

Abstract
We report a recurrent microdeletion syndrome causing mental retardation, epilepsy and variable facial and digital dysmorphisms. We describe nine patients, including six probands; two with de novo deletions, two who inherited the deletion from an affected parent, and two with unknown inheritance. The proximal breakpoint of the largest deletion is contiguous with breakpoint 3 (BP3) of the Prader-Willi and Angelman syndrome region extending 3.95 Mb distally to BP5. A smaller 1.5 Mb deletion has proximal breakpoint within the larger deletion (BP4) and shares the same distal BP5. This recurrent 1.5 Mb deletion contains six genes, including a candidate gene for epilepsy (CHRNA7) that is likely responsible for the observed seizure phenotype. The BP4-BP5 region undergoes frequent inversion, suggesting a possible link between this inversion polymorphism and recurrent deletion. The frequency of these microdeletions in mental retardation cases is ~0.3% (6/2082 tested), a prevalence comparable to that of the Williams, Angelman, and Prader-Willi syndromes.
Keywords: genomic disorder, array CGH, epilepsy, segmental duplication, CHRNA7
Genomic disorders result from DNA rearrangements mediated by non-allelic homologous recombination (NAHR) between large, highly homologous flanking segmental duplications1. The clinical features of many of the known genomic disorders include mental retardation and developmental delay; and several recent studies of individuals with mental retardation have led to the identification of novel recurrent genomic disorders2. By whole-genome array CGH screening of 757 patients with mental retardation and/or congenital anomalies, we identified two unrelated individuals with mild to moderate MR, dysmorphic features, and abnormal EEG findings who both have identical de novo 1.5 Mb deletions of 15q13.3. These two deletions share the same distal breakpoint (BP5) with a previously reported 3.95 Mb deletion of 15q133 (Figure 1, Figure 2a), with a proximal breakpoint (BP4) within this larger deletion (Figure 1, Figure 2b, 2c and 2d). The shared 1.5 Mb region contains six known genes. Our array CGH screening also detected a single patient with a proximal deletion breakpoint corresponding to breakpoint region 3 (BP3) of the Prader-Willi and Angelman syndrome region and a distal breakpoint at BP4 (Patient 543/06, Figure 1). However, the deletion was also detected in the patient’s unaffected father. We therefore interpret this BP3-BP4 deletion as likely representing a benign copy number variation, although we cannot exclude that it may instead be a pathogenic deletion with incomplete penetrance.
Figure 1.
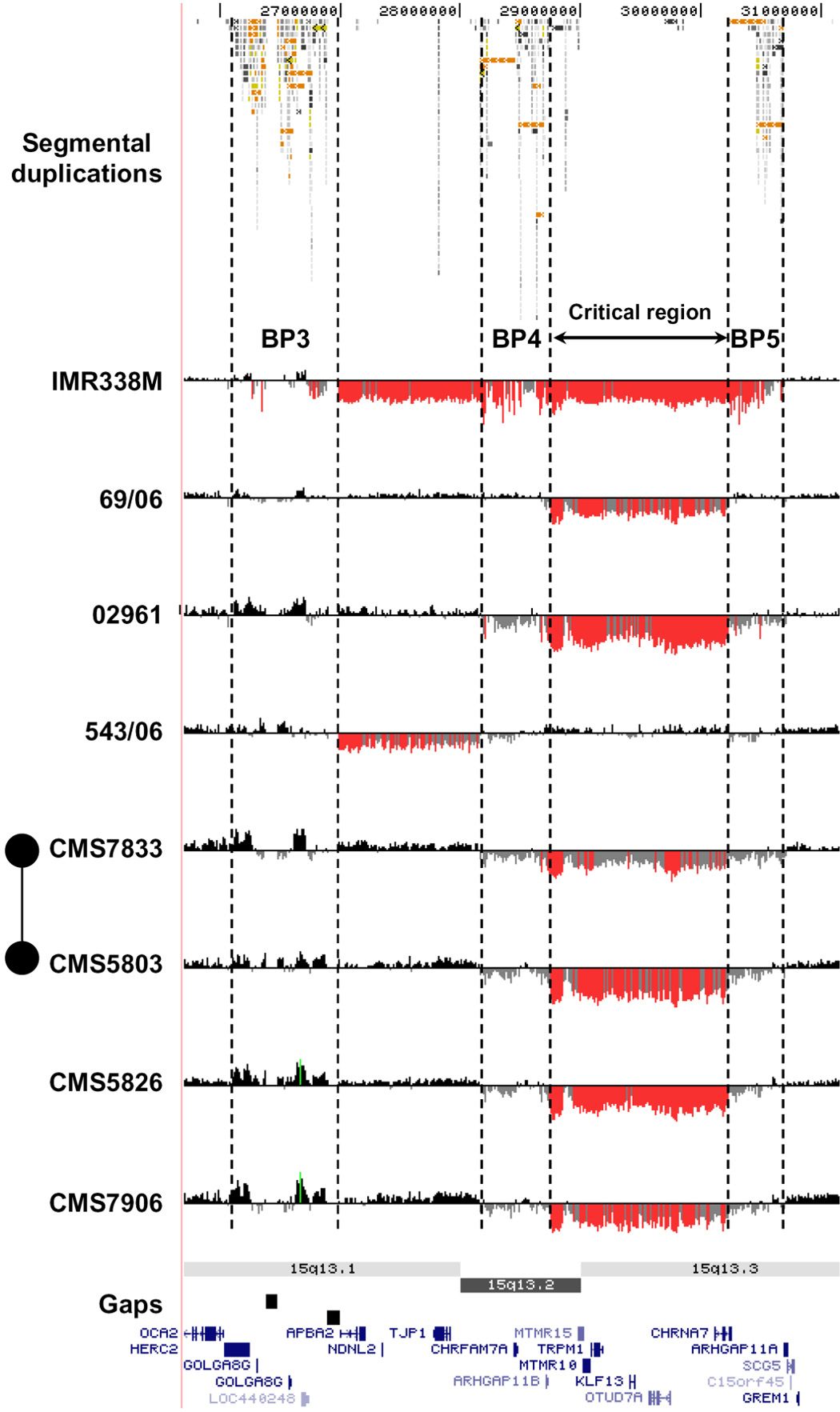
High-resolution oligonucleotide array mapping of 15q12-q13.3 rearrangements (chr15:25,700,000–31,400,000). Although there appears to be variation in the exact location of breakpoints, all map to large blocks of segmental duplication at BP3, BP4, and BP5 (indicated by dashed lines). For each individual, deviations of probe log2 ratios from zero are depicted by grey/black lines, with those exceeding a threshold of 1.5 standard deviations from the mean probe ratio coloured green and red to represent relative gains and losses, respectively. Segmental duplications of increasing similarity (90–98%, 98–99%, and >99%) are represented by grey/yellow/orange bars, respectively. A number of other 15q rearrangements with breakpoints mapping to BP3, BP4, and BP5 are shown in Supplementary Figure 5.
Figure 2.
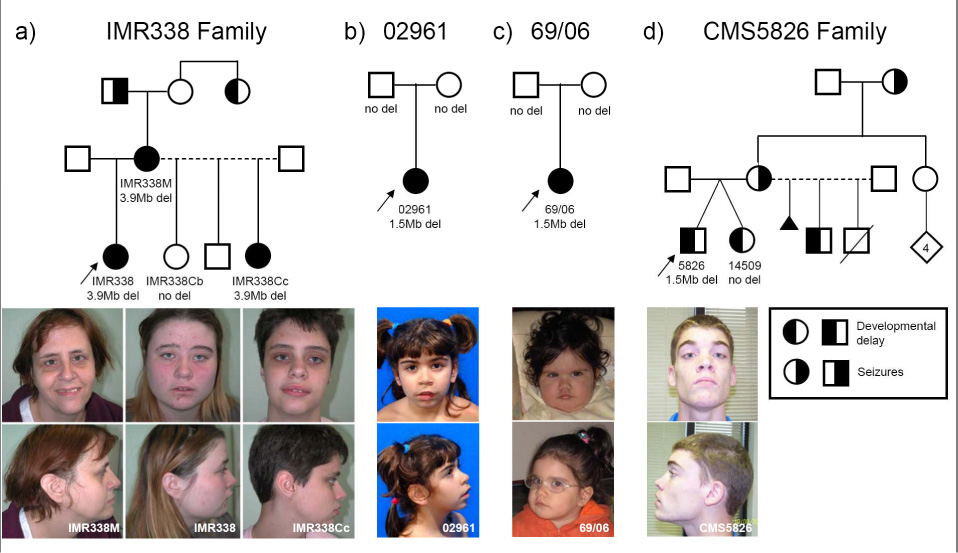
Pedigrees and patient photographs of 15q13 deletions. Developmental delay and seizure phenotype is indicated by left- and right-half shaded symbols, respectively. Presence or absence of 15q13 deletions is shown below each symbol in all individuals tested (absence of text indicates unavailable for testing). Photographs of affected family members are below each pedigree. We obtained consent to publish photographs from each individual included in this figure. (a) Family of proband IMR338. All affected individuals have 3.9 Mb deletions. Note the full everted lips and deep-set eyes evident in affected individuals. IMR338Cb is unaffected and does not have the deletion. (b) Patient 02961 (de novo deletion); note hypertelorism, synophrys, prominent philtrum, everted upper lip, and hypotonic facies. (c) Patient 69/06 (de novo deletion); note the prominent philtrum, everted upper lip, hypertelorism, and hypotonic facies. (d) Family of proband CMS5826. Note upslanting palpebral fissures and prominent philtrum in the patient.
In order to rapidly screen a large collection of affected individuals for deletions in the shared 1.5 Mb interval between BP4 and BP5, we developed two TaqMan quantitative PCR (qPCR) assays targeted to this region and screened 1040 individuals with mental retardation of unknown etiology. This cohort, obtained from the Greenwood Genetic Center (South Carolina), consists of an approximately equal number of individuals of Caucasian and African American descent. qPCR analyses identified four individuals as potentially harboring a deletion of the interval BP4-BP5. Samples were subsequently validated by BAC array comparative genomic hybridization (data not shown) and a custom oligonucleotide array (Figure 1). Review of pedigrees showed evidence of multiple affected individuals for each case (Figure 2, Supplementary Figure 1), and review of the sample collection revealed that, although the series was thought to have only unrelated individuals, two of the patients we identified with deletions are mother (CMS7833) and son (CMS5803).
Review of the phenotypes observed in the nine individuals identified with deletions of 15q13 shows a number of phenotypic features (summarized in Table 1 and Figure 2, with full phenotype details in Supplementary Note 1). The most consistent features among the patients in our series are mild to moderate mental retardation (9/9 cases) and epilepsy and/or abnormal EEG findings, which were noted in seven of the nine individuals. All patients also have mild facial dysmorphism, though these features are variable. Features shared among three or more patients include hypertelorism, upslanting palpebral fissures, prominent philtrum with full everted lips, and short and/or curved fifth finger and short fourth metacarpals. Related to the latter, it is noteworthy that skeletal and/or joint defects of the hand were noted in seven of nine patients (Table 1). Testing for the 15q13.3 deletion should therefore be considered in patients with unexplained mental retardation, seizures, and mild dysmorphic features.
Table 1.
Phenotypic features of nine patients with deletions of 15q13
| Patient ID / inheritance | Cognitive | Growth | Facial features | Hands | Neurologic | Other |
|---|---|---|---|---|---|---|
| 69/06 | Moderate delays; | BW 3rd centile | Mild hypertelorism; | Moderate to severe | EEG: unusual delta-theta | |
| de novo, | Brunet-Lezine global | OFC (birth) 25th centile | prominent philtrum; | hypotonia; | activity with sporadic slow | |
| maternal | score 0.37 | At 2 yrs: | everted upper lip; | stereotypic hand | waves. | |
| origin | Wt 75th centile | small L eye; strabismus; | movements; | Normal brain MRI scan | ||
| Ht 50th centile | astigmatism | abnormal EEG | Right-side heart defect | |||
| OFC 10th centile | ||||||
| 02961 | Mild MR; | Wt 25th centile | Brachycephaly; hypertelorism; | Tapering fingers; | ADHD; | EEG: 3 c/s Sharps-Waves |
| de novo, | speech delay | Ht 3rd–10th centile | astigmatism; synophris; | 5th-finger | hypotonia; | complexes, high voltage spikes |
| paternal | OFC 10th–25th centile | wide nasal bridge; anteverted nares; | clinodactyly | myoclonic epilepsy | in frontal region. | |
| origin | short thick philtrum; full, everted | Normal brain MRI scan. | ||||
| upper lip; low-set ears | Early pubarche; hypertrichosis | |||||
| IMR338 | Dev. delay; | Ht 97th centile | Round flat face; upslanting PF; | 5th-finger | Absence seizures; | EEG: abnormal spike and wave |
| maternal | speech difficulties | OFC >97th centile | epicanthic folds; optic pit vs. | clinodactyly; | one tonic clonic | activity, focus in left frontal |
| inheritance | coloboma; everted upper lip; nasal | lax thumb joint; | seizure | area. MRI: patchy change in | ||
| speech | single palmar | white matter adjacent to left | ||||
| crease | ventricle. Hyperphagia, obesity, | |||||
| type II DM | ||||||
| IMR338M | Dev. delay; | Squint | Lax thumb joint | Grand mal | Normal head CT scan | |
| unknown | mild learning | seizures | ||||
| inheritance | difficulties | |||||
| IMR338Cc | Dev. delay; | OFC 90th centile | Deep-set eyes; squint; | Lax thumb joint; | Intractable | |
| maternal | moderate to severe | everted upper lip | long fingers | epilepsy | ||
| inheritance | learning difficulties | |||||
| CMS7906 | Mild to moderate MR | Ht 30th centile | Full round face; upslanting PF; | Short 4th MCPs; | Single seizure, 12 | |
| unknown | IQ 52 | OFC 35th centile | widely-spaced teeth | limited elbow | yrs old | |
| inheritance | extension | |||||
| CMS5826 | Mild to moderate MR | BW 50th centile OFC | Long face; upslanting PF; | Short 5th finger | Mild autism | Normal brain MRI; |
| unknown | IQ 62 at 4 yrs | 30th centile | prominent philtrum; depressed nasal | Aggressive behavior, rage | ||
| inheritance | IQ 44 at 15 yrs | bridge; anteverted nares; thick ear | ||||
| Speech delay | helices | |||||
| CMS5803 | Mild MR | Ht 5th–10th centile | Short philtrum; full lips; everted | Short 4th MCPs; | Hypotonia | |
| maternal | IQ 56 | OFC <3rd centile | lower lip | stiff fingers | ||
| inhertance | ||||||
| CMS7833 | Moderate MR | Ht 5th centile | Round face; depressed nasal bridge; | Short 4th MCPs; | Seizures under | |
| unknown | IQ 34–46 | OFC 3rd centile | smooth philtrum; everted lower lip | stiff fingers | good control with | |
| inheritance | medication | |||||
BW, birth weight; Wt, weight; Ht, height; OFC, occipitofrontal circumference; PF, palpebral fissures; MCP, metacarpals; ADHD, Attention Deficit Hyperactivity Disorder; DM, diabetes mellitus; bold indicates features shared by three or more patients.
Each breakpoint region corresponds to a complex set of segmental duplications, termed duplication blocks. BP4 is an 818 kb duplication block with three large regions of homology to BP5 (95 kb, 140 kb, and 218 kb with 99.6% identity, http://genome.ucsc.edu/), each of which lie in an inverted orientation relative to those at BP5 (Figure 3, Supplementary Table 1 and Supplementary Figure 2). Consistent with NAHR as the mechanism underlying rearrangements of 15q13.3, our data localized the breakpoints of all five BP4-BP5 deletions and a BP4-BP5 duplication to these large paralogous sequences (Figure 1, Figure 4, Supplementary Figure 3). The BP3 region, which is the common distal breakpoint in Prader-Willi/Angelman syndrome deletions, corresponds to an 843 kb duplication block (hr17/Build 35, chr15:26,053,472–26,896,735). Duplicated sequence within BP3 shows relatively small stretches of homology to BP4 (17 kb with 93% identity) and BP5 (30 kb with 98% identity and 11 kb with 93% identity). BP3 also contains two assembly gaps, and our data from IMR338 and 543/06 suggest that the proximal breakpoint in these deletions occur within the distal gap. Given the likelihood that NAHR underlies rearrangements of 15q13, we suggest that this region contain as-yet uncharacterized segmental duplications paralogous to those at BP4 and BP5 which catalyze recurrent rearrangements of 15q13. Thus, the increased prevalence of rearrangements observed between BP4-BP5 relative to those involving BP3-BP4/BP5 is consistent with the size and homology of duplicons in 15q13. It is interesting that most of the breakpoint regions associated with genomic disorders on chromosome 15 correspond to duplication blocks which harbor copies of the GOLGA gene family4, 5. Within the limits of oligonucleotide array comparative genomic hybridization, breakpoint regions appear to overlap with these specific duplicons (Supplementary Figure 4).
Figure 3.
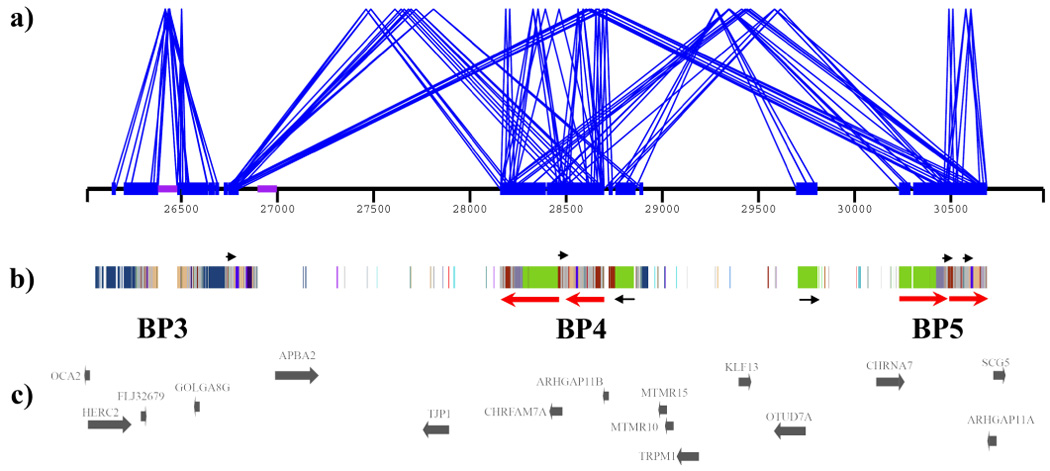
Duplication architecture of 15q13 breakpoint regions. (a) Paralogy between large (≥10 kb), highly identical (≥95%) segmental duplications (blue bars) is shown between BP3, BP4 and BP5 as pairwise alignments (blue lines). Sequence assembly gaps are shown as purple bars. (b) The underlying duplicon structure (blocks of identical colour represent those that share the same evolutionary origin) and orientation (red/black arrows) of pairwise alignments between the blocks9. (c) RefSeq genes. BP3-BP4 and BP3-BP5 share fewer large, high-identity duplications when compared to BP4-BP5 (BP4-BP5, total aligned bp=571.8 kb, mean identity=98.6%) (Supplementary Table 1). Most notable are three segmental duplications each with >99.4% identity ranging in length from 96 kb to 218 kb, in which the breakpoints of all recurrent 1.5 Mb deletions we describe occur (red arrows). All three of these large duplications lie in an opposing orientation between BP4 and BP5 in the reference assembly. As a result, inversions of this region could result in these duplications being placed in a direct orientation, creating a configuration predisposed to microdeletion by NAHR.
Figure 4.
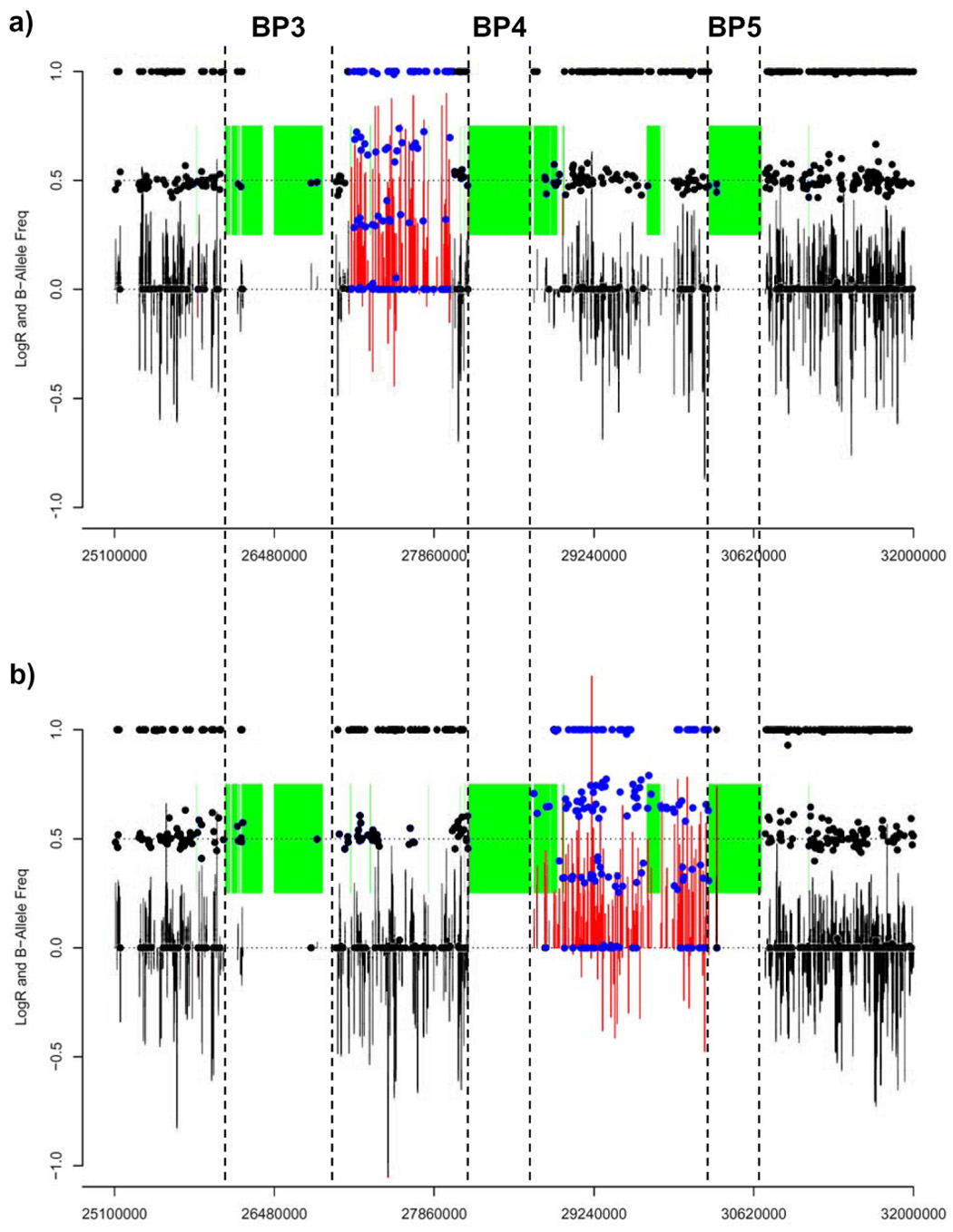
Duplications of (a) 15q13.1-q13.2 (BP3-BP4), and (b) 15q13.3 (BP4-BP5) identified in 2 of 960 normal control samples using the HumanHap300 Genotyping BeadChip (Illumina, San Diego, CA). Data shows probe position (x-axis) against smoothed LogR intensity ratio (red/black bars) and B-allele frequency (blue/black circles) (y-axis) for probes on chromosome 153. Data points within the duplicated regions are shown in colour, while those outside are shown in black. Green shaded boxes indicated segmental duplications. Within the limits of resolution of this data, the BP4-BP5 duplication appears to be the reciprocal event to the recurrent deletion shown in Figure 1. However, the BP3-BP4 duplication is clearly smaller than the deletion identified in patient 543/06, with breakpoints located in unique sequence (not in segmental duplications). No deletions of BP3-BP4 or BP4-BP5 were observed in this control population.
Observations in several genomic disorders suggest that microdeletions arise preferentially from chromosomes carrying an inversion of that same region3, 6–9. Given the opposing orientation of the duplicons at BP4 relative to BP5 in the reference assembly (Supplementary Figure 2), we hypothesized that 15q13.3 might also be a site of inversion polymorphism which could create a configuration predisposed to recurrent rearrangement. In order to investigate this possibility, we utilized FISH to assay the BP4-BP5 region. Testing of eight HapMap individuals of varied ethnicities showed the presence of a common inversion of this interval, which was present on 7 of the 16 chromosomes assayed (frequency of inversion relative to reference assembly 0.44, 95% CI 0.19–0.68) (Figure 5, Supplementary Table 2). In the two cases where we were able to study the parent-of-origin of BP4-BP5 deletions (mother of Patient 69/06 and father of Patient 02961), we found both to be heterozygous for the inversion. Although the high frequency of this inversion in the normal population and the lack of sufficient numbers of parental samples mean that we are currently unable to verify this hypothesis, our observations are consistent with a model in which inversion polymorphism of the BP4-BP5 region results in these flanking duplicons being placed in direct orientation, creating a configuration predisposed to microdeletion/duplication by NAHR. These data strengthen the growing link between the occurrence of large inversion polymorphisms and genomic disorders (reviewed in Sharp et al. 2006)10.
Figure 5.
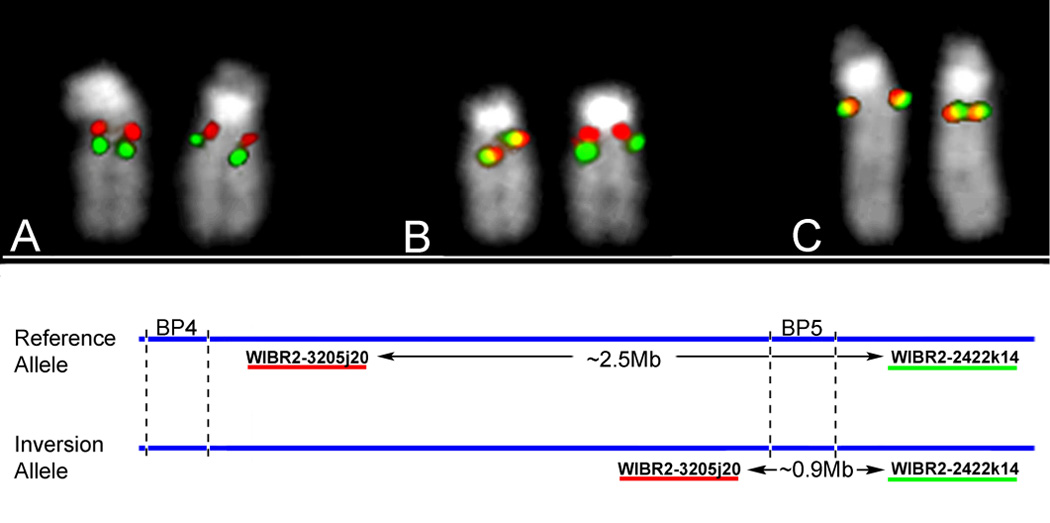
Identification of a common inversion polymorphism in 15q13.3. To detect inversions of the BP4-BP5 microdeletion region, we performed FISH mapping using fosmid probes located proximal and distal to BP5. The separation of these probes in the reference assembly is ~2.5 Mb, enough to visualize these as two separate signals on metaphase chromosomes. Inversion of the region BP4-BP5 moves the two probes within close proximity to each other, visualized as overlapping yellow signals. Using this assay on 8 HapMap individuals of different ethnicities, we observed the inversion on 7/16 chromosomes (~44% of the population, Supplementary Table 2). We also tested the mother of patient 69/06 (in whose germline the BP4-BP5 deletion arose), who was found to be heterozygous. Shown are individuals (a) homozygous for the reference allele, (b) heterozygous, or (c) homozygous for the inversion allele.
In order to assess the frequency of copy number changes between BP3, BP4, and BP5 in the general population, we screened 960 control individuals at this locus (n=960 unrelated Caucasians genotyped with a high density SNP Genotyping BeadChip11 for variable efficacy of statin drug response on cardiovascular disease [R. Krauss and D. Nickerson, personal communication]). We identified two individuals with duplications of 15q13: one individual with a duplication within the interval BP3-BP4, and a second with a duplication between BP4 and BP5 (Figure 3). Within the limits of resolution of these data, the BP4-BP5 duplication appears to be reciprocal to the deletions shown in Figure 1, providing further evidence that NAHR likely underlies rearrangements of 15q13. However, the BP3-BP4 duplication is smaller than the deletion identified in patient 543/06, with breakpoints located within unique sequence (i.e. not within the segmental duplications). We did not detect any large deletions of 15q13 in this control set. While some smaller regions of copy number polymorphism are present within 15q13 (mostly corresponding to the segmental duplication blocks at BP3, BP4, and BP5), deletions or duplications comparable to those that we describe have not been found in previous studies of a further 2002 control individuals12–19. Together, these results suggest that deletions of BP4-BP5 are pathogenic (6/2082 probands with MR vs. 0/2962 controls, p=0.005, Fisher’s Exact test). In contrast, our limited data suggest that duplication events of BP3-BP4 and BP4-BP5 are likely either (i) benign copy number variants, or (ii) are frequently associated with milder phenotypic abnormalities, although additional studies are necessary to confirm this interpretation. Parental origin studies in the two de novo cases showed paternal origin of the deletion in Patient 02961 and maternal origin in Patient 69/06. In addition, no imprinted genes have been identified to date within BP3-BP5 region (http://www.geneimprint.com/site/genes-by-species), suggesting that imprinting is unlikely to significantly affect the phenotype of these patients (Supplementary Figure 5).
Interestingly, seven of nine deletion carriers presented with seizures or abnormal EEG findings (Table 1). One of the genes within the critical region is CHRNA7 (cholinergic receptor, neuronal nicotinic, alpha polypeptide 7), a synaptic ion channel protein that mediates neuronal signal transmission. Linkage studies have suggested CHRNA7 as a susceptibility factor for both juvenile myoclonic epilepsy20 and benign epilepsy of childhood with centrotemporal spikes21. Further, it has been reported that mice with a knockout of CHRNA7 show a hypersynchronous neocortical EEG phenotype22. As a result, CHRNA7 represents an excellent candidate gene, haplo-insufficiency for which may underlie the epilepsy/seizure phenotype seen in the patients we describe. As copy-number variation of a region including CHRNA7 has also been observed in the general population13–16, the possible involvement of this gene as a genetic risk factor for epilepsy warrants further investigation.
Although we present the first description of recurrent BP4-BP5 microdeletions, there are single reports of apparently similar deletions at BP3-BP4-BP5 (http://decipher.sanger.ac.uk/). Several previous studies have also described a variety of different structural rearrangements involving 15q13, suggesting that this is a highly unstable genomic region. BAC array CGH studies of isodicentric(15q) chromosomes, marker(15) chromosomes, atypical deletions associated with Angelman syndrome, and deletions of 15q14 have suggested that the breakpoints of these rearrangements often map to the segmental duplication blocks at BP3, BP4, and BP523–26. We therefore investigated a number of other rearrangements of chromosome 15 using high resolution oligonucleotide array CGH. Consistent with previous studies25, data obtained in two unrelated inv dup(15) carriers were similar. Results showed that both inv dup(15) chromosomes are complex, being composed of two copies of the region 15cen-BP4 and a single copy of region BP4-BP5. This was subsequently confirmed by FISH (data not shown). Results in a patient with an Angelman syndrome class II deletion and in a second case with a mar(15) showed that in both cases the distal breakpoints mapped to the large segmental duplication block at BP3 (Supplementary Figure 6).
Our screen of 2082 patients with idiopathic mental retardation identified six unrelated individuals with 15q13.3 deletions, suggesting that this microdeletion accounts for ~0.3% of mental retardation of unknown etiology. The 1040 individuals screened by qPCR are a subset of a larger population of patients who receive services through the South Carolina Department of Disabilities and Special Needs27. We compared the frequency of the 15q13.3 deletion to that of other known microdeletions resulting in mental retardation in the larger cohort from which our population was obtained (10,997 patients of both European and African ancestry from South Carolina). Within the larger cohort, Prader-Willi syndrome was diagnosed in 0.22%, Angelman syndrome in 0.34%, and Williams syndrome in 0.31%27. In comparison, we identified 15q13.3 deletions in 0.29% of the subgroup (3 unrelated probands of 1040 tested) and 0.29% of the European populations tested by array CGH (3 unrelated probands of 1042 tested), suggesting the frequency of this 15q13.3 microdeletion syndrome may be comparable to that of the disorders listed above (estimated frequency in MR patients 0.29%; 95% CI 0.06–0.52%). Two pedigrees were of African American descent with the remainder of Caucasian origin, indicating this syndrome is found in patients of different ethnic backgrounds. Given that this microdeletion is well below the resolution of conventional cytogenetics—it could only be detected using techniques such as array CGH—we anticipate that the increasing resolution of these studies will lead to the future identification of additional microdeletion syndromes. Assuming a prevalence of moderate mental retardation in the general population of ~0.8%28, we estimate an approximate population incidence for this 15q13.3 microdeletion of 1/40,000. Given this relatively high frequency and the multiethnic distribution, we recommend testing for this disorder in individuals with features similar to the patients presented here.
Methods
DNA Samples
DNA samples were obtained from the following cohorts after obtaining informed consent: (i) children and young adults from a variety of UK clinical genetics centers, community learning disability teams, and other sources, including hospital neuro-pediatricians; common causes of mental retardation including karyotype and subtelomeric abnormalities and Fragile X were previously excluded (n=394)3; (ii) karyotypically normal individuals presenting with mental retardation and/or dysmorphism collected by the University of Pavia (n=510); (iii) individuals admitted to the IRCCS Associazione Oasi Maria Santissima (an Institute for Research and Care in Mental Retardation and Brain Aging) were screened for mental retardation according to the DSM-IV-TR criteria, and the common causes of mental retardation were excluded (n=138, all Caucasian, 75 female, 63 male); (iv) individuals who received services through the South Carolina Department of Disabilities and Special Needs and for whom common causes of mental retardation had been excluded by fragile X testing, chromosome analysis, amino acid and organic analyses, and urinary metabolic screening (n=1040, 540 female, 500 male, 562 African American, 448 Caucasian, 30 unstated or other ethnicity; all females and all but 10 of the males had IQ scores <70)29. Two control groups were used to assess the extent of normal copy number variation. The first control population consisted of 316 unrelated individuals that had been tested using the same BAC duplication microarray13, 15. A second control population, comprised of 960 unrelated Caucasian adults from the USA (age 40–70 years), were genotyped using the HumanHap300 Genotyping BeadChips (Illumina, San Diego, CA), comprising ~317,000 HapMap SNPs spread throughout the genome. Each individual was enrolled in the PARC study which aims to identify genetic contributors to the variable efficacy of statin drugs on cardiovascular disease risk (http://www.pharmgkb.org/network/members/parc.jsp#team). Hybridizations, data analysis, and copy number analysis, with particular reference to the chromosome 15q13 (155 probes between BP3-BP4, 167 probes between BP4-BP5), were performed according to published protocols11.
Array CGH
Patients from the UK (n=394) were hybridised to a custom BAC array consisting of 2007 clones targeted to regions of the genome flanked by segmental duplications, as described previously15. This array includes all regions associated with known genomic disorders and an additional 105 regions with similar genomic architecture. Because of the targeted nature of this array, it has reduced power to detect rearrangements not mediated by segmental duplications15. Regions were scored as copy number variant if the log2 ratio of two or more consecutive clones each exceeded twice the standard deviation of the autosomal clones in dye-swap replicate experiments7. Patients from Italy (n=648) were molecularly karyotyped using Agilent 244A Human Genome CGH Microarrays (Agilent Technologies, Santa Clara, CA). Parents of probands showing cryptic deletions were also analyzed to exclude inherited imbalances. Rearrangements of 15q were analysed utilising a custom oligonucleotide array (NimbleGen Systems, Madison, WI) consisting of 166,000 isothermal probes (length 45–75 bp; mean probe density, 1 probe/130 bp) covering a number of chromosomal regions, including 42,698 probes covering a 6 Mb region of chromosome 15q13 (chr15:25,500,000–31,500,000). Hybridizations were performed as described previously3 and utilized a single normal male as a reference (GM15724, Coriell, Camden, NJ). Microsatellite analysis to determine parental origin of the deletions utilised markers STS6_chr15, D15S1031, and D15S165 located within the interval BP4-BP5.
TaqMan quantitative PCR
Patients from the USA (n=1040) were assayed for copy number of the region BP4-BP5 using two TaqMan Gene Copy Number Assays. Primers and probes were designed from genomic sequence (hg18/Build 36) using Applied Biosytems proprietary software. Each assay was run as a duplex TaqMan real-time PCR reaction, utilizing a FAM dye-based assay targeted to 15q13.3 and a VIC dye-based assay for the reference gene, RNase P (PN 4316844 from Applied Biosystems, Foster City, CA). Each PCR assay was performed in quadruplicate and comprised 10 ng gDNA, 1×TaqMan probe/primer mix in 1×TaqMan Universal Master Mix in a 10 µl reaction amplified using an Applied Biosystems 7900HT SDS instrument. Cycling conditions were 2 mins at 50°C, 10 mins at 95°C, followed by 40 cycles of 15 secs at 92°C, and 60 secs at 60°C. Real-time data were collected by the SDS 2.3 software. The method involves relative quantification of the test sequence versus a reference gene known to have two copies per diploid genome. Relative quantity is determined by the ΔΔCt [(FAM Ct- VIC Ct)sample - (FAM Ct – VIC Ct)calibrator] method, where a reference sample or calibrator known to have two copies of the test sequence is used as the basis for comparative results. Gene copy number is 2× the relative quantity30. The two regions assayed were (1) chr15:29,000,001–29,000,077 and (2) chr15:29,239,994–29,240,092.
FISH inversion assay
Metaphase spreads were obtained from lymphoblast cell lines from 8 HapMap individuals (Coriell Cell Repository, Camden, NJ), and cultured peripheral blood lymphocytes from the mother of patient 69/06. FISH was performed using fosmid WIBR2-3205j20 (chr15:29,009,842–29,052,657) directly labeled by nick-translation with Cy3-dUTP (Perkin-Elmer) and WIBR2-2422k14 (chr15:31,531,310–31,570,294) labeled with fluorescein-dUTP (Enzo). Each hybridization utilized 300 ng of labeled probe, 5µg COT1 DNA (Roche), and 3µg sonicated salmon sperm DNA at 37°C in 10µl 2×SSC/50% formamide/10% dextran sulphate, followed by three posthybridization washes at 60°C in 0.1×SSC. Nuclei were stained with DAPI and digital images obtained using a Leica DMRXA2 epifluorescence microscope equipped with a cooled CCD camera (Princeton Instruments).
Supplementary Material
Acknowledgements
We are grateful to Dr. Ronald Krauss and the Pharmacogenomics and Risk of Cardiovascular Disease (PARC) project for the use and analysis of Illumina SNP genotyping data, funded by NIH grant U01 HL069757. This work was supported in part by grants from the NIH (HD043569, EEE), the South Carolina Department of Disabilities and Special Needs (CS, RES, RJS, and CES), Oxford Genetics Knowledge Park and the Oxford NIHR Biomedical Research Centre (RR and SJLK), Fondazione Mariani, CARIPLO and PRIN 2005 (OZ), and the Italian Ministry of Health (CR, PF, LC, and MF). EEE is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Accession Number Microarray data have been deposited in the Gene Expression Omnibus database under accession number GSE10189.
URLs University of California Santa Cruz genome browser, http://genome.ucsc.edu; Genomic Imprinting Wesite, http://www.geneimprint.com/site/gene-by-species, Sanger DECIPHER, http://decipher.sanger.ac.uk/; PARC study, http://www.pharmgkb.org/network/members/parc.jsp#team.
Conflict of interest statement K. Li, A. J. Broomer, Y. Wang, C. Xiao, C. Barbacioru, and C. Chen are employees of Applied Biosystems, Inc. and have stock options in the company.
References
- 1.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 2.de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal S, Nillesen WM, Huys EH, Leeuw N, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet. 2007;39:1361–1368. doi: 10.1038/ng.2007.9. [DOI] [PubMed] [Google Scholar]
- 5.Zody MC, Garber M, Sharpe T, Young SK, Rowen L, O'Neill K, Whittaker CA, Kamal M, Chang JL, Cuomo CA, et al. Analysis of the DNA sequence and duplication history of human chromosome 15. Nature. 2006;440:671–675. doi: 10.1038/nature04601. [DOI] [PubMed] [Google Scholar]
- 6.Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, et al. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12:849–858. doi: 10.1093/hmg/ddg101. [DOI] [PubMed] [Google Scholar]
- 7.Kurotaki N, Stankiewicz P, Wakui K, Niikawa N, Lupski JR. Sotos syndrome common deletion is mediated by directly oriented subunits within inverted Sos-REP low-copy repeats. Hum Mol Genet. 2005;14:535–542. doi: 10.1093/hmg/ddi050. [DOI] [PubMed] [Google Scholar]
- 8.Lupski JR. Genome structural variation and sporadic disease traits. Nat Genet. 2006;38:974–976. doi: 10.1038/ng0906-974. [DOI] [PubMed] [Google Scholar]
- 9.Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, et al. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–325. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp AJ, Cheng Z, Eichler EE. Structural variation of the human genome. Annu Rev Genomics Hum Genet. 2006;7:407–442. doi: 10.1146/annurev.genom.7.080505.115618. [DOI] [PubMed] [Google Scholar]
- 11.Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 13.Locke DP, Sharp AJ, McCarroll SA, McGrath SD, Newman TL, Cheng Z, Schwartz S, Albertson DG, Pinkel D, Altshuler DM, et al. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am J Hum Genet. 2006;79:275–290. doi: 10.1086/505653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 15.Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon-Sanchez J, Scholz S, Fung HC, Matarin M, Hernandez D, Gibbs JR, Britton A, de Vrieze FW, Peckham E, Gwinn-Hardy K, et al. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet. 2007;16:1–14. doi: 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]
- 17.Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, Eichler EE, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Smith AJ, Tsalenko A, Sampas N, Scheffer A, Yamada NA, Tsang P, Ben-Dor A, Yakhini Z, Ellis RJ, Bruhn L, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet. 2007;16:2783–2794. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- 19.Zogopoulos G, Ha KC, Naqib F, Moore S, Kim H, Montpetit A, Robidoux F, Laflamme P, Cotterchio M, Greenwood C, et al. Germ-line DNA copy number variation frequencies in a large North American population. Hum Genet. 2007;122:345–353. doi: 10.1007/s00439-007-0404-5. [DOI] [PubMed] [Google Scholar]
- 20.Elmslie FV, Rees M, Williamson MP, Kerr M, Kjeldsen MJ, Pang KA, Sundqvist A, Friis ML, Chadwick D, Richens A, et al. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet. 1997;6:1329–1334. doi: 10.1093/hmg/6.8.1329. [DOI] [PubMed] [Google Scholar]
- 21.Neubauer BA, Fiedler B, Himmelein B, Kampfer F, Lassker U, Schwabe G, Spanier I, Tams D, Bretscher C, Moldenhauer K, et al. Centrotemporal spikes in families with rolandic epilepsy: linkage to chromosome 15q14. Neurology. 1998;51:1608–1612. doi: 10.1212/wnl.51.6.1608. [DOI] [PubMed] [Google Scholar]
- 22.Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL, et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, Anselm I, Waisbren S, Beaudet AL, Peters SU. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. 2007;15:943–949. doi: 10.1038/sj.ejhg.5201859. [DOI] [PubMed] [Google Scholar]
- 24.Sahoo T, Shaw CA, Young AS, Whitehouse NL, Schroer RJ, Stevenson RE, Beaudet AL. Array-based comparative genomic hybridization analysis of recurrent chromosome 15q rearrangements. Am J Med Genet A. 2005;139:106–113. doi: 10.1002/ajmg.a.31000. [DOI] [PubMed] [Google Scholar]
- 25.Wang NJ, Liu D, Parokonny AS, Schanen NC. High-resolution molecular characterization of 15q11-q13 rearrangements by array comparative genomic hybridization (array CGH) with detection of gene dosage. Am J Hum Genet. 2004;75:267–281. doi: 10.1086/422854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erdogan F, Ullmann R, Chen W, Schubert M, Adolph S, Hultschig C, Kalscheuer V, Ropers HH, Spaich C, Tzschach A. Characterization of a 5.3 Mb deletion in 15q14 by comparative genomic hybridization using a whole genome "tiling path" BAC array in a girl with heart defect, cleft palate, and developmental delay. Am J Med Genet A. 2007;143:172–178. doi: 10.1002/ajmg.a.31541. [DOI] [PubMed] [Google Scholar]
- 27.Stevenson RE, Procopio-Allen AM, Schroer RJ, Collins JS. Genetic syndromes among individuals with mental retardation. Am J Med Genet A. 2003;123:29–32. doi: 10.1002/ajmg.a.20492. [DOI] [PubMed] [Google Scholar]
- 28.Larson SA, Lakin KC, Anderson L, Kwak N, Lee JH, Anderson D. Prevalence of mental retardation and developmental disabilities: estimates from the 1994/1995 National Health Interview Survey Disability Supplements. Am J Ment Retard. 2001;106:231–252. [PubMed] [Google Scholar]
- 29.Stevenson RE, Schroer RJ. Mental retardation in South Carolina: Characteristics of the study population; Proceedings of the Greenwood Genetic Center; 1996. [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.