant from the National Cancer Institute (K22CA109577) to R.L., and grants from National Cancer Institute (RO1CA109860 and PO1CA78582) to T.H.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Begemann G, Paricio N, Artero R, Kiss I, Perez-Alonso M, Mlodzik M. Muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development. 1997;124:4321–4331. doi: 10.1242/dev.124.21.4321. [DOI] [PubMed] [Google Scholar]
- 2.Artero R, et al. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev Biol. 1998;195:131–143. doi: 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- 3.Miller JW, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fardaei M, Larkin K, Brook JD, Hamshere MG. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001;29:2766–2771. doi: 10.1093/nar/29.13.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fardaei M, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 6.Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum Mol Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 7.Ladd AN, Stenberg MG, Swanson MS, Cooper TA. Dynamic balance between activation and repression, regulates pre-mRNA alternative splicing during heart development. Dev Dyn. 2005;233:783–793. doi: 10.1002/dvdy.20382. [DOI] [PubMed] [Google Scholar]
- 8.Fukushima Y, Ohnishi T, Arita N, Hayakawa T, Sekiguchi K. Integrin α3β1-mediated interaction with laminin-5 stimulates adhesion, migration and invasion of malignant glioma cells. Int J Cancer. 1998;76:63–72. doi: 10.1002/(sici)1097-0215(19980330)76:1<63::aid-ijc11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 9.Lohi J. Laminin-5 in the progression of carcinomas. Int J Cancer. 2001;94:763–767. doi: 10.1002/ijc.1539. [DOI] [PubMed] [Google Scholar]
- 10.Santoro MM, Gaudino G, Pier Carlo Marchisio PC. The MSP receptor regulates α6β4 and α3β1 integrins via 14-3-3 proteins in keratinocyte migration. Dev Cell. 2003;5:257–271. doi: 10.1016/s1534-5807(03)00201-6. [DOI] [PubMed] [Google Scholar]
- 11.Kislauskis EH, Li Z, Singer RH, Taneja KL. Isoform-specific 3′-untranslated sequences sort α-cardiac and β-cytoplasmic actin messenger RNAs to different cytoplasmic compartments. J Cell Biol. 1993;123:165–172. doi: 10.1083/jcb.123.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bassell GJ, et al. Sorting of β-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18:251–265. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu W, Pan F, Zhang H, Bassell GJ, Singer RH. A predominantly nuclear protein affecting cytoplasmic localization of β-actin mRNA in fibroblasts and neurons. J Cell Biol. 2002;156:41–51. doi: 10.1083/jcb.200105133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Micklem DR. mRNA localisation during development. Dev Biol. 1995;172:377–395. doi: 10.1006/dbio.1995.8048. [DOI] [PubMed] [Google Scholar]
- 15.Johnstone O, Lasko P. Translational regulation and RNA localization in Drosophila oocytes and embryos. Annu Rev Genet. 2001;35:365–406. doi: 10.1146/annurev.genet.35.102401.090756. [DOI] [PubMed] [Google Scholar]
- 16.Fulton AB. Spatial organization of the synthesis of cytoskeletal proteins. J Cell Biochem. 1993;52:148–152. doi: 10.1002/jcb.240520206. [DOI] [PubMed] [Google Scholar]
- 17.Kislauskis EH, Singer RH. Determinants of mRNA localization. Curr Opin Cell Biol. 1992;4:975–978. doi: 10.1016/0955-0674(92)90128-y. [DOI] [PubMed] [Google Scholar]
- 18.Singer RH. The cytoskeleton and mRNA localization. Curr Opin Cell Biol. 1992;4:15–19. doi: 10.1016/0955-0674(92)90053-f. [DOI] [PubMed] [Google Scholar]
- 19.Ross AF, Oleynikov Y, Kislauskis EH, Taneja KL, Singer RH. Characterization of a β-actin mRNA zipcode-binding protein. Mol Cell Biol. 1997;17:2158–2165. doi: 10.1128/mcb.17.4.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Hoog CL, Foster LJ, Mann M. RNA and RNA binding proteins participate in early stages of cell spreading through spreading initiation centers. Cell. 2004;117:649–662. doi: 10.1016/s0092-8674(04)00456-8. [DOI] [PubMed] [Google Scholar]
- 21.Jüschke C, Ferring D, Jansen RP, Seedorf M. A novel transport pathway for a yeast plasma membrane protein encoded by localized mRNA. Curr Biol. 2004;14:406–411. doi: 10.1016/j.cub.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 22.Waterman-Storer CM, Salmon ED. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr Biol. 1998;8:798–806. doi: 10.1016/s0960-9822(98)70321-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary Information is available on the Nature Cell Biology website.