

Abstract
Melatonin is a recognized antioxidant with high potential as a protective agent in many conditions related to oxidative stress such as neurodegenerative diseases, ischemia/reperfusion syndromes, sepsis and aging. These processes may be favorably affected by melatonin through its radical scavenging properties and/or antiapoptotic activity. Also, there is increasing evidence that these effects of melatonin could be relevant in keratinocytes, the main cell population of the skin where it would contribute to protection against damage induced by ultraviolet radiation (UVR). We therefore investigated the kinetics of UVR-induced apoptosis in cultured keratinocytes characterizing the morphological and mitochondrial changes, the caspases-dependent apoptotic pathways and involvement of poly(ADP-ribose) polymerase (PARP) activation as well as the protective effects of melatonin. When irradiated with UVB radiation (50 mJ/cm2), melatonin treated, cultured keratinocytes were more confluent, showed less cell blebbing, more uniform shape and less nuclear condensation as compared to irradiated, nonmelatonin-treated controls. Preincubation with melatonin also led to normalization of the decreased UVR-induced mitochondrial membrane potential. These melatonin effects were followed by suppression of the activation of mitochondrial pathway-related initiator caspase 9 (casp-9), but not of death receptor-dependent casp-8 between 24 and 48 hr after UVR exposure. Melatonin down-regulated effector caspases (casp-3/casp-7) at 24–48 hr post-UV irradiation and reduced PARP activation at 24 hr. Thus, melatonin is particularly active in UV-irradiated keratinocytes maintaining the mitochondrial membrane potential, inhibiting the consecutive activation of the intrinsic apoptotic pathway and reducing PARP activation. In conclusion, these data provide detailed evidence for specific antiapoptotic mechanisms of melatonin in UVR-induced damage of human keratinocytes.
Keywords: antioxidant, apoptosis, caspases, keratinocytes, melatonin, mitochondria, poly(ADP-ribose) polymerase, ultraviolet radiation
Introduction
The human skin is exposed lifelong to ultraviolet radiation (UVR) and its damaging effects are represented predominantly by skin aging and skin cancer [1–9]. An important strategy for the protection of the skin against these UVR-induced alterations has been the development of potent sun damage-preventing substances. However, as UVR is also required for photosynthesis of vitamin D3, endogenous protective agents would be highly desirable. As oxidative stress is known to be the key factor in UVR-related damage [3, 10–14], substances with antioxidant activity, such as melatonin, could be effective for the prevention of short- and long-term UVR damage [2, 15–19]. Melatonin was originally identified as the chief secretory product of the pineal gland [20] and later defined as a substance being also synthesized in a variety of extrapineal sites with bioactivity in a number of targets in single cells, animals and humans [21–29]. Thus, melatonin acts as a receptor-independent antioxidant [30–33], as an antiaging substance [34, 35] and as an anticarcinogenic factor [36, 37] within a wide range of concentrations [38].
Melatonin may also play a role in cutaneous biology as a melatoninergic system has been shown to be fully expressed in rodent and human skin [28, 29, 39–42]. The human skin is also capable of producing melatonin and melatonin metabolites, all with strong antioxidant activity [29, 40, 43–45]; therefore, this organ represents another site expressing the free radical scavenging cascade of melatonin as shown in many other biological systems [43]. Melatonin metabolism is in fact stimulated by UV irradiation, generating a ‘melatoninergic antioxidative system’ (MAS) in the skin [44]. Functional data suggest that melatonin exerts growth stimulatory effects in human and rodent keratinocytes, inhibitory effects in melanoma cells [41, 46–48], and also suppresses UVR-induced reactive oxygen species (ROS) formation [49–52]. Moreover, UV-induced cell damage and cell death in human keratinocytes can be effectively prevented by melatonin by ensuring short-term cell survival as well as long-term survival shown by increased cell colony formation [45]. The mechanism underlying these cell protective effects was identified as prevention of apoptosis represented by reduced apoptotic DNA-fragmentation [45]. Thus, the melatoninergic system in the skin appears to counteract the effects of the environmental stressor UVR by preserving the organ's functional integrity and maintaining its homeostasis [28, 29, 44].
Apoptosis is a well defined, highly regulated form of programmed cell death that is evident in normal skin (e.g. during keratinocytes differentiation) as well as in pathological conditions [53]. Ultraviolet B (UVB)-induced apoptosis is mediated by apoptotic signals, activating two main pathways: intrinsic (mitochondrial) and extrinsic (death receptor-dependent). The extrinsic pathway is initiated by activation of cell membrane-bound death receptors (tumor necrosis factor-alpha [54, 55] Fas [56]) followed by generation of Fas-associated death domain protein and death-inducing signaling complex to activate the key enzyme, casp-8 [57, 58]. Activation of the intrinsic pathway is initiated by mitochondrial damage leading to release of cytochrome c [59], formation of apoptotic protease-activating factor (Apaf-1) and activation of the initiator casp-9 [53, 60, 61].
In recent years, the role of mitochondrial damage has been enhanced, being perhaps the most sensitive detector of apoptotic signals [12, 62, 63]. Poly(ADP-ribose) polymerase (PARP) is an enzyme activated by single-strand DNA breaks, ROS or disruption of mitochondrial membrane potential [64–68] and its detection is an early marker of apoptosis [65].
In the present study, we investigated the effects of UVR and pretreatment with melatonin in keratinocytes. We evaluated cell morphology and characterized the extrinsic and intrinsic pathway of apoptosis determining mitochondrial membrane potential, activation of initiator (casp-8/casp-9) and effector caspases (casp-3/casp-7) as well as PARP activity.
Materials and methods
Cell culture
HaCaT keratinocytes were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with glucose, l-glutamine, pyridoxine hydrochloride (Gibco, Invitrogen Life Technologies, Carlsbad, CA, USA), 10% fetal bovine serum (FBS) (Mediatech Inc., Herndon, VA, USA) and 1% penicillin/streptomycin/amphotericin (Sigma Chemical Co., St Louis, MO, USA). Cells were removed from culture flasks by trypsinization and seeded in Petri dishes of 10 cm diameter at a density of 106 cells/dish (Corning Inc., Corning, NY, USA). Confluence of 80–90% was reached the next day; media were removed and replaced by melatonin-containing media or media without melatonin as a control. These pretreated keratinocytes were then submitted to UV irradiation and further used for morphological analysis and assessment of caspase activity by immunoblotting and mitochondrial membrane potential by confocal microscopy.
UV irradiation
Irradiation experiments were performed with a Biorad UV transluminator 2000 (Bio-Rad Laboratories, Hercules, CA, USA) calibrated as described previously [45]. Before each experiment, wavelength calibration and gain were established or verified. The light emitted by the UV source consisted primarily of UVB (wavelengths 280–320 nm; ∼60%), with minor components in the UVA (320–400 nm) and UVC (120–280 nm) range (∼30% and ∼10%, respectively). Keratinocytes were irradiated at the UVR-dose of 50 mJ/cm2 or were sham-irradiated (controls).
Melatonin treatment
Melatonin (Sigma) was dissolved in ethanol, diluted with phosphate-buffered saline (PBS) (final concentration of ethanol <0.2%) and added to the medium at the concentrations to be tested. After overnight incubation, cell media were removed and replaced with fresh media containing melatonin 10−3 m, the maximum effective concentration as determined in previous studies [50, 51] or with fresh media without melatonin (control). After preincubation with melatonin added at 24 and 12 hr before irradiation, melatonin-containing media were removed, cells were then washed once with PBS to remove remnants of media and melatonin, and PBS was added another time in amounts sufficient to keep cells immersed in PBS during UVR exposure. The Petri dishes containing keratinocytes were irradiated with UVR from below, and immediately after irradiation PBS was replaced with fresh culture media that was left for the next 24 or 48 hr. In parallel experiments, cells were incubated with or without melatonin, and subjected to sham-irradiation (not exposed to UVR) to control for the effects of melatonin alone.
Morphology analysis
Digital pictures from six to ten randomly chosen fields per each Petri dish from every experimental condition were acquired after UV exposure (at 0, 24 and 48 hr) with a NIKON Eclipse TE300 microscope (Melville, NY, USA). At 24 and 48 hr after UV irradiation, pictures were taken first of detached cells, then the detached cells were removed and another set of pictures was acquired to assess the degree of confluency of the cells which were still attached to the bottom of the culture dish. Pictures were recorded and analysed with MetaVue software (Molecular Devices Corporation, Downingtown, PA, USA). Keratinocytes were then harvested from Petri dishes by trypsinization, washed three times with ice-cold PBS and frozen at −80°C until further processing.
Measurement of mitochondrial membrane potential (Δψ) using JC-1
Mitochondrial inner membrane potential (Δψ) in immortalized HaCaT keratinocytes was measured using confocal microscopy with 5,5′,6,6′tetrachloro-1,1′,3,3′-tetraethyl-benzimidazol-carbocyanine iodide (JC-1) (Molecular Probes, Carlsbad, CA, USA) [69]. JC-1 selectively enters the mitochondria where it aggregates when the membrane potential exceeds 80–100 mV, causing a shift in fluorescence from 530 nm (green) to 590 nm (red). For these experiments, human HaCaT keratinocytes were seeded in Lab-Tek II 8 well-chambered cover glass (Nalge Nunc Inc., Naperville, IL, USA) and grown until 90–100% of confluence. Culture media were removed and the cells were washed with PBS, and incubated with melatonin for 30 or 120 min using the concentrations as shown in Fig. 3A,B. After incubation, cells were washed twice with PBS and subjected to irradiation with UV (50 mJ/cm2) or incubated with serum-free medium containing H2O2 (1 mm) for 1 hr. After irradiation, the cells were incubated in DMEM containing 5% FBS and supplemented with JC-1 (2.5 μg/mL) for 30 min at 37°C. The cells were then washed with serum-containing medium and slides were observed with laser scanning confocal fluorescent microscope (LSM 510; Carl Zeiss GmbH, Jena, Germany) equipped with Plan-Neofluor oil immersion 40 × objective with suitable filter setup. Images were acquired from 6 to 18 randomly chosen fields for each experimental condition showing nuclear cross-section. Green and red channels were merged and the ratio of red to green channel, shown in blue, was recorded.
Fig. 3.
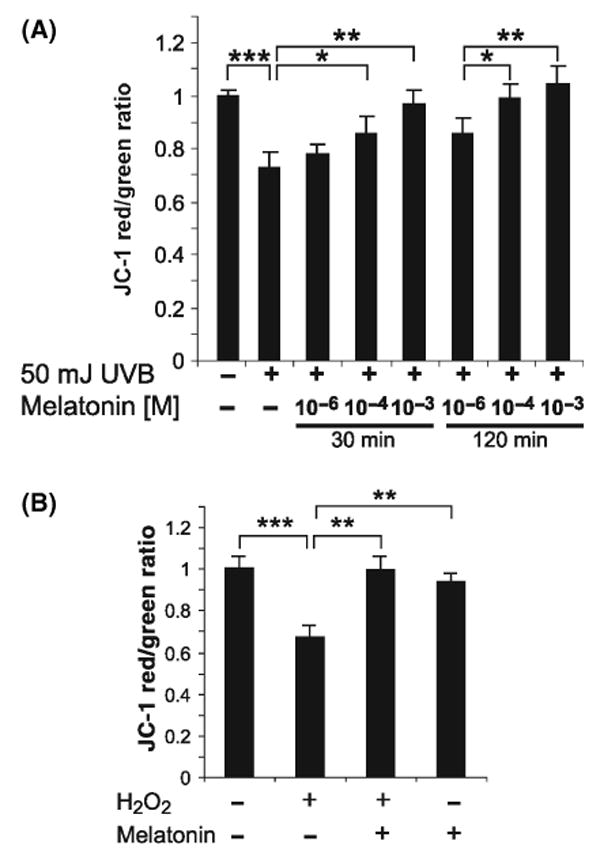
Changes in mitochondrial potential (Δψ) induced by UV (A) or H2O2 (B) are prevented by pretreatment with melatonin. HaCaT keratinocytes were preincubated with melatonin at the concentration of 10−3, 10−4 and 10−6 m for 30 or 120 min followed by irradiation with UVB (50 mJ/cm2) (A) or by treatment with H2O2 (1 mm, 60 min) (B). Mitochondrial potential (Δψ) is expressed as the ratio of J-monomer/J-aggregate fluorescence (red/green); lower values represent stronger reduction of membrane potential. *P < 0.05, **P < 0.005, ***P < 0.0005 versus UV-treated cells (no melatonin) (Panel A); **P < 0.005, ***P < 0.0005 versus H2O2-treated cells (no melatonin) (Panel B); (n = 6–18).
Immunoblot
Cell pellets were mixed with lysis buffer (PBS-containing Triton X 100 0.2% and 1 μL protease inhibitor per 100 μL buffer) and left on ice for 30 min. After centrifugation for 5 min at 9000 g the supernatant was taken for protein determination with the BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA).
Aliquots of cell lysates (four samples per condition) were used for immunoblotting. For each immunoblot, lysates containing 50 μg protein were mixed with loading buffer, boiled for 5 min at 95°C and separated on a 12% or 15% SDS-PAGE gel (PAGEr Duramide Precast Gel; Cambrex BioScience, Rockland, ME, USA). A parallel biotinylated protein ladder (Cell Signaling Technology Inc., Danvers, MA, USA) was used as marker. The separated proteins were then blotted onto an Immobilon-P polyvinylidene fluoride (PVDF) membrane (Millipore Corp., Bedford, MA, USA). After blotting, membranes were blocked with 5% nonfat dry milk in TBS-Tween 0.1% for 1 hr with gentle shaking and then washed three times with TBS-Tween 20 alone. The membranes were then incubated overnight at 4°C with the specific primary antibody dissolved in 5% nonfat dry milk. The antibodies used were as follows: rabbit anticaspase 3, 7 and 9 antibody (1:1000), rabbit antibody specific against the cleaved forms of casp-3, -7 and -9 (1:500) and rabbit PARP and cleaved PARP antibody (1:1000). Casp-8 was detected with a mouse antibody against casp-8 (1:1000) (all antibodies from Cell Signaling Technology). After incubation with the primary antibodies, membranes were incubated with secondary goat antirabbit or antimouse horseradish peroxidase (HRP)-linked IgG antibody (1:2000) in presence of antibiotin HRP-linked antibody for the protein ladder (1:2000) at room temperature for 1.5 hr. Bands were visualized with SuperSignal West Pico reagents (Pierce Biotechnology) and chemiluminescence was analysed by Fluor-S Multi-Imager using Quantity One software (both Bio-Rad Laboratories). The membranes were also applied to autoradiography film being developed with photodeveloper and fixer (Kodak, Rochester, NY, USA). Densitometry was performed with Scion Image analysis software (NIH, Bethesda, MN, USA).
Statistical analysis
Bands of immunoblots were evaluated by measurement of density with Scion Image analysis software (NIH). Density values from two or three separate experiments were taken for calculation of means and standard deviations. Analysis of mitochondrial membrane potential was performed using images showing nuclear cross-section, acquired from 6 to 18 randomly chosen fields for each experimental condition. Ratio of red/green emission was calculated with ImageJ software (NIH). Differences were analysed with the Student's t-test and considered significant at the P-value < 0.05.
Results
Keratinocytes in the nonirradiated condition were morphologically similar in the melatonin-treated and untreated groups, and confluency increased continuously over the 48 hr study period (Fig. 1A). UV irradiation (50 mJ/cm2) resulted in significant decrease in cell confluency when compared to nonirradiated cells with the occurrence of cell detachment and consequent empty spaces (ES) (Fig. 1B). At higher magnification (40 × ), the ES became more obvious and cell blebbing (CB) was observed (Fig. 1C). In melatonin-treated culture dishes, confluency was more prominent than in nontreated controls and ES were not seen (Fig. 1B,C). The number of detached cells was higher in nonmelatonin-treated Petri dishes compared with melatonin treated. The detached cells from nonmelatonin-treated dishes showed dysmorphic cell shape, more CB and expressed a higher degree of nuclear condensation (NC) indicative for apoptosis than samples treated with melatonin (Fig. 1D).
Fig. 1.
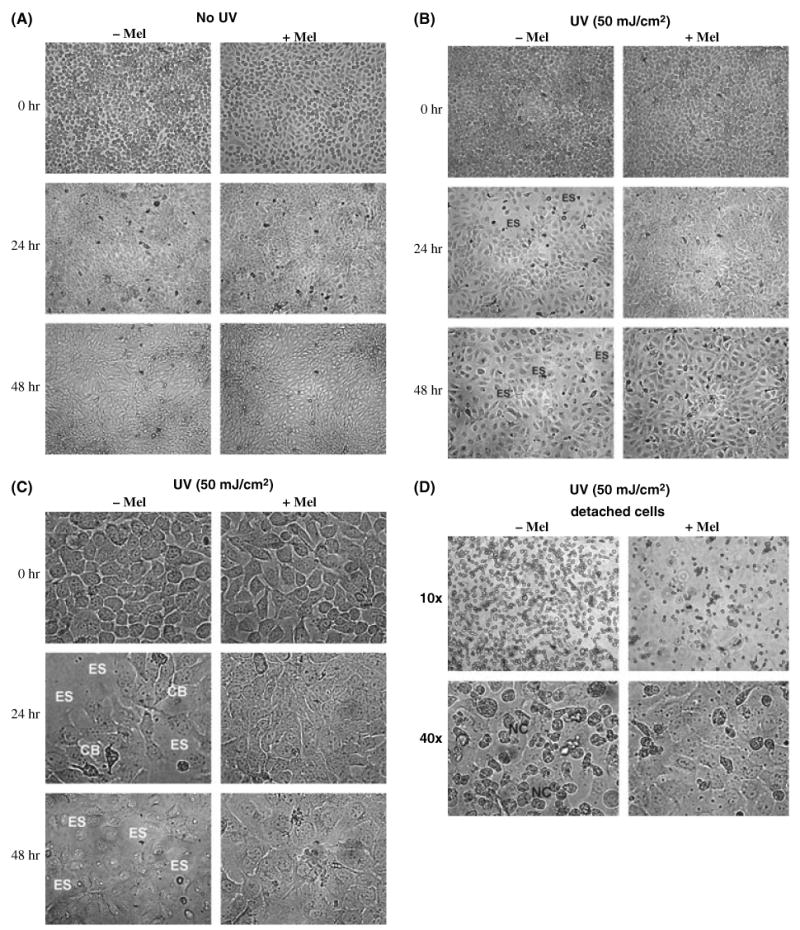
Protective effect of melatonin on keratinocytes morphology changes induced by ultraviolet radiation (UVR). Control represented by nonirradiated keratinocytes (either melatonin treated or untreated) is shown in (A) showing no influence of melatonin on cell morphology and growth characteristics. Immediately after irradiation with 50 mJ/cm2 (B) no differences between melatonin-and untreated keratinocytes are visible (0 hr, upper panel), whereas at 24 hr (mid panel) and 48 hr (lower panel) after UV exposure untreated cultures show empty spaces (ES), which are absent in melatonin-treated keratinocytes. At higher magnification (40 × ) (C), cell blebbing (CB) is seen and ES become more obvious. A higher number of detached cells showing dysmorphic shape are seen in nonmelatonin-treated cells as compared to less detached cells with more normomorphic shape in cell cultures pretreated with melatonin (D) at 24 hrs.
Regarding the mitochondrial membrane potential, the JC-1 probe detected intense red fluorescence, co-localized with mitochondria in nonirradiated, nonmelatonin-treated cells representing the membrane potential under physiological conditions (control) (Fig. 2A). To test whether melatonin alone (without exposure to UVR) could influence mitochondrial membrane potential, cells were incubated with melatonin at the concentration of 10−4 m; mitochondrial membrane potential remained unchanged (Fig. 2B). Irradiation with 50 mJ/cm2 of keratinocytes not treated with melatonin showed decrease of the red fluorescence and development of green fluorescence (mostly cytoplasmatic) indicative for loss of mitochondrial membrane potential (Fig. 2C). Preincubation with melatonin at the concentration of 10−4 m preserved the mitochondrial membrane potential, representing reduction of UVR-induced damage to the mitochondria (Fig. 2D). The decrease in concentration of aggregated form of JC-1 (red, left panel) was reciprocal to the accumulation of JC-1 monomer (green, middle panel) and was also expressed as the red/green ratio shown in blue (Fig. 2, right panel).
Fig. 2.
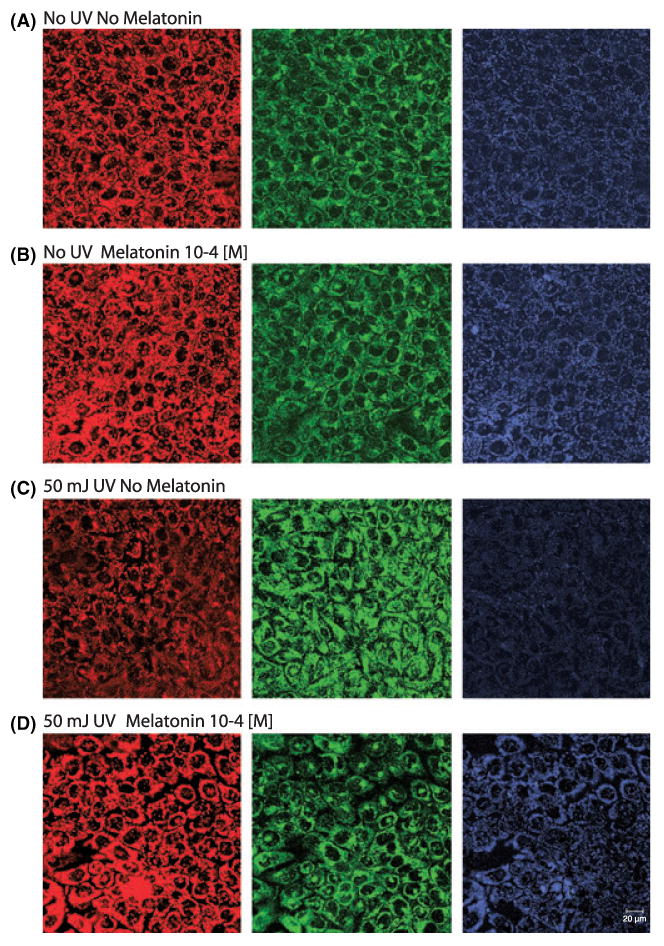
UVR-induced changes in mitochondrial potential (Δψ) and its prevention by pretreatment with melatonin. Representative images of mitochondrial membrane potential of control (A), non-irradiated HaCaT keratinocytes incubated with melatonin (10−4 m) (B) and keratinocytes, irradiated with UVB (50 mJ/cm2) without preincubation with melatonin (C) or parallel pretreatment with melatonin (10−4 m) (D). Nuclear cross-sections were acquired with confocal microscopy. Mitochondrial membrane potential is indicated by JC-1 red fluorescence (left panels). Relative changes in mitochondrial membrane potential are expressed as shifts from red to green fluorescence (middle panels) and presented as the red to green ratio that produces blue fluorescence (right panel). Bar, 20 μm.
To confirm the observed microscopic ratio between J-monomer/J-aggregate, fluorescence ratio (red/green) was also calculated directly. This indicated that UV irradiation induced significant mitochondrial membrane potential reduction and that this effect was attenuated by pretreatment with melatonin (10−6, 10−4, 10−3 m) in a dose-dependent manner. Statistical evaluation of J-monomer/J-aggregate ratios confirmed that the action of melatonin became significant at the concentrations of 10−4 and 10−3 m. Moreover, the protective effects of melatonin were slightly stronger when incubation was performed for 120 min compared with 30 min (Fig. 3A). To control for the direct effect of oxidative stress, we tested the action of H2O2 on mitochondrial potential (Δψ). H2O2 significantly reduced mitochondrial membrane potential, which was also counteracted by melatonin at the concentration of 10−4 m (Fig. 3B). Melatonin alone (without prior UV irradiation) did not influence mitochondrial membrane potential.
Activation of casp-8 was observed already at 24 hr after UV irradiation as shown by appearance of the 57 kDa product and its cleaved (activated) form of 43 kDa; melatonin treatment was without any effect on this process (data not shown). Casp-9 was also strongly activated at 24 hr after UV irradiation, while melatonin pretreated samples showed weaker expression of the corresponding cleaved forms of 35 and 17 kDa (Fig. 4A,E). Notably, at 48 hr post-UVR, the cleavage products of casp-9 were still present, even though at lower levels. Regarding casp-3, at 24 hr, the specific antibody detected the 35 kDa product, whereas the specific antibody against cleaved casp-3 detected the activated form of 17 kDa. At 48 hr after UV-treatment, further cleavage of casp-3 was observed that clearly was reduced by pretreatment with melatonin (Fig. 4B lower panel, 4F). The antibody against the effector caspase (casp-7) detected the relevant protein at expression levels that were similar at 24 and 48 hr after irradiation (Fig. 4C,G). The specific antibody against the cleaved form of casp-7 showed positive staining for the 20 kDa product, which was reduced in melatonin pretreated keratinocytes. The antibody against PARP detected the protein at 24 hr after UV exposure while its levels were also reduced by pretreatment with melatonin (Fig. 4D,H). PARP was detected with PARP antibody as a 89 kDa protein and the specific antibody revealed the cleaved form of PARP (Fig. 4D, lower panel); pretreatment with melatonin was associated with inhibition of expression and cleavage of the protein at both time points.
Fig. 4.
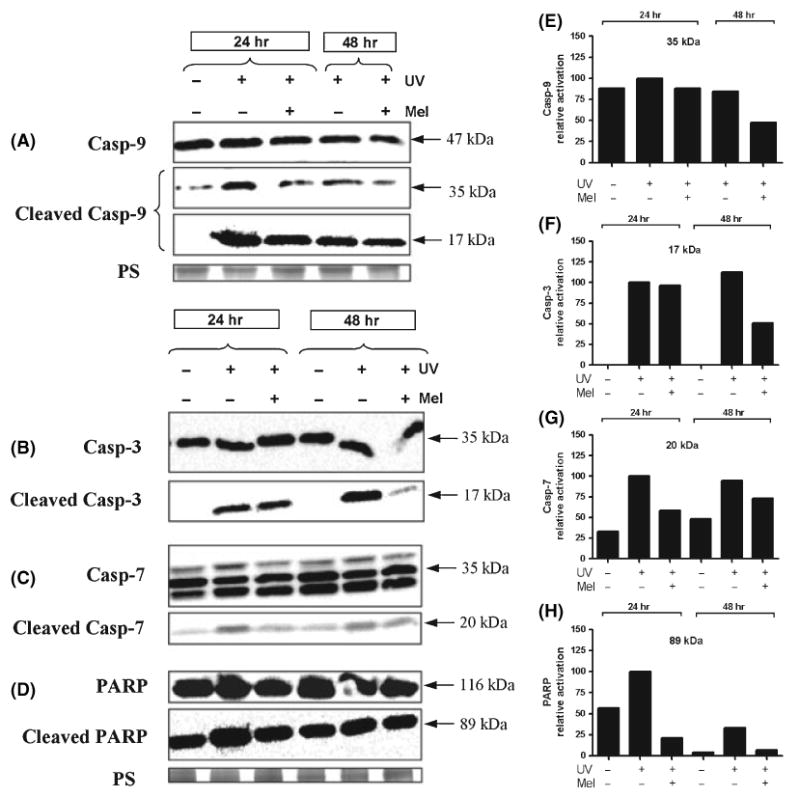
Melatonin attenuates the UV-induced activation of initiator and effector caspases and poly(ADP-ribose) polymerase (PARP). Decreased activation of caspase 9 (casp-9) in melatonin-treated samples compared to untreated cultures as represented by lower expression of cleaved form of casp-9 of 35 and 17 kDa (A,E). UV-induced caspases activation is stronger at 24 hr than 48 hr after UV exposure. The peak of the cleaved form of effector caspases 3 (17 kDa) (B,F) and casp-7 (20 kDa) (C,G) is delayed to 48 hr post-UVR and their activation is similarly reduced by melatonin pretreatment. Activated PARP (89 kDa) is detected as early as 24 hr after UV irradiation and loss of its activation is induced by pretreatment with melatonin (D,H). Protein samples were loaded equally as confirmed by protein staining (PS) with Coomassie Blue.
Discussion
This study demonstrated a specific protective effect of melatonin on UVR-induced changes in keratinocytes. The UVR attenuating effect of melatonin was expressed morphologically and biochemically by inhibition of UV-induced mitochondrial apoptotic pathway, e.g. attenuation of mitochondrial membrane potential reduction, reduced activation of initiator casp-9 and effector caspases (casp-3/casp-7) and reduced PARP activation.
In previous studies, we had demonstrated that UVR at 50 mJ/cm2 causes considerable reduction of cell viability, clonogenic cell growth and DNA-fragmentation, which was prevented by melatonin [45]. Recently, melatonin was also reported to reduce apoptosis and cell cycle regulators as well as antiapoptotic enzymes in UVB-irradiated HaCaT keratinocytes [70]. This evidence suggests involvement of apoptotic signaling events, influenced by melatonin. Other studies have shown that the dose of 50 mJ/cm2 but not 15 mJ/cm2 was most constantly associated with induction of apoptosis and consecutive activation of casp-9, casp-3, casp-7 and PARP [71, 72]. The same UV-dose also led to cytochrome c release [73]. Sitailo et al. [61] used a UV source emission spectrum (UVB ∼65%; UVA ∼34%) similar to the present study – albeit at a slightly lower UV-dose of 30 mJ/cm2 – and found greatest activation of casp-3 with additional activation of casp-9 stronger than casp-8 in normal human keratinocytes and HaCaT keratinocytes. This indicated that the intrinsic (mitochondrial) pathway would represent a major contributor to the UV-induced apoptosis in keratinocytes. It must be mentioned that some authors have instead assumed that casp-8 activation would be a bystander effect of the mitochondrial apoptotic pathway and that casp-8 is activated downstream of casp-9 and casp-3 activation [53]. However, more recent highly targeted studies have shown that activation of casp-8 is a UVB-specific event, independent of changes in casp-9 and casp-3.
In the current study, we tested different regulatory circuits of apoptosis and their modification by treatment with melatonin. Thus, we showed the activation of casp-8 at 24 hr after UV irradiation that is not affected by melatonin. Most likely, this is related to the insensitivity of this pathway to activation by ROS, hence, the antioxidant melatonin would be ineffective. In contrast, the mitochondrial pathway, which is activated by mitochondrial ROS (mROS), was strongly influenced by melatonin. This mechanism of antiapoptotic action of melatonin is in accordance with previous studies showing down-regulation of mitochondrial cytochrome c release and inhibition of activation of casp-9 and casp-3 in homocysteine-induced rat hippocampus apoptosis [74]. Thus, the present study showed mitochondrial membrane potential reduction upon exposure to UVR, whereas melatonin prevented the drop in membrane potential when added at the concentration of 10−3, 10−4, or 10−6 m, consistent with its antiapoptotic effects in HaCaT keratinocytes as previously described [45]. Moreover, the mitochondrial pathway initiator casp-9 was also activated, and followed by activation of the downstream effector caspases (casp-3/casp-7). Of these, casp-3 seems to be the most relevant, as casp-3 deficient mouse embryonic stem cells do not undergo UV-induced apoptosis [75, 76].
In our study, UV-induced activation of both effector caspases was significantly suppressed in keratinocytes preincubated with melatonin, which could be a consequence of reduced activation of upstream caspases. Support for this hypothesis comes from the apparent earlier activation of casp-9 compared to casp-3 and casp-7, consistent with the chronologic sequence of the caspase-related apoptotic activation cascade. Alternatively, melatonin could also inhibit directly the effector casp-3, as it has been shown that melatonin can prevent neuronal death in a mouse brain ischemia model by direct inhibition of casp-3 [77]. Also, casp-3 is directly reduced by melatonin in aflatoxin B1-treated liver cells [78].
In contrast, PARP was activated already at 24 hr post-UVR representing an early event in UV-induced apoptosis. PARP is a Zn-finger nuclear protein, activated by single-strand DNA breaks, ROS or disruption of mitochondrial membrane potential [64–68]. PAR, the resulting product of PARP activation induced by UVR, is an early marker of apoptosis that becomes positive in HaCaT keratinocytes at 24 hr post-UV irradiation [65]. This is in agreement with our own results showing stronger expression at 24 hr compared to 48 hr. PARP is needed for DNA repair upon ROS-induced damage [79], and PARP expression positively correlates with the number of single-strand DNA breaks [68]. Thus, reduction of PARP activation by melatonin would result from reduced DNA damage. Saldeen et al. [64] have shown that PARP cleavage can also be directly induced by disruption of mitochondrial membrane potential. Moreover, Baydas et al. [74] have already observed that melatonin is indeed able to reduce PARP activation and subsequent DNA-fragmentation in rat hippocampus neuronal cell apoptosis. Therefore, the reduction of PARP activation by melatonin in the current study investigating UV-induced apoptosis may be accounted by the reduced mitochondrial damage and/or by reduction of ROS generation through intramitochondrial/cytosolic actions of melatonin, as melatonin is a potent radical scavenger [30, 51].
In earlier studies, we found that UV-induced DNA-fragmentation in keratinocytes is indeed successfully reduced by pretreatment with melatonin [45]. Thus, the combined diminished mitochondrial membrane potential reduction, followed by reduced activation of casp-3, -7 and -9 together with reduced degree of DNA damage reflected by attenuated PARP cleavage result in the survival of a ‘healthy’ cell population (cells which do not undergo mutation and tumour promotion, but stay ‘benign’) in keratinocytes pretreated with melatonin before UVR exposure.
We also determined the effect of UVR on mitochondrial membrane potential. UVR produces oxidative stress and concomitant formation of mROS leading to calcium influx into the mitochondria with consecutive opening of the mitochondrial permeability transition pore (MPTP) [80] and depolarization of the mitochondrial membrane potential [59] as the end result of UV-induced mitochondrial damage. Melatonin has been shown to act as antiapoptotic agent at the mitochondrial level through direct inhibition of MPTP opening, a newly identified mechanism influenced by melatonin [80]. Melatonin interferes with this pathway by reducing mROS generation and calcium release as well as inhibiting the opening of the MPTP as shown in rat brain astrocytes [59], mouse striatal neurons [80] and rat cerebellar granule neurons [81]. In the current study, we provide the first evidence in keratinocytes – the main cell population of the skin – that melatonin prevents mitochondrial membrane potential from UVR-induced reduction. As mitochondrial damage is a very sensitive marker, as well as early event in UVR-induced apoptosis, this fact underlines – similar to the observed PARP inhibition by melatonin – the strong and pleiotropic protective effects of melatonin which interferes with the two main apoptotic pathways in UV-exposed keratinocytes.
To conclude, the present study shows for the first time that keratinocytes, the cutaneous cell population in which UV radiation is most relevant, are responsive to the UVR protective effects of melatonin that blocks activation of apoptotic pathways induced by 50 mJ/cm2 UVB (Fig. 5). Thus, melatonin stabilizes mitochondria by maintaining its membrane potential, whose reduction is a key-event in early apoptosis development. Melatonin also inhibits the consequent activation of the caspases-mediated intrinsic apoptotic pathway, and reduces PARP activation, which is associated with DNA damage.
Fig. 5.
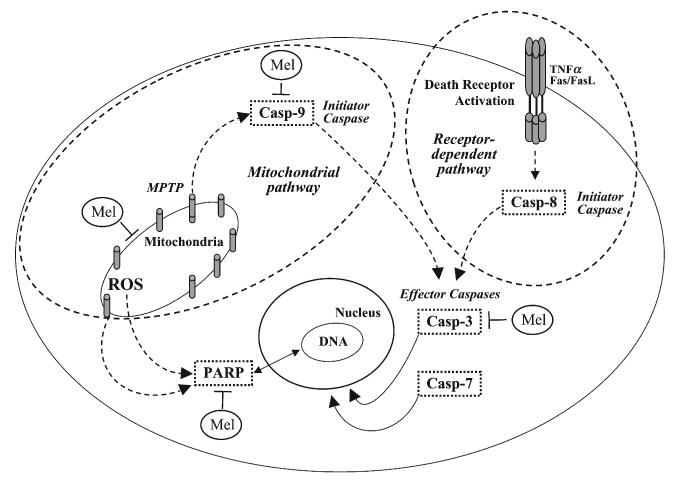
Putative interaction of melatonin with cellular pathways of apoptosis activated by UVR in keratinocytes. Added melatonin acts at the mitochondrial level reducing/scavenging ROS, enhancing reduced mitochondrial membrane potential to normal levels and inhibiting consecutive activation of early initiator caspases in the mitochondrial (casp-9) pathway. Later downstream events such as activation of effector caspases (casp-3/casp-7) are also reduced in melatonin-treated cells, and PARP activation is also attenuated by melatonin. Melatonin's stabilizing actions at the mitochondrion level may be direct or indirect, secondary to protection of DNA integrity. The death-receptor-mediated extrinsic pathway through casp-8 is not affected by melatonin. Mel, melatonin; MPTP, mitochondrial permeability transition pore; ROS, reactive oxygen species.
Acknowledgments
We thank Dr Robert M. Sayre, Division of Dermatology, Department of Medicine, UTHSC for calibration of the UV source. This work was supported by ‘German Academy of Natural Scientists Leopoldina’ with funds from the ‘Federal Ministry of Education and Research’ Ref-No. BMBF-LPD 9901/8-113 to TWF, University of Tennessee Cancer Center Pilot Grant to AS and TWF, and NIH grant AR052190 to AS. We further thank the Foundation ‘Rene Touraine’ for supporting TWF with a Short-Term International Fellowship. Confocal microscopy was performed on the equipment obtained through Shared Instrumentation Grant from National Center for Research Purposes at the National Institute of Health (S10RR13725-01).
References
- 1.Yaar M, Gilchrest BA. Cellular and molecular mechanisms of cutaneous aging. J Dermatol Surg Oncol. 1990;16:915–922. doi: 10.1111/j.1524-4725.1990.tb01555.x. [DOI] [PubMed] [Google Scholar]
- 2.Sander CS, Chang H, Hamm F, et al. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int J Dermatol. 2004;43:326–335. doi: 10.1111/j.1365-4632.2004.02222.x. [DOI] [PubMed] [Google Scholar]
- 3.Emerit I. Free radicals and aging of the skin. EXS. 1992;62:328–341. doi: 10.1007/978-3-0348-7460-1_33. [DOI] [PubMed] [Google Scholar]
- 4.Cleaver JE, Crowley E. UV damage, DNA repair and skin carcinogenesis. Front Biosci. 2002;7:d1024–d1043. doi: 10.2741/A829. [DOI] [PubMed] [Google Scholar]
- 5.Yaar M, Gilchrest BA. Ageing and photoageing of keratinocytes and melanocytes. Clin Exp Dermatol. 2001;26:583–591. doi: 10.1046/j.1365-2230.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- 6.Hadshiew IM, Eller MS, Gilchrest BA. Skin aging and photoaging: the role of DNA damage and repair. Am J Contact Dermat. 2000;11:19–25. doi: 10.1016/s1046-199x(00)90028-9. [DOI] [PubMed] [Google Scholar]
- 7.Gilchrest BA. Skin aging 2003: recent advances and current concepts. Cutis. 2003;72:5–10. [PubMed] [Google Scholar]
- 8.Berneburg M, Plettenberg H, Krutmann J. Photoaging of human skin. Photodermatol Photoimmunol Photomed. 2000;16:239–244. doi: 10.1034/j.1600-0781.2000.160601.x. [DOI] [PubMed] [Google Scholar]
- 9.Slominski APJ. Animals under the sun: effects of ultraviolet radiation on mammalian skin. Clin Dermatol. 1998;16:503–515. doi: 10.1016/s0738-081x(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 10.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 11.Ogura R, Sugiyama M, Nishi J, et al. Mechanism of lipid radical formation following exposure of epidermal homogenate to ultraviolet light. J Invest Dermatol. 1991;97:1044–1047. doi: 10.1111/1523-1747.ep12492553. [DOI] [PubMed] [Google Scholar]
- 12.Wang CB, Huang MQ, Tao GL, et al. Polypeptide from Chlamys farreri protects HaCaT cells from UVB-induced apoptosis. Chem Biol Interact. 2004;147:119–127. doi: 10.1016/j.cbi.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Kulms D, Zeise E, Poppelmann B, et al. DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene. 2002;21:5844–5851. doi: 10.1038/sj.onc.1205743. [DOI] [PubMed] [Google Scholar]
- 14.Gilchrest BA, Bohr VA. Aging processes, DNA damage, and repair. FASEB J. 1997;11:322–330. doi: 10.1096/fasebj.11.5.9141498. [DOI] [PubMed] [Google Scholar]
- 15.Wenk JBP, Meewes C, Wlaschek M, et al. UV-induced oxidative stress and photoaging. In: Thiele JEP, editor. Current Problems in Dermatology: Oxidants and Antioxidants in Cutaneous Biology. Karger; Basel: 2001. pp. 83–94. [DOI] [PubMed] [Google Scholar]
- 16.Thiele JJ, Hsieh SN, Ekanayake-Mudiyanselage S. Vitamin E: critical review of its current use in cosmetic and clinical dermatology. Dermatol Surg. 2005;31:805–813. doi: 10.1111/j.1524-4725.2005.31724. [DOI] [PubMed] [Google Scholar]
- 17.Grether-Beck S, Wlaschek M, Krutmann J, et al. Photodamage and photoaging – prevention and treatment] J Dtsch Dermatol Ges. 2005;3 2:S19–S25. doi: 10.1111/j.1610-0387.2005.04394.x. [DOI] [PubMed] [Google Scholar]
- 18.Biesalski HK, Berneburg M, Grune T, et al. Hohenheimer consensus talk. Oxidative and premature skin ageing. Exp Dermatol. 2003;12 3:3–15. doi: 10.1111/j.0906-6705.2003.00148.x. [DOI] [PubMed] [Google Scholar]
- 19.Krutmann J. New developments in photoprotection of human skin. Skin Pharmacol Appl Skin Physiol. 2001;14:401–407. doi: 10.1159/000056374. [DOI] [PubMed] [Google Scholar]
- 20.Lerner AB, Case JD, Takahashi Y. Isolation of melatonin, a pineal factor that lightens melanocytes. J Am Chem Soc. 1958;80:2587. [Google Scholar]
- 21.Bubenik GA. Gastrointestinal melatonin: localization, function, and clinical relevance. Dig Dis Sci. 2002;47:2336–2348. doi: 10.1023/a:1020107915919. [DOI] [PubMed] [Google Scholar]
- 22.Cahill GM, Besharse JC. Light-sensitive melatonin synthesis by Xenopus photoreceptors after destruction of the inner retina. Vis Neurosci. 1992;8:487–490. doi: 10.1017/s0952523800005009. [DOI] [PubMed] [Google Scholar]
- 23.Carrillo-Vico A, Calvo JR, Abreu P, et al. Evidence of melatonin synthesis by human lymphocytes and its physiological significance: possible role as intracrine, autocrine, and/or paracrine substance. FASEB J. 2004;18:537–539. doi: 10.1096/fj.03-0694fje. [DOI] [PubMed] [Google Scholar]
- 24.Itoh MT, Ishizuka B, Kuribayashi Y, et al. Melatonin, its precursors, and synthesizing enzyme activities in the human ovary. Mol Hum Reprod. 1999;5:402–408. doi: 10.1093/molehr/5.5.402. [DOI] [PubMed] [Google Scholar]
- 25.Tan DX, Manchester LC, Reiter RJ, et al. High physiological levels of melatonin in the bile of mammals. Life Sci. 1999;65:2523–2529. doi: 10.1016/s0024-3205(99)00519-6. [DOI] [PubMed] [Google Scholar]
- 26.Tan DX, Manchester LC, Reiter RJ, et al. Identification of highly elevated levels of melatonin in bone marrow: its origin and significance. Biochim Biophys Acta. 1999;1472:206–214. doi: 10.1016/s0304-4165(99)00125-7. [DOI] [PubMed] [Google Scholar]
- 27.Reiter RJ, Tan DX. Melatonin: an antioxidant in edible plants. Ann N Y Acad Sci. 2002;957:341–344. doi: 10.1111/j.1749-6632.2002.tb02938.x. [DOI] [PubMed] [Google Scholar]
- 28.Slominski A, Fischer TW, Zmijewski MA, et al. On the role of melatonin in skin physiology and pathology. Endocrine. 2005;27:137–148. doi: 10.1385/ENDO:27:2:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slominski A, Wortsman J, Tobin DJ. The cutaneous serotoninergic/melatoninergic system: securing a place under the sun. FASEB J. 2005;19:176–194. doi: 10.1096/fj.04-2079rev. [DOI] [PubMed] [Google Scholar]
- 30.Tan DX, Chen LD, Poeggeler B, et al. Melatonin: a potent, endogenous hydroxyl radical scavenger. Endocr J. 1993;1:57–60. [Google Scholar]
- 31.Tan DX, Manchester LC, Hardeland R, et al. Melatonin: a hormone, a tissue factor, an autocoid, a paracoid, and an antioxidant vitamin. J Pineal Res. 2003;34:75–78. doi: 10.1034/j.1600-079x.2003.02111.x. [DOI] [PubMed] [Google Scholar]
- 32.Tan DX, Reiter RJ, Manchester LC, et al. Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem. 2002;2:181–197. doi: 10.2174/1568026023394443. [DOI] [PubMed] [Google Scholar]
- 33.Maestroni GJ. The immunotherapeutic potential of melatonin. Expert Opin Investig Drugs. 2001;10:467–476. doi: 10.1517/13543784.10.3.467. [DOI] [PubMed] [Google Scholar]
- 34.Karasek M, Reiter RJ. Melatonin and aging. Neuroendocrinol Lett. 2002;23 1:14–16. [PubMed] [Google Scholar]
- 35.Reiter RJ, Tan DX, Poeggeler B, et al. Melatonin as a free radical scavenger: implications for aging and age-related diseases. Ann N Y Acad Sci. 1994;719:1–12. doi: 10.1111/j.1749-6632.1994.tb56817.x. [DOI] [PubMed] [Google Scholar]
- 36.Karbownik M. Potential anticarcinogenic action of melatonin and other antioxidants mediated by antioxidative mechanisms. Neuroendocrinol Lett. 2002;23 1:39–44. [PubMed] [Google Scholar]
- 37.Fischer TW, Zmijewski MA, Zbytek B, et al. Oncostatic effects of the indole melatonin and expression of its cytosolic and nuclear receptors in cultured human melanoma cell lines. Int J Oncol. 2006;29:665–672. doi: 10.3892/ijo.29.3.665. [DOI] [PubMed] [Google Scholar]
- 38.Reiter RJ, Tan DX, Maldonado MD. Melatonin as an antioxidant: physiology versus pharmacology. J Pineal Res. 2005;39:215–216. doi: 10.1111/j.1600-079X.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- 39.Slominski A, Baker J, Rosano TG, et al. Metabolism of serotonin to N-acetylserotonin, melatonin, and 5-methoxytryptamine in hamster skin culture. J Biol Chem. 1996;271:12281–12286. doi: 10.1074/jbc.271.21.12281. [DOI] [PubMed] [Google Scholar]
- 40.Slominski A, Pisarchik A, Semak I, et al. Serotoninergic and melatoninergic systems are fully expressed in human skin. FASEB J. 2002;16:896–898. doi: 10.1096/fj.01-0952fje. [DOI] [PubMed] [Google Scholar]
- 41.Slominski A, Pisarchik A, Zbytek B, et al. Functional activity of serotoninergic and melatoninergic systems expressed in the skin. J Cell Physiol. 2003;196:144–153. doi: 10.1002/jcp.10287. [DOI] [PubMed] [Google Scholar]
- 42.Slominski A, Semak I, Pisarchik A, et al. Conversion of l-tryptophan to serotonin and melatonin in human melanoma cells. FEBS Lett. 2002;511:102–106. doi: 10.1016/s0014-5793(01)03319-1. [DOI] [PubMed] [Google Scholar]
- 43.Tan DX, Manchester LC, Terron MP, et al. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42:28–42. doi: 10.1111/j.1600-079X.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 44.Fischer TW, Sweatman TW, Semak I, et al. Constitutive and UV-induced metabolism of melatonin in keratinocytes and cell-free systems. FASEB J. 2006;20:1564–1566. doi: 10.1096/fj.05-5227fje. [DOI] [PubMed] [Google Scholar]
- 45.Fischer TW, Zbytek B, Sayre RM, et al. Melatonin increases survival of HaCaT keratinocytes by suppressing UV-induced apoptosis. J Pineal Res. 2006;40:18–26. doi: 10.1111/j.1600-079X.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 46.Hipler UC, Fischer TW, Elsner P. HaCaT cell proliferation influenced by melatonin. Skin Pharmacol Appl Skin Physiol. 2003;16:379–385. doi: 10.1159/000072933. [DOI] [PubMed] [Google Scholar]
- 47.Slominski A, Pruski D. Melatonin inhibits proliferation and melanogenesis in rodent melanoma cells. Exp Cell Res. 1993;206:189–194. doi: 10.1006/excr.1993.1137. [DOI] [PubMed] [Google Scholar]
- 48.Slominski A, Chassalevris N, Mazurkiewicz J, et al. Murine skin as a target for melatonin bioregulation. Exp Dermatol. 1994;3:45–50. doi: 10.1111/j.1600-0625.1994.tb00265.x. [DOI] [PubMed] [Google Scholar]
- 49.Fischer TW, Elsner P. The antioxidative potential of melatonin in the skin. Curr Probl Dermatol. 2001;29:165–174. doi: 10.1159/000060665. [DOI] [PubMed] [Google Scholar]
- 50.Fischer TW, Scholz G, Knoll B, et al. Melatonin reduces UV-induced reactive oxygen species in a dose-dependent manner in IL-3-stimulated leukocytes. J Pineal Res. 2001;31:39–45. doi: 10.1034/j.1600-079x.2001.310106.x. [DOI] [PubMed] [Google Scholar]
- 51.Fischer TW, Scholz G, Knoll B, et al. Melatonin suppresses reactive oxygen species in UV-irradiated leukocytes more than vitamin C and trolox. Skin Pharmacol Appl Skin Physiol. 2002;15:367–373. doi: 10.1159/000064543. [DOI] [PubMed] [Google Scholar]
- 52.Fischer TW, Scholz G, Knoll B, et al. Melatonin suppresses reactive oxygen species induced by UV irradiation in leukocytes. J Pineal Res. 2004;37:107–112. doi: 10.1111/j.1600-079X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 53.Assefa Z, Van Laethem A, Garmyn M, et al. Ultraviolet radiation-induced apoptosis in keratinocytes: on the role of cytosolic factors. Biochim Biophys Acta. 2005;1755:90–106. doi: 10.1016/j.bbcan.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 54.Stander S, Schwarz T. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is expressed in normal skin and cutaneous inflammatory diseases, but not in chronically UV-exposed skin and non-melanoma skin cancer. Am J Dermatopathol. 2005;27:116–121. doi: 10.1097/01.dad.0000154401.45465.ee. [DOI] [PubMed] [Google Scholar]
- 55.Poppelmann B, Klimmek K, Strozyk E, et al. NF{kappa}B-dependent down-regulation of tumor necrosis factor receptor-associated proteins contributes to interleukin-1-mediated enhancement of ultraviolet B-induced apoptosis. J Biol Chem. 2005;280:15635–15643. doi: 10.1074/jbc.M413006200. [DOI] [PubMed] [Google Scholar]
- 56.Leverkus M, Yaar M, Gilchrest BA. Fas/Fas ligand interaction contributes to UV-induced apoptosis in human keratinocytes. Exp Cell Res. 1997;232:255–262. doi: 10.1006/excr.1997.3514. [DOI] [PubMed] [Google Scholar]
- 57.Lieman JH, Worley LA, Harbour JW. Loss of Rb-E2F repression results in caspase-8-mediated apoptosis through inactivation of focal adhesion kinase. J Biol Chem. 2005;280:10484–10490. doi: 10.1074/jbc.M409371200. [DOI] [PubMed] [Google Scholar]
- 58.Takasawa R, Nakamura H, Mori T, et al. Differential apoptotic pathways in human keratinocyte HaCaT cells exposed to UVB and UVC. Apoptosis. 2005;10:1121–1130. doi: 10.1007/s10495-005-0901-8. [DOI] [PubMed] [Google Scholar]
- 59.Jou MJ, Peng TI, Reiter RJ, et al. Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J Pineal Res. 2004;37:55–70. doi: 10.1111/j.1600-079X.2004.00140.x. [DOI] [PubMed] [Google Scholar]
- 60.Leon J, Acuna-Castroviejo D, Escames G, et al. Melatonin mitigates mitochondrial malfunction. J Pineal Res. 2005;38:1–9. doi: 10.1111/j.1600-079X.2004.00181.x. [DOI] [PubMed] [Google Scholar]
- 61.Sitailo LA, Tibudan SS, Denning MF. Activation of caspase-9 is required for UV-induced apoptosis of human keratinocytes. J Biol Chem. 2002;277:19346–19352. doi: 10.1074/jbc.M200401200. [DOI] [PubMed] [Google Scholar]
- 62.Peng TI, Jou MJ. Mitochondrial swelling and generation of reactive oxygen species induced by photoirradiation are heterogeneously distributed. Ann N Y Acad Sci. 2004;1011:112–122. doi: 10.1007/978-3-662-41088-2_12. [DOI] [PubMed] [Google Scholar]
- 63.Jou MJ, Jou SB, Chen HM, et al. Critical role of mitochondrial reactive oxygen species formation in visible laser irradiation-induced apoptosis in rat brain astrocytes (RBA-1) J Biomed Sci. 2002;9:507–516. doi: 10.1159/000064723. [DOI] [PubMed] [Google Scholar]
- 64.Saldeen J, Tillmar L, Karlsson E, et al. Nicotinamide- and caspase-mediated inhibition of poly(ADP-ribose) polymerase are associated with p53-independent cell cycle (G2) arrest and apoptosis. Mol Cell Biochem. 2003;243:113–122. doi: 10.1023/a:1021651811345. [DOI] [PubMed] [Google Scholar]
- 65.Chang H, Sander CS, Muller CS, et al. Detection of poly(ADP-ribose) by immunocytochemistry: a sensitive new method for the early identification of UVB- and H2O2-induced apoptosis in keratinocytes. Biol Chem. 2002;383:703–708. doi: 10.1515/BC.2002.072. [DOI] [PubMed] [Google Scholar]
- 66.Alvarez-Gonzalez R, Spring H, Muller M, et al. Selective loss of poly(ADP-ribose) and the 85-kDa fragment of poly(ADP-ribose) polymerase in nucleoli during alkylation-induced apoptosis of HeLa cells. J Biol Chem. 1999;274:32122–32126. doi: 10.1074/jbc.274.45.32122. [DOI] [PubMed] [Google Scholar]
- 67.Scovassi AI, Denegri M, Donzelli M, et al. Poly(ADP-ribose) synthesis in cells undergoing apoptosis: an attempt to face death before PARP degradation. Eur J Histochem. 1998;42:251–258. [PubMed] [Google Scholar]
- 68.Farkas B, Magyarlaki M, Csete B, et al. Reduction of acute photodamage in skin by topical application of a novel PARP inhibitor. Biochem Pharmacol. 2002;63:921–932. doi: 10.1016/s0006-2952(01)00929-7. [DOI] [PubMed] [Google Scholar]
- 69.Smiley ST, Reers M, Mottola-Hartshorn C, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cho JW, Kim CW, Lee KS. Modification of gene expression by melatonin in UVB-irradiated HaCaT keratinocyte cell lines using a cDNA microarray. Oncol Rep. 2007;17:573–577. [PubMed] [Google Scholar]
- 71.Takasawa R, Tanuma S. Sustained release of Smac/DIABLO from mitochondria commits to undergo UVB-induced apoptosis. Apoptosis. 2003;8:291–299. doi: 10.1023/a:1023629023696. [DOI] [PubMed] [Google Scholar]
- 72.Lee CH, Yu CL, Liao WT, et al. Effects and interactions of low doses of arsenic and UVB on keratinocyte apoptosis. Chem Res Toxicol. 2004;17:1199–1205. doi: 10.1021/tx049938m. [DOI] [PubMed] [Google Scholar]
- 73.Wang HQ, Quan T, He T, et al. Epidermal growth factor receptor-dependent, NF-kappaB-independent activation of the phosphatidylinositol 3-kinase/Akt pathway inhibits ultraviolet irradiation-induced caspases-3, -8, and -9 in human keratinocytes. J Biol Chem. 2003;278:45737–45745. doi: 10.1074/jbc.M300574200. [DOI] [PubMed] [Google Scholar]
- 74.Baydas G, Reiter RJ, Akbulut M, et al. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and anti-apoptotic protein levels. Neuroscience. 2005;135:879–886. doi: 10.1016/j.neuroscience.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 75.Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320–7326. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 76.Martin SA, Ouchi T. BRCA1 phosphorylation regulates caspase-3 activation in UV-induced apoptosis. Cancer Res. 2005;65:10657–10662. doi: 10.1158/0008-5472.CAN-05-2087. [DOI] [PubMed] [Google Scholar]
- 77.Kilic E, Kilic U, Yulug B, et al. Melatonin reduces disseminate neuronal death after mild focal ischemia in mice via inhibition of caspase-3 and is suitable as an add-on treatment to tissue-plasminogen activator. J Pineal Res. 2004;36:171–176. doi: 10.1046/j.1600-079x.2003.00115.x. [DOI] [PubMed] [Google Scholar]
- 78.Meki AR, Esmail Eel D, Hussein AA, et al. Caspase-3 and heat shock protein-70 in rat liver treated with aflatoxin B1: effect of melatonin. Toxicon. 2004;43:93–100. doi: 10.1016/j.toxicon.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 79.Scovassi AI. Mitochondrial poly(ADP-ribosylation): from old data to new perspectives. FASEB J. 2004;18:1487–1488. doi: 10.1096/fj.04-1841rev. [DOI] [PubMed] [Google Scholar]
- 80.Andrabi SA, Sayeed I, Siemen D, et al. Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004;18:869–871. doi: 10.1096/fj.03-1031fje. [DOI] [PubMed] [Google Scholar]
- 81.Han YX, Zhang SH, Wang XM, et al. Inhibition of mitochondria responsible for the anti-apoptotic effects of melatonin during ischemia-reperfusion. J Zhejiang Univ Sci B. 2006;7:142–147. doi: 10.1631/jzus.2006.B0142. [DOI] [PMC free article] [PubMed] [Google Scholar]