

Abstract
Integrin proteins are critical for stabilization of hippocampal long-term potentiation but the mechanisms by which integrin activities are involved in synaptic transmission are not known. The present study tested whether activation of α-amino-3-hydroxy-5-methylisoxazole-4-proprionate (AMPA) class glutamate receptors increases surface expression of α5β1 integrin implicated in synaptic potentiation. Surface protein biotinylation assays demonstrated that AMPA treatment of COS7 cells expressing GluR1 homomeric AMPA receptors increased membrane insertion and steady-state surface levels of α5 and β1 subunits. Treated cells exhibited increased adhesion to fibronectin- and anti-α5-coated substrates and tyrosine kinase signaling elicited by fibronectin-substrate adhesion, as expected if new surface receptors are functional. Increased surface expression did not occur in calcium-free medium and was blocked by the protein kinase C inhibitor chelerythrine chloride and the exocytosis inhibitor brefeldin A. AMPA treatment similarly increased α5 and β1 surface expression in dissociated neurons and cultured hippocampal slices. In both neuronal preparations AMPA-induced integrin trafficking was blocked by combined antagonism of NMDA receptor and L-type voltage-sensitive calcium channel activities but was not induced by NMDA treatment alone. These results provide the first evidence that glutamate receptor activation increases integrin surface expression and function, and suggest a novel mechanism by which synaptic activity can engage a volley of new integrin signaling in coordination with, and probably involved in, stabilization of synaptic potentiation.
Keywords: biotinylation, exocytosis, glutamate receptor, integrin, long-term potentiation, trafficking
Integrins are transmembrane, heterodimeric (αβ) receptors that mediate cell–extracellular matrix and cell–cell adhesion throughout the body (Alpin et al. 1998). The 18 α and nine β subunits so far identified combine in limited combinations to form over 20 different integrins with subunit composition determining both ligand specificity and signaling properties of the receptor (Hynes 1992). Integrins cross-link their extracellular matrix ligands (e.g. fibronectin, LI) (Plow et al. 2000; Previtali et al. 2001) with the actin cytoskeleton (Geiger et al. 2001) but also mediate what has been termed ‘outside in’ and ‘inside out’ signaling (Hynes 1992; Dedhar and Hannigan 1996). In the former instance, integrin ligand binding, or integrin clustering, leads to conformational changes that propagate from the extracellular to the cytoplasmic domain of the receptor and, by changing the spatial arrangements of associated signaling proteins, initiate signaling activities including tyrosine kinase and Rho GTPase cascades (Howe et al. 1998; Vuori 1998; Giancotti and Ruoslahti 1999; Parsons et al. 2000). This integrin signaling influences an impressive array of basic cellular functions (e.g. gene expression, cell cycling, calcium influx, among others) (Yamada and Miyamoto 1995; Lafrenie and Yamada 1996). ‘Inside out’ signaling refers to the process of integrin activation. Transmembrane integrin receptors can exist in states of low or high affinity for ligand binding; intracellular events [e.g. changes in protein kinase C (PKC) activity or divalent cation concentrations] can increase ligand affinity and thereby ‘activate’ the receptor to enable ligand binding (Mould 1996; Hughes and Pfaff 1998).
Although studies of integrin function in neurons have largely focused on developmental phenomena (Milner and Campbell 2002), recent work has shown that integrins are broadly expressed in adult brain (Pinkstaff et al. 1999) and influence mature synaptic responses (Gall and Lynch 2004). In particular, manipulation of integrin function by treatment with soluble integrin ligands (Staubli et al. 1990, Staubli 1998; LeBaron et al. 2003), function blocking antibodies (Chun et al. 2001; Kramár et al. 2002) and high-affinity toxins (Chun et al. 2001) blocks the stabilization of long-term potentiation (LTP) at glutamatergic hippocampal synapses. Studies with antibodies have implicated specific integrins in the stabilization process and in particular suggested the involvement of α5β1, α3β1 and αvβ3, all of which are expressed in hippocampus (Pinkstaff et al. 1999). Genetic suppression of α5β1 and α3β1 together perturbs hippocampal LTP and, in addition, produces severe impairments in hippocampus-dependent forms of learning (Chan et al. 2003). These findings accord with evidence that integrin ligand treatment blocks rapid kindling in rat hippocampal slices (Grooms and Jones 1997) and activity-induced synaptic plasticity in Drosophila (Rohrbough et al. 2000).
As best characterized in non-neural cells, integrin signaling regulates the activity of a variety of kinases (Vuori 1998; Avraham et al. 2000; Parsons et al. 2000) and a subset of these [e.g. focal adhesion kinase (FAK), Src, calcium–calmodulin-dependent kinase II] have otherwise been implicated in LTP (Malenka and Nicoll 1999; Ali and Salter 2001; Yang et al. 2003). Recent studies confirm that synaptic integrins regulate these same kinases as well as the phosphorylation of glutamate receptors (Kramár et al. 2002; Bernard et al. 2005) and the synaptic currents they mediate (Lin et al. 2003). These effects provide links between integrin signaling and the events that express and stabilize LTP.
Less progress has been made in understanding how integrin signaling is engaged by, or coordinated with, processes that induce activity-dependent synaptic plasticity. One possibility is that intense glutamatergic activity (the trigger for LTP) increases the abundance or availability of integrin ligands, as thought to occur with activity-regulated extracellular proteolysis (Davis et al. 2000). Alternatively, or in association with this, synaptic activity may increase integrin surface expression or activate latent transmembrane integrins, leading to new integrin binding to extant matrix ligands and a wave of increased integrin signaling. The latter idea seems reasonable given evidence that synaptic, or more specifically glutamatergic, activity drives other receptor types [e.g. α-amino-3-hydroxy-5-methylisoxazole-4-proprionate (AMPA) receptors, neurotrophin receptors] into the membrane at the region of the synapse (Du et al. 2000; Shi et al. 2001; Tomita et al. 2001). The present studies tested whether glutamate receptor stimulation increases integrin surface expression using surface protein biotinylation techniques and three experimental preparations. Initial studies employed COS7 cells transfected to express GluR1 homomeric AMPA receptors; COS7 cells naturally express high levels of the α5 and β1 integrin subunits that together form the α5β1 dimer implicated in LTP (Chun et al. 2001), and this and other clonal cell lines have proven valuable for studies of the parameters and mechanisms of receptor trafficking as occur in neurons (Liang and Huganir 2001; Passafaro et al. 2001; Shi et al. 2001). The GluR1 subunit was chosen for transfection because, among the AMPA receptor subunits, GluR1 has most convincingly been demonstrated to play a critical role in LTP (Vanderklish et al. 1992; Lee et al. 2003; Meng et al 2003; Schmitt et al. 2005). Subsequent experiments used cultured forebrain neurons and cultured hippocampal slices to test whether effects described in COS7 cells were similarly elicited in neurons. Together, the results from these studies demonstrate that stimulation of AMPA class glutamate receptors does indeed increase integrin surface expression in a time frame that is appropriate for a contributor to LTP consolidation.
Materials and methods
COS7 cell culture and transient transfection
Effects of AMPA receptor activation on integrin surface expression were evaluated in COS7 cells transfected to transiently express GluR1 giving rise to GluR1 homomeric AMPA receptors. COS7 cells (CRL-1651; American Type Culture Collection, Manassas VA, USA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen Life Tech, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine and 100 units/mL each of penicillin and streptomycin in a humidified 5% CO2 incubator at 37°C. Every 2–3 days the cells were subcultured by trypsinization (0.05% w/v trypsin, with 1% EDTA) followed by plating on to uncoated dishes (Hynes 1992). For experimental use, cells were transiently transfected with GluR1/pRK5 (Verdoorn et al. 1991) (a gift from Dr Kathy Partin and Dr Peter Seeburg) using Lipofectamine 2000 reagent (Invitrogen). At 2–3 days after transfection, GluR1 expression (total and cell surface) was verified by western blotting using techniques and antisera described below.
Cultured hippocampal slices
Hippocampal slices were cultured from Sprague-Dawley rats (9–10 days postnatal (P); Simonsen Laboratories, Gilroy, CA, USA) as described previously (Lauterborn et al. 2000). Slices were cut at a thickness of 400 μm perpendicular to the long axis of the hippocampus, and included hippocampus, entorhinal cortex and portions of adjacent neocortex. Slices were explanted on to Millicell-CM biomembrane inserts (Millipore Corporation, Bedford, MA, USA) in a six-well culture plate (Corning Inc., Acton, MA, USA) (six slices/well) containing 1 mL/well medium consisting of minimum essential medium, 30 mM dextrose, 1 mg/L insulin, 20% horse serum (pH 7.2), 30 mM HEPES, 5 mM NaHCO3, 3 mM glutamine, 0.5 mM ascorbic acid, 2 mM CaCl2 and 2.5 mM MgSO4 (all reagents from Sigma, St Louis, MO, USA unless otherwise noted); each pup generated four wells within one culture plate. Slices were maintained in a humidified, 5% CO2 incubator at 37°C with medium changed every other day for 12–18 days in vitro before experimental use. For surface biotinylation studies described below, 18 slices from three separate culture plates were pooled after treatment for each experimental condition.
In all cases, experiments were performed in accordance with National Institutes of Health guidelines and protocols approved by the Institutional Animal Care and Use Committee, taking care to minimize distress to the animals and the number of animals used.
Primary neuronal culture
Dissociated neurons from P1 Sprague–Dawley rat hippocampus and neocortex were cultured on to six-well culture plates (Corning Inc.) coated with poly-D-lysine (Sigma) at 5–10 × 106 cells/well as described by Li et al. (1997). Cells were fed twice per week with Neurobasal media containing B27 supplement (Invitrogen; medium preconditioned on astroglial feeder cells for 24 h) and the mitotic inhibitor 5-fluoro-2-deoxyuridine (to minimize non-neuronal cells in the cultures). Mature neurons (~ 20 days in vitro) from two culture wells were used for each experimental condition for integrin surface expression experiments.
Measures of integrin cell surface expression
Procedures for measuring new cell surface expression (i.e. membrane insertion and steady-state surface expression) were adapted from Liang and Huganir (2001). Similar procedures were applied to cultured GluR1-transfected COS7 cells (to be referred to as GluR1/COS7 cells), hippocampal slices and dissociated neurons. To measure proteins newly inserted into the membrane (membrane ‘insertion’ assay), cultures were treated with sulfo-N-hydroxysuccinimide (NHS)-acetate (4°C, three times each for 20 min; Pierce Biotechnology, Rockford IL, USA) to block pre-existing cell surface proteins from biotinylation. Cultures were washed twice with 0.1 M sodium phosphate buffer with 0.9% NaCl (phosphate-buffered saline, PBS) supplemented with 100 mM glycine and twice with PBS alone at 4°C, and then incubated with 50 μM AMPA (Tocris Cookson, Ellisville MO, USA) or vehicle at 37°C (10 min). In some experiments, parallel cultures were similarly treated but an antagonist was added to the medium 20 min before and during AMPA exposure; antagonists included the AMPA receptor antagonist 6,7 dinitroquinoxaline-2,3-dione (DNQX; 20 μM), the NMDA receptor antagonist DL-2-amino-5-phosphonovaleric acid (APV; 50 μM) and the L-type voltage-sensitive channel blocker nifedipine (Nif; 10 μM) (Tocris Cookson). To assess ‘steady-state’ integrin surface expression, cultures were similarly treated with AMPA (or AMPA + DNQX or AMPA + APV/Nif) alone; i.e. the sulfo-NHS-acetate blocking and glycine rinse steps were omitted. After AMPA treatment, cells/ explants were shifted to 4°C for surface biotinylated with 1 mg/mL sulfo-NHS-S-S-biotin (Pierce) for 60 min, washed twice with PBS supplemented with 100 mM glycine at 4°C (20 min), washed with PBS alone, and then lysed and tip sonicated within RIPA buffer containing 10 mM Tris, pH 7.2, 158 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 1% Triton X-100, 1 mM Na3VO4 and Complete Protease Inhibitor Cocktail (Roche Diagnostics, Indianapolis, IN, USA). Protein levels in the lysate were determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA) and volumes were adjusted to normalize protein density. An aliquot of the normalized lysate was used for western blot assessment of total integrin protein levels. From the remaining samples biotinylated proteins were precipitated with NeutrAvidin beads (Pierce) and processed for western blot analysis of β1 and α5 integrin subunit immunoreactivities using antisera AB1952 and AB1921, respectively, and of actin immunoreactivity using MAB3128 (Chemicon, Temecula, CA, USA).
Immunoreactive bands were measured using enhanced chemiluminescence ECL Plus kit and film from Amersham Biosciences, Inc. (Arlington Heights, IL, USA) and densitometric analysis of films using the AIS image analysis system (Imaging Research, St Catherines, ON, Canada). For these and subsequent analyses, the significance of the effects of treatment was assessed using (1) the paired or unpaired t-test for comparison of two groups or (2) one- or two-way ANOVA followed by the Student–Newman–Keuls (SNK) post hoc test for paired comparisons for the analysis of three or more groups. The 95% confidence level (p < 0.05) was considered statistically significant.
Adhesion assays
Two different assay systems were used to evaluate GluR1/COS7 cell adhesion to fibronectin- and anti-α5 integrin-coated substrates. For the fibronectin adhesion assay (Dedhar and Hannigan 1996), tissue culture plates were coated with 10 μg/mL full-length human fibronectin (Invitrogen) in PBS overnight at 4°C and blocked with 1% bovine serum albumin before use. Near-confluent cells were serum starved overnight in DMEM containing 0.1% FBS. Cells were then incubated at 37°C in DMEM containing 0.1% FBS with or without AMPA (50 μM) for 10 min, washed with PBS, and then trypsinized in 0.01% trypsin–EDTA (Invitrogen) containing 5 mM EDTA for 5 min at 4°C. Trypsinization was terminated with 1 mg/mL soybean trypsin inhibitor (Invitrogen) in PBS containing 5 mM EDTA. Cells were collected, washed in PBS/5 mM EDTA, and resuspended in serum-free DMEM for at least 30 min at 4°C. Cells were then placed on fibronectin-coated plates at a density of 3–4 × 106 cells per 10-cm plate at 37°C in a CO2 incubator for 10 or 30 min. Cells were then washed once with DMEM to remove non-adherent cells and lysed on the plate in RIPA buffer (as above); lysate protein levels were assayed (Bio-Rad).
The second adhesion assay used the α5 Integrin-Mediated Cell Adhesion Kit from Oncogene Research Products (La Jolla, CA, USA). Briefly, near-confluent GluR1/COS7 cells were serum starved and treated with AMPA (or vehicle) for 10 min and then resuspended in serum-free DMEM (4°C for 30 min to 1 h) as above. The cell suspension was then distributed across wells of 96-well plates previously coated with murine monoclonal antiserum to the extracellular domain of α5 integrin (JA7673; Oncogene Research Products) as per kit instructions; negative control wells were similarly treated with 100 μL of assay diluent only. To each well, 150 μL of 1 × 105 cells per mL suspension was added and incubated at 37°C for 10 or 30 min. The medium and non-adherent cells were then removed and wells were washed gently with PBS containing 1 mM CaCl2 and 1 mM MgCl2. The kit cell staining solution (100 μL/well) was then added for 15 min at room temperature (22–24°C) and removed. Wells were washed with PBS containing calcium and magnesium (as above) and allowed to air dry. Finally, 100 μL/well of the kit stain extraction solution was then applied for 10–20 min to solubilize the cell-bound stain. The optical density of each well was determined at 595 nm using a microtiter plate reader and SoftMax Pro 4.0 software (Molecular Devices, Sunnyvale, CA, USA).
Adhesion-induced signaling
Evaluation of cellular mechanisms mediating treatment effects on adhesion-induced signaling employed the fibronectin substrate attachment preparation described above. In separate preparations, GluR1/COS7 cells were treated with 50 μM AMPA alone or in the presence of cycloheximide (10 μg/mL for 30 min; Sigma), brefeldin A (BFA; 10 μg/mL for 30 min; Sigma), or chelerythrine chloride (CCl; 500 nM for 30 min; Calbiochem, La Jolla, CA). Control cells were treated with normal medium for the same interval. Cells were washed and resuspended as described above and either left in suspension for 30 min or placed on fibronectin-coated plates at a density of 3–4 × 106 cells per 10-cm plate for 10 or 30 min. Suspended (non-adherent) cells were pelleted then lysed in RIPA. Adherent cells were washed once with DMEM and lysed in RIPA on the plate. Activation of integrin signaling intermediaries in suspended and adherent samples was examined by western blotting with tyrosine (Y) or threonine (T) phospho-specific antibodies to phosphorylated FAK (Y397) (Sanders and Basson 2000; Bongiorno-Borbone et al. 2002), Src (Y418) (Sanna et al. 2000), paxillin (Y31) (Schaller et al. 1999) (Biosource Int., Camarillo CA, USA) and p44/42 mitogen-activated protein kinase (MAPK) (T202/Y204) (Cell Signaling Technology, Beverly, MA, USA); all primary antisera were used at 1 : 1100 dilution in Tris-buffered saline supplemented with 5% bovine serum albumin. Western blot analysis of actin immunoreactivity used MAB3128 (Chemicon). Immunoreactive bands were detected using enhanced chemiluminesence techniques described above. Measures of protein levels reflect band relative optical density × area.
Results
AMPA receptor activation increases α5 and β1 integrin surface expression in GluR1/COS7 cells
COS7 cells transfected to express GluR1 homomeric receptors were used to measure the concentration of newly inserted copies of the α5 and β1 integrin subunits. Preliminary studies using surface protein biotinylation followed by precipitation with NeutrAvidin beads and western blot analysis of the precipitates confirmed that GluR1/COS7 cells expressed GluR1 protein at the cell surface (data not shown). To measure stimulus-driven membrane insertion, the transfected cells were pretreated with membrane-impermeable sulfo-NHS-acetate to block biotinylation of pre-existing cell surface proteins, and then treated for 10 min with AMPA (50 μM, 37°C) followed by the membrane-impermeable sulfo-NHS-S-S-biotin. Under these conditions only newly inserted plasma membrane proteins are biotinylated. Biotinylated proteins were harvested by NeutrAvidin bead precipitation and then subjected to western blot analysis to assess levels of biotinylated α5 and β1 integrin protein. AMPA receptor stimulation dramatically increased insertion of α5 and β1 integrin relative to levels in cells processed without AMPA treatment. The immunoblots on the left and right side of Fig. 1(a) respectively show relative levels of (1) biotinylated protein, newly inserted into the cell membrane and (2) total protein within the lysate for one representative experiment. As shown, the agonist increased the insertion of both α5 and β1 proteins, and both effects were almost completely blocked by co-treatment with the AMPA receptor antagonist DNQX. In contrast, AMPA treatment did not detectably alter total levels of either α5 or β1 integrin protein (Fig. 1a, right side). There was no measurable biotinylation of the intracellular protein actin, which was readily labeled in cell lysates (Fig. 1a, bottom panel), indicating that the sulfo-NHS-S-S-biotin did not penetrate the plasma membrane and attached only to proteins on the cell surface. Figure 1(b) summarizes results from densitometric measurements of western blot band densities from four independent experiments. AMPA treatment caused an approximately seven-fold increase in membrane insertion of α5 and an eight-fold increase in β1, both effects being blocked by DNQX. Figures 1(c) and (d) show the effect of the AMPA receptor agonist on new membrane insertion of β1 and α5 integrin subunits in non-transfected COS7 cells; measurements from three independent experiments demonstrated that neither AMPA or AMPA + DNQX influenced β1 or α5 integrin membrane insertion in cells lacking GluR1 expression.
Fig. 1.
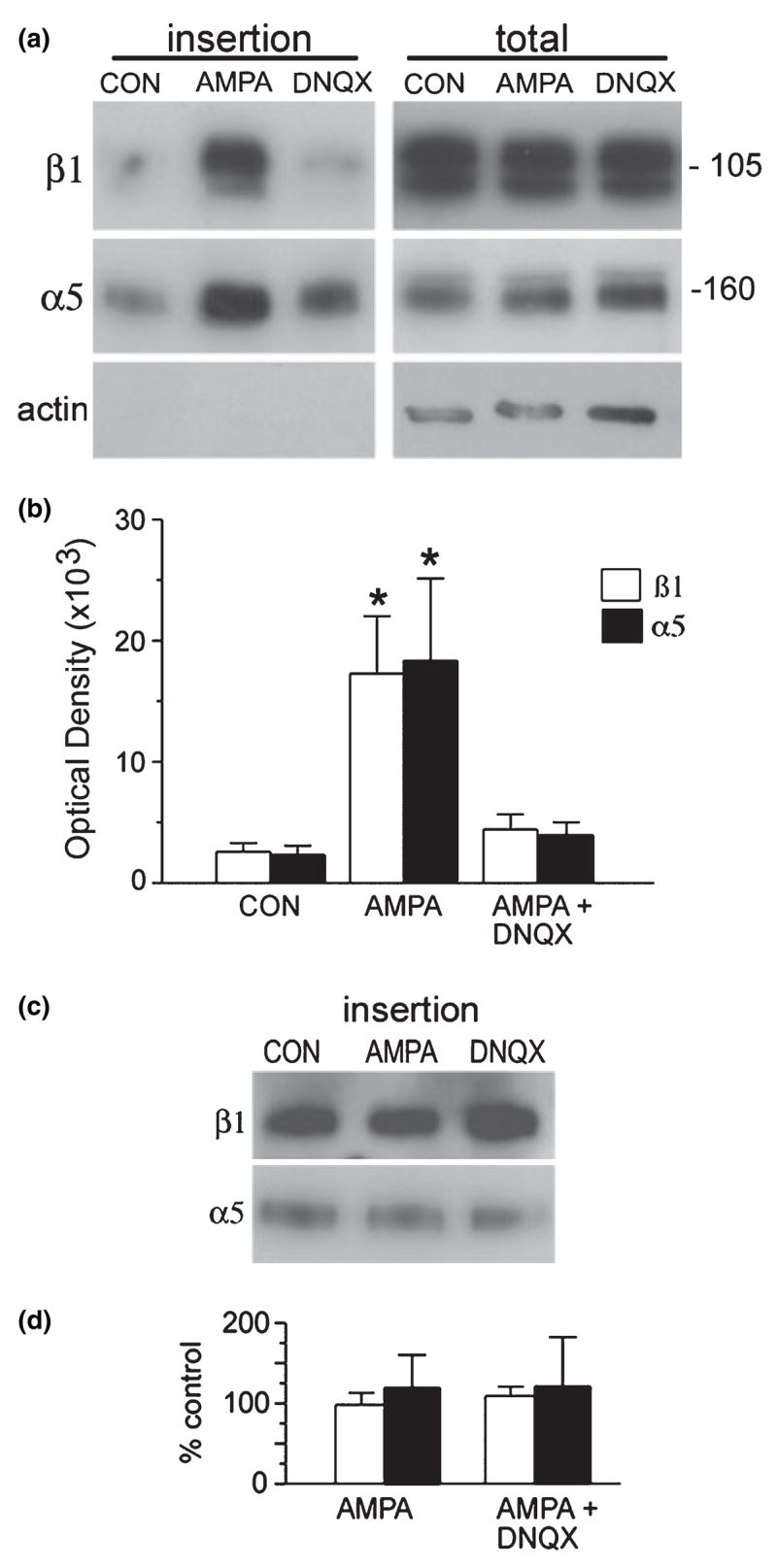
AMPA stimulation increases surface expression of β1 and α5 in GluR1/COS7 cells. GluR1-transfected COS7 cells were pretreated with sulfo-NHS-acetate then incubated for 10 min in the absence (control; CON) or presence (AMPA) of 50 mM AMPA. Parallel sets of cells were treated with DNQX before and during AMPA treatment. Newly exposed cell surface proteins were then biotinylated and precipitated, and western blots were used to determine α5 and β1 levels in the biotinylated fraction (‘insertion’ samples) and the total cell lysate (‘total’ samples), (a) Western blots show immunoreactivity within the insertion (left) and total lysate (right) pools for a single experiment. AMPA induced large increases in newly inserted α5 and β1 immunoreactivities that were blocked by DNQX co-treatment. Blots on the right show that AMPA and AMPA + DNQX (DNQX lane) had no obvious effect on total α5 or β1 protein levels. Lower blots in each column show levels of actin immunoreactivity. (b) Bar graph shows quantification of newly inserted α5 and β1 immunoreactivities as determined from densitometric analysis of western blots (mean ± SEM optical density values; n = 4). AMPA treatment reliably increased α5 and β1 membrane insertion (p = 0.01, one-way ANOVA; *p < 0.05 versus CON, SNK test) that was blocked by DNQX. (c) Western blot shows levels of newly inserted β1 and α5 immunoreactivities in non-transfected COS7 cells treated with normal medium (CON), 50 μM AMPA or AMPA + DNQX. AMPA treatment did not increase surface expression in non-transfected cells, (d) Bar graph shows quantification of newly inserted α5 and β1 immunoreactivities in non-transfected cells treated with AMPA or AMPA + DNQX (expressed as percentage of paired CON values; mean ± SEM; n = 3).
In the experiments illustrated in Fig. 2, GluR1/COS7 cells were processed for analysis of the time course of AMPA effects on new membrane insertion and steady-state surface levels of the α5 and β1 integrin subunits. Cells were exposed to AMPA for 1 min (pulse treatment) or continuously until the time of harvest. For the membrane insertion assay cells were treated with sulfo-NHS-acetate to mask surface proteins to future labeling, treated with 50 μM AMPA, and harvested 1,3, 5, 8, 10 and 20 min after the onset of AMPA exposure for surface biotinylation. To assess AMPA treatment effects on steady-state α5 and β1 surface levels, cells were similarly processed but without the sulfo-NHS-acetate masking treatment before AMPA exposure. As illustrated in Fig. 2(a), with continuous AMPA treatment there was a clear, progressive increase in both newly inserted and steady-state levels of α5 and β1 immunoreactivity on the cell surface. For α5, increased insertion was evident by 3 min and reached peak levels at 8–20 min; increases in steady-state levels (which should reflect both pre-existing and newly exposed surface immunoreactivities) were evident at 5 min and more markedly at 8 min after the start of treatment, although steady-state levels continued to increase up to the 20-min time point. Effects of continuous AMPA treatment on surface β1 immunoreactivity were similar, with increases in both insertion and steady-state levels evident at 5 min and the rate of new insertion peaking at 8–10 min after treatment onset. Steady-state β1 levels were dramatically increased at 8–20 min of treatment.
Fig. 2.
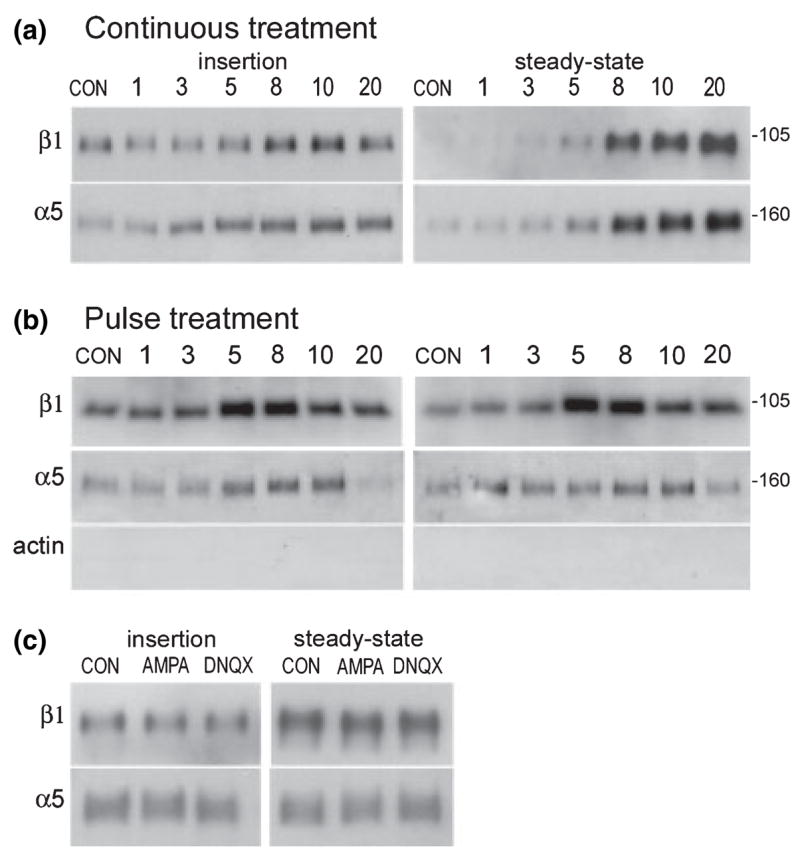
AMPA-induced increases in integrin surface expression: time course and calcium dependence. (a,b) GluR1/COS7 cells were treated with 50 μM AMPA continuously for 1–20 min (a) or for 1 min and then harvested from 1 to 20 min after treatment onset (b) and processed for surface biotinylation/western blot analysis of new membrane ‘insertion’ (left panel) and ‘steady-state’ surface expression (right panel) of β1 and α5 immunoreactivities present as bands at 105 kDa and 160 kDa respectively. (Note, blot for the continuous-treatment steady-state assay was exposed to film more briefly than the others to avoid saturatation of labeling at the later time points. Consequently low levels of surface β1 in control samples are not evident in the panel shown). With both continuous and pulse AMPA treatment, increases in newly inserted and steady-state α5 and β1 surface expression were evident in 5–8 min; with pulse AMPA application, new insertion and steady-state levels then declined, (c) GluR1/COS7 cells in calcium-free medium were treated with 50 μM AMPA for 10 min and processed for either the new membrane insertion or steady-state surface protein assay. In the absence of extracellular calcium AMPA treatment had no effect on α5 or β1 membrane insertion or steady-state surface levels.
Results were similar with AMPA applied in a 1-min pulse and cell harvest at a range of time points thereafter. As shown in Fig. 2(b), with pulse treatment both newly inserted and steady-state surface β1 immunoreactivity was abruptly increased between 3 and 5 min after treatment onset, peaked at 5–8 min, and then declined but remained above control levels at the 20-min time point. Pulse treatment increased α5 insertion and steady-state levels most clearly by 5 and 8 min respectively, with a return to control values at the 20-min time point.
Effects of AMPA receptor activation on glutamate receptor surface expression are dependent upon calcium influx as demonstrated by the fact that they are not observed in calcium-free medium. To determine whether AMPA-induced increases in integrin surface expression involve similar mechanisms, GluR1/COS7 cells were treated with AMPA in calcium-free medium and effects on integrin membrane insertion and steady-state surface expression were examined. As shown in Fig. 2(c), newly inserted and steady-state β1 and α5 surface levels were unaffected by AMPA or AMPA + DNQX treatment under these conditions.
AMPA receptor activation increases cell-substrate adhesion
Although the above experiments indicate that stimulation of AMPA-type glutamate receptors increases the surface expression of α5 and β1 integrins, they leave open the questions of whether the newly exposed subunits form functional fibronectin receptors and subserve increased adhesive function. To test this possibility, assays were conducted to determine whether the pronounced increases in membrane insertion are accompanied by equivalent changes in matrix adhesion by GluR1/COS7 cells.
Adhesion was assayed by plating cells on to substrates coated with either fibronectin or an antibody directed against the extracellular domain of α5 integrin. Figure 3(a) summarizes mean results for six independent experiments in which adhesion to fibronectin was compared in stimulated and yoked control populations of cells. In all cases cells were exposed to the glutamate receptor agonist AMPA (or control solution) for 10 min, washed and resuspended for 30 min, and then plated on to the test substrate for the indicated periods. At harvest the level of adhesion was assessed by measures of substrate adherent protein. Adhesion by control and experimental populations was highly correlated across experiments (r = 0.98) and, as shown in Fig. 3(a), the predicted difference was obtained in paired statistical comparisons. Adhesion measures for AMPA-treated cells were 26% greater than controls at 10 min after plating (p = 0.029, t-test) and were more prominent still (+ 60%; p = 0.019) after 30 min of attachment. Similar results, with less between-experiment variability in control binding, were obtained using immobilized antibodies directed to the α5 integrin as substrate (Fig. 3b). AMPA stimulation increased adhesion by 78% of paired control values at 10 min after plating (p < 0.001) and by 77% at 30 min (p < 0.01). The AMPA receptor antagonist DNQX had no effect on adhesion when applied alone but fully blocked AMPA effects on adhesion when applied 10 min before and during agonist treatment.
Fig. 3.
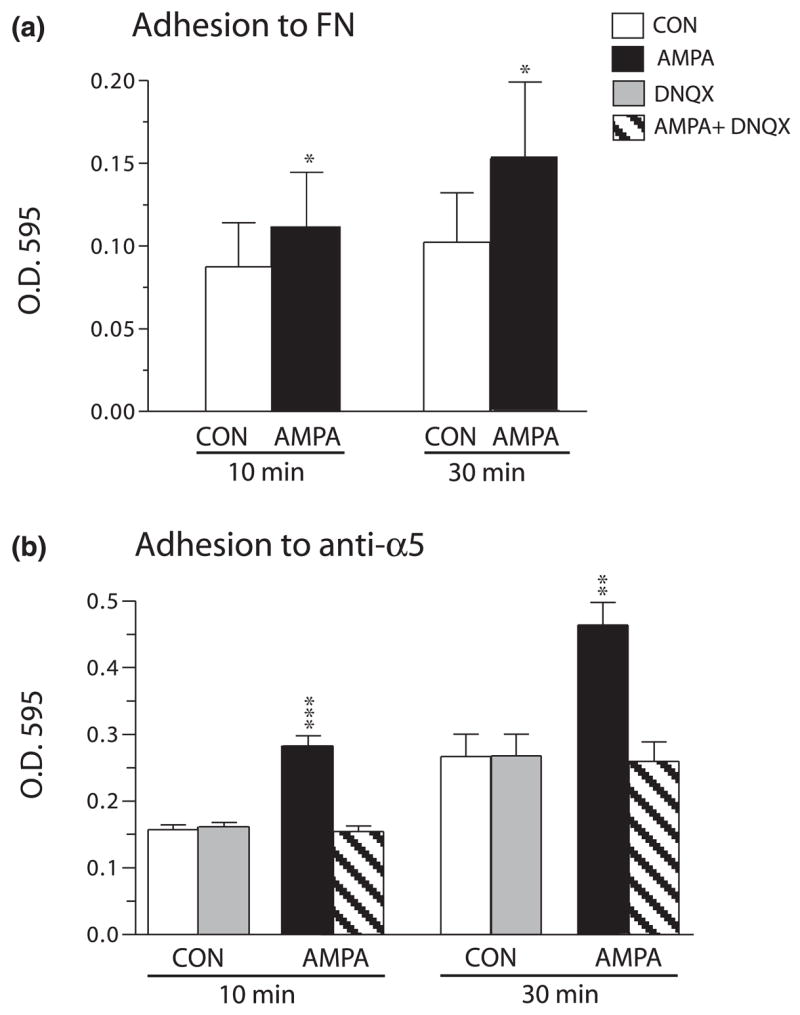
AMPA treatment increases GluR1/COS7 cell adhesion to fibronectin- and anti-α5-coated substrates. GluR1/COS7 cells were treated for 10 min, plated on to either fibronectin (FN)-coated (a) or anti-α5 integrin-coated (b) substrates, and adherent protein levels were assessed at 10 and 30 min after plating. Graphs show mean ± SEM values for six and three experiments in (a) and (b) respectively, (a) AMPA treatment increased GluR1/COS7 adhesion to FN at both 10 and 30 min after plating [at 10 min: *p<0.03 versus control (CON) (one-tailed paired ttest; correlation coefficient = 0.98, effectiveness of pairing p = 0.004); at 30 min: *p = 0.019 versus CON (correlation coefficient = 0.98, effectiveness of pairing p = 0.0002)]. (b) AMPA treatment increased adhesion to anti-α5-coated substrate (p < 0.05 and p < 0.001 for main effect of treatment, separate one-way ANOVA analyses for 10- and 30-min time points respectively; **p < 0.01 and ***p < 0.001 versus yoked control values, SNK post hoc test).
AMPA pretreatment enhances integrin-mediated signaling
Following binding to extracellular matrix ligands, the longer cytoplasmic tails of β integrin subunits directly associate with FAK and/or its homologue proline-rich tyrosine kinase 2 (Pyk2) (Schlaepfer and Hunter 1998; Schuster et al. 2001; Loeser et al. 2003). FAK and Pyk2 then become tyrosine phosphorylated, events that lead to binding and activation of Src family kinases as well as FAK phosphorylation at secondary sites (Vuori 1998; Parsons et al. 2000). Integrin ligation also results in tyrosine phosphorylation of paxillin, which binds both α and β integrin subunits (Liu et al. 2000; Parsons et al. 2000), and to downstream increases in activities of p44/42 MAPK (also known as ERK 1/2), Rho family GTPases and phophatidylinositol 3-kinase (Vuori 1998; Giancotti and Ruoslahti 1999; Schwartz and Ginsberg 2002). These downstream events provide ample targets with which to determine whether increased insertion due to glutamatergic stimulation results in surface integrins that are not only competent for adhesion but are also capable of setting in motion the signaling sequences through which adhesion modifies cell structure and function. Accordingly, we tested whether AMPA stimulation of GluR1/COS7 cells potentiates key steps in the abovementioned integrin-driven phosphorylation cascades.
GluR1/COS7 cells were treated with 50 μM AMPA for 10 min, resuspended, and then plated on to a fibronectin-coated substrate for 10 or 30 min, after which cells adhering to the substrate or remaining in suspension were harvested separately and processed for immunoblot analysis of levels of phospho (p)-FAK (Y397), p-Src (Y418), p-paxillin (Y31) and p-p44/42MAPK (T202/Y204) using phosphorylation site-specific antibodies. Results of a representative experiment are shown in Fig. 4(a). Phospho-protein levels were low in control cells that had been plated for just 10 min, indicating that, although substantial adhesion had occurred by this point (see Fig. 3), signaling that follows from it was still in development. However, cells pretreated with AMPA had substantially higher levels of all four phospho-proteins even at 10 min (Fig. 4a). The difference between the two groups of cells was still very much in evidence at 30 min after plating, a time point at which signaling was well under way in control cells. Actin immunoreactivity was comparable at the two time points and was not detectably affected by AMPA treatment (Fig. 4a, bottom panel). The cells retained in suspension had negligible phospho-protein levels in the control group and, with the exception of very low levels of p-pMAPK, in the pretreated group; this confirms that activation of signaling is adhesion mediated and that AMPA receptor stimulation has little effect on the target proteins independent of adhesion. Quantification of treatment effects on p-FAK immunoreactivity across four independent experiments confirmed these observations (Fig. 4b). With AMPA pretreatment, levels of p-FAK (Y397) were significantly greater than the low levels found in yoked control cells at 10 min after plating on to the fibronectin substrate (p = 0.014; paired Mann-Whitney U-test); moreover, these large differences were again found at 30 min after plating, a point at which control levels were readily measured. Prior AMPA stimulation had no effect on p-FAK in cells in suspension (Fig. 4b, right bars). The same basic response profile was obtained with the protein synthesis inhibitor cycloheximide (10 μM) applied at 30 min before and during AMPA treatment. Thus AMPA treatment potentiated adhesion-induced phosphorylation of all signaling intermediaries; among adherent cells p-FAK levels were significantly greater in AMPA-treated compared with non-pretreated cells at both 10 and 30 min after plating (p < 0.05; paired t-test, n = 4).
Fig. 4.
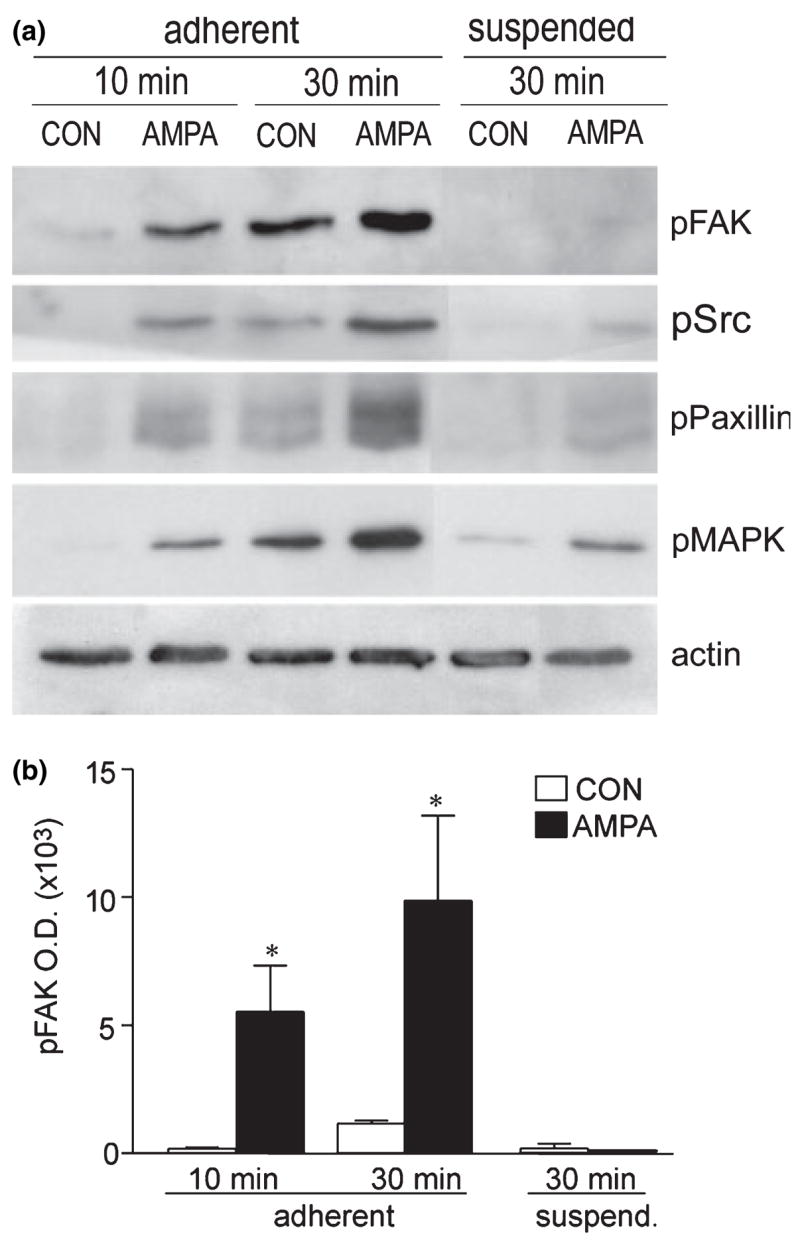
AMPA pretreatment increases adhesion-induced signaling. GluR1/COS7 cells were treated with normal medium (CON) or 50 μM AMPA for 10 min then retained in suspension or plated on to fibronectin for 10 or 30 min. Cell lysates were processed for western blot analyses using phospho (p)-specific antibodies to p-FAK (Y397), p-Src (Y418), p-paxillin (Y31) and p-p44/42MAPK (T202/Y204). Aliquots were also assessed for actin immunoreactivity to verify comparable sample protein content, (a) Blots show results for one experiment (aliquotsfrom the same samples, all rows). For adherent cells, levels of p-FAK, p-Src, p-paxillin and p-pMAPK were all higher in AMPA-pretreated (AMPA) than in control (CON) samples at 10 and 30 min after plating. In contrast, phospho-protein levels were extremely low in suspended cells although p-p44/42MAPK was more abundant in AMPA-pretreated than CON cells, (b) The same pattern of results was obtained across four experiments as illustrated by the quantification of p-FAK band densities (mean ± SEM; n = 4 per group). AMPA pretreatment significantly increased adhesion-induced p-FAK levels at 10 and 30 min after plating (*p < 0.05 vs. CON, one-tailed paired Mest). ANOVA of 30-min values (among adherent and suspended cells) demonstrated significant effects of AMPA (p < 0.05) and adhesion (p < 0.01), and significant interactions between the two groups (p < 0.05).
AMPA receptor-driven increases in α5 and β1 surface expression involve PKC and BFA-sensitive exocytotic mechanisms
PKC has been implicated in both integrin trafficking to the membrane (Ng et al. 1999; Wang et al. 2002) and inside out integrin activation leading to a high-affinity ligand state (Hynes 1992; Mould et al. 1995; Hughes and Pfaff 1998; Parsons et al. 2000). We used the PKC inhibitor CCl to test whether the kinase also plays a role in AMPA-induced increases in integrin surface expression and adhesion-dependent signaling. As shown in Figs 5(a) and (b), AMPA treatment alone induced a robust increase in new membrane insertion of α5 and β1 as indexed by surface protein biotinylation (as in above experiments); this increase was completely blocked by CCl which, by itself, had no measurable effect on surface (biotinylated) integrin levels.
Fig. 5.
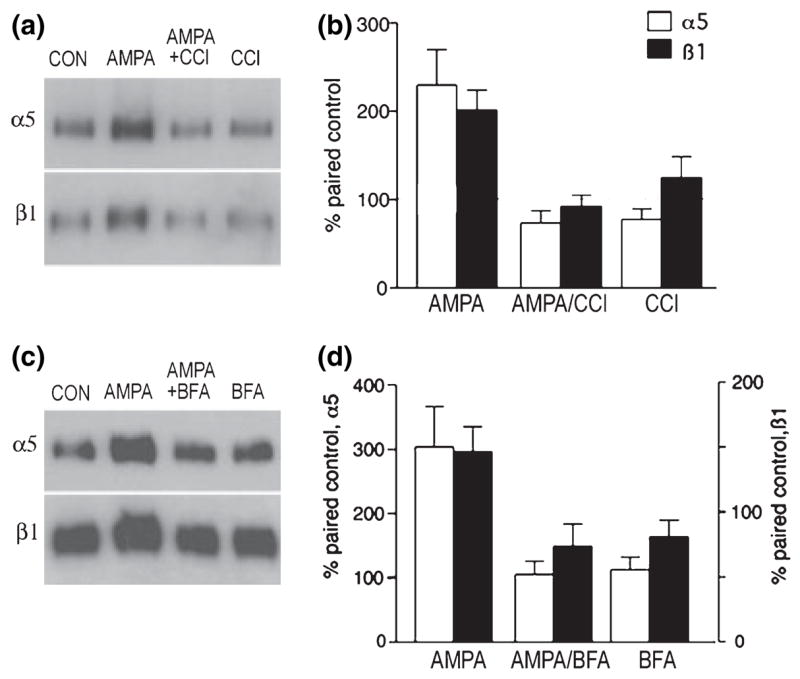
CCl and BFA block AMPA-induced increases in α5 and β1 integrin membrane insertion. GluR1/COS7 cells were pretreated with membrane-impermeable sulfo-NHS-acetate and then treated with normal medium (CON), 50 μM AMPA for 10 min, 500 nM CCl or 10 ng/mL BFA for 30 min before and during AMPA treatment, or CCl or BFA alone for 40 min. Newly exposed cell surface proteins were then biotinylated, precipitated, and processed for western blot analysis of α5 and β1 levels, (a) Blots from one experiment using CCl demonstrate that AMPA alone induced marked increases in biotinylated (i.e. newly inserted) α5 and β1 levels that were blocked by CCl co-treatment, (b) Quantification of band densities for the four treatment groups in three experiments (mean ± SEM) demonstrates that CCl co-treatment blocked AMPA-induced increases in newly inserted α5 and β1 levels (AMPA vs. CON, p < 0.01 for both α5 and β1; other groups not significantly different; SNK test), (c) Blot from one experiment showing that AMPA treatment increased levels of newly inserted α5 and β1 levels that were blocked by BFA co-treatment, (d) Graph showing mean band densities in two experiments using BFA.
Figures 6(a) and (b) illustrate the effect of CCl on AMPA modulation of integrin signaling activities in three experiments. GluR1/COS7 cells were treated with vehicle plus the inhibitor or with AMPA plus inhibitor, suspended and washed, and then plated for 10 or 30 min. As in the previous experiment (see Fig. 4b), levels of p-FAK increased dramatically from 10 to 30 min after plating and were higher in adherent cells than in cells in suspension (Fig. 6b), indicating that some level of adhesion-dependent signaling remained intact in the presence of the PKC antagonist. However, in marked contrast to the effects of AMPA treatment alone, AMPA treatment in the presence of CCl did not potentiate adhesion-induced signaling as assessed at 10 or 30 min after plating (Figs 6a and b).
Fig. 6.
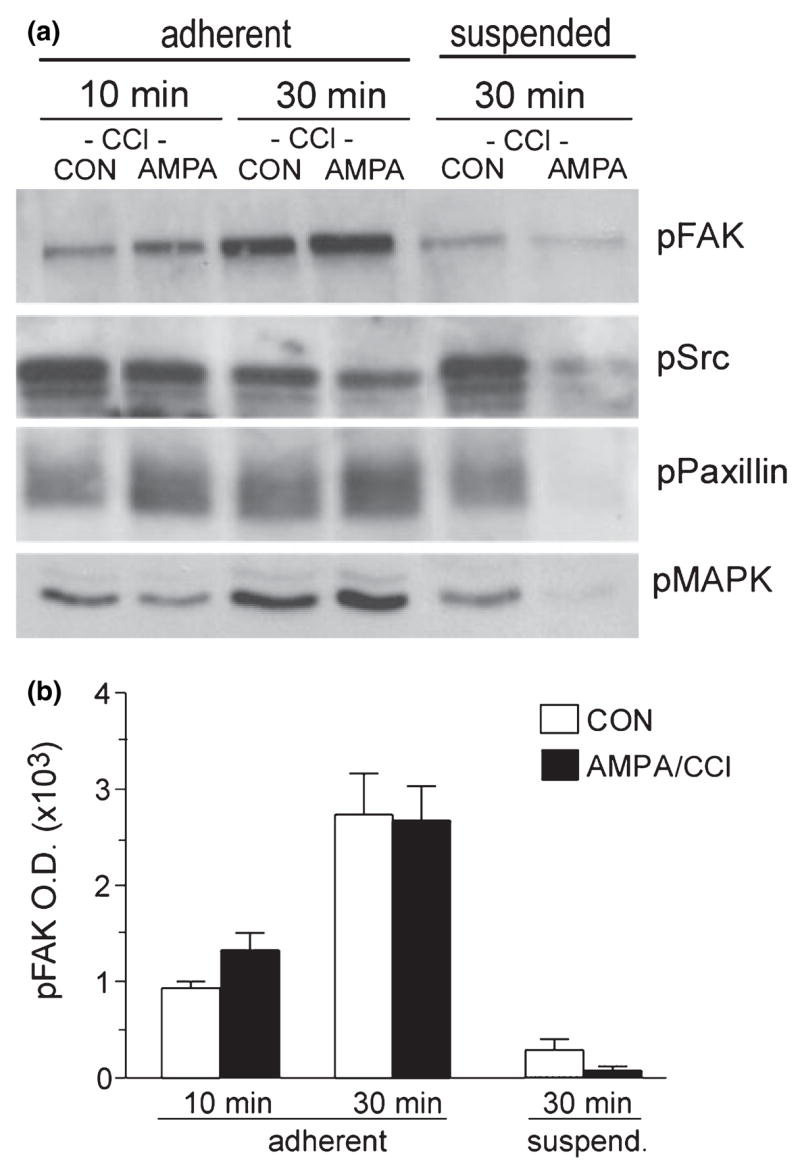
CCl blocks AMPA enhancement of adhesion-induced signaling. GluR1/COS7 cells were treated with CCl (500 niui) 30 min before and during treatment with AMPA (50 μM, 10 min) or normal medium (CON), and then processed for western blot analysis of adhesion-induced increases in phosphoprotein levels at 10 and 30 min after plating on a fibronectin substrate (‘adherent’ groups) or after 30 min in suspension (‘suspended’ groups), (a) Western blots show levels of phospho (p)-FAK (Y397), p-Src (Y418), p-paxillin (Y31) and p-p44/ 42MAPK (T202/Y204) in samples from an individual experiment. In the presence of CCl, AMPA treatment only very modestly enhanced the adhesion-induced increase in p-FAK and p-paxillin at 10 and 30 min after plating. CCl eliminated AMPA enhancement of adherent cell p-Src and p-p44/42MAPK levels, (b) Bar graph shows quantification of p-FAK band densities across four experiments (mean ± SEM). CCl blocked the effects of AMPA but not of substrate adhesion on levels of this phosphoprotein (for 30-min groups, p < 0.001 for effects of adhesion; effects of AMPA not significant, interaction between AMPA and adhesion not significant; two-way ANOVA).
BFA inhibits exocytosis by disruption of protein trafficking from the Golgi–endoplasmic reticulum (ER) network and recycling of endocytotic vesicles (Klausner et al. 1992; Shah and Klausner 1993). As a consequence, with BFA treatment newly synthesized proteins are retained in the ER and secretory activities are blocked (Shah and Klausner 1993). Recent work has shown that integrin-mediated adhesion and cellular migration can be controlled by proteins known to function in membrane trafficking and, in particular, involve processes sensitive to the effects of BFA (Schultz and Armant 1995; Wang et al. 2002). As this type of rapid trafficking might account for the speed at which increased expression occurs in AMPA-treated cells, we tested the effects of BFA on surface concentrations of α5 and β1 integrins. The blot in Fig. 5(c) shows that stimulation of AMPA receptors again increased levels of biotinylated (newly inserted) α5 and β1 proteins exposed at the cell surface and that this effect was eliminated by 10 μg/mL BFA co-treatment (Fig. 5c); BFA alone had no evident effects on surface expression. Figure 5(d) shows quantification of treatment effects on new insertion in two experiments. Tests of the effects of BFA on AMPA-driven increases in adhesion-induced kinase signaling (as in Fig. 6) demonstrated unanticipated responses to the antagonist applied alone: i.e. BFA increased p-FAK, p-Src and p-paxillin in both suspended and adherent cells. Hence, with BFA this assay was not considered useful as a secondary measure of AMPA effects on integrin surface function.
AMPA treatment increases neuronal α5 and β1 surface expression
Results of the above studies demonstrate that stimulation of GluR1 homomeric AMPA receptors increases integrin surface expression in GluR1/COS7 cells. To test whether native AMPA receptors similarly regulate integrin surface expression in forebrain neurons, protein biotinylation techniques were used to examine the effects of glutamate receptor stimulation on α5 and β1 surface expression in primary cultures of cortical/hippocampal neurons and in cultured hippocampal/entorhinal slices. Pilot studies indicated that the surface biotinylation assays could be used to measure surface proteins in either preparation with relatively modest changes from the protocols used in the GluR1/COS7 experiments.
Effects of AMPA treatment of cultured neurons and hippocampal slices are shown in Fig. 7. The blots in Fig. 7(a) (left panel) show that in primary neuronal cultures AMPA treatment for 10 min induced marked increases in both new α5 and β1 membrane insertion (57–200% increase above paired control values in four independent measures across two experiments) and steady-state surface expression (64–171% increase above control values); both effects were absent when DNQX was applied before and during AMPA treatment. Blots in the right panel of Fig. 7(a) show there were no treatment effects on total β1 and α5 protein levels. Results with cultured hippocampal slices were similar (Fig. 7b). AMPA treatment reliably increased β1 and α5 membrane insertion and α5 steady-state membrane expression across three experiments. In this set, effects of treatment on steady-state β1 surface expression were not reliably obtained. As with dissociated cell experiments, effects of AMPA treatment on α5 and β1 surface expression were blocked or attenuated by DNQX.
Fig. 7.
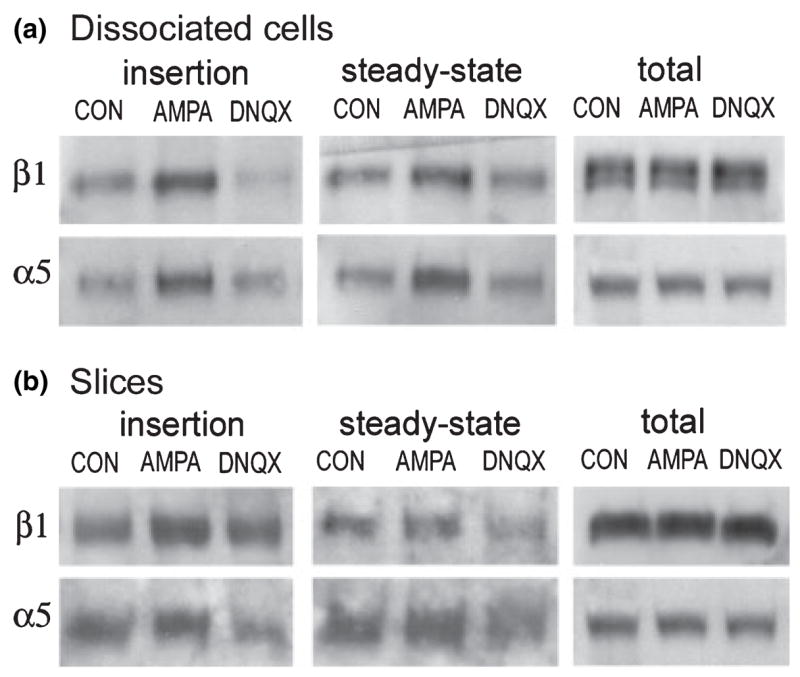
AMPA stimulation increases neuronal β1 and α5 integrin surface expression. Dissociated cortical/hippocampal neurons (a) and cultured hippocampal slices (b) were treated for 10 min with normal medium (CON), 100 μM AMPA, or AMPA plus 40 μM DNQX (DNQX lanes), and α5 and β1 immunoreactivities were measured in the newly inserted membrane protein (left panels), steady-state surface protein (middle panels) and total cell lysate (right panels), (a) Western blots from a representative dissociated cell experiment show that, relative to control levels, AMPA treatment induced increases in new membrane insertion and steady-state membrane levels of α5 and β1 integrin immunoreactivities that were blocked by DNQX co-treatment. Blots in the ‘total’ column show that AMPA and AMPA + DNQX had no obvious effect on total integrin protein levels, (b) Western blots show that in cultured hippocampal slices AMPA treatment increased β1 and α5 membrane insertion and α5 steady-state surface expression. For both subunits, DNQX attenuated or blocked increases in insertion and steady-state surface expression. AMPA and AMPA + DNQX had no effect on total β1 and α5 protein levels in cultured slices.
In experiments using COS7 cells described above, AMPA effects on integrin surface expression were dependent on extracellular calcium. The GluR1 homomeric AMPA receptors expressed in the COS7 cell studies are known to pass calcium (Hollmann et al. 1991; Jonas and Burnashev 1995) and most probably mediated the calcium influx that supported integrin trafficking. The great majority of native, neuronal AMPA receptors contain a GluR2 subunit and, consequently, do not pass calcium (Jonas and Burnashev 1995). Thus, if neuronal integrin trafficking is similarly dependent on calcium influx, the activities of native AMPA receptors may regulate integrin trafficking, at least in part, through depolarization-induced increases in calcium influx via voltage-gated calcium channels and NMDA receptors. To test for involvement of these secondary channels in integrin trafficking, neurons were treated with AMPA alone or with AMPA in the presence of the NMDA receptor antagonist APV and the L-type voltage-sensitive calcium channel (VSCC) blocker Nif. As shown in blots from a representative experiment in Figs 8(a) and (b), treatment with AMPA alone induced clear increases in α5 and β1 membrane insertion (Fig. 8a) and steady-state membrane levels (Fig. 8b). These increases were blocked with AMPA applied in the presence of APV and Nif, whereas treatment with APV and Nif alone did not alter either measure of surface expression. Quantification of western blot band densities across three such experiments demonstrated that effects of treatment on α5 insertion were statistically significant (p = 0.046, p < 0.05 and p = 0.053 for effects of AMPA, APV/Nif, and an interaction between AMPA and APV/Nif respectively, two-way ANOVA; p < 0.05 for control vs. AMPA and for AMPA vs. AMPA + APV/Nif, SNK test). For β1 there were significant interactions between effects of AMPA and APV/Nif treatment on membrane insertion (p = 0.042) although, with the small number of cases, post hoc tests did not identify significant group differences. Effects of treatments were more robust on measures of total, steady-state surface expression (Fig. 8d). There were significant effects of AMPA treatment on both α5 and β1 (p = 0.046 and p = 0.038 respectively, two-way ANOVA; p < 0.01 and p < 0.05 for control vs. AMPA and for AMPA vs. AMPA + APV/Nif for α5 and β1 respectively, SNK test). For α5, interactions between AMPA and APV/Nif treatment were also significant (p = 0.012 and p = 0.06 for interactions between AMPA and APV/Nif on α5 and β1 steady-state membrane levels respectively).
Fig. 8.
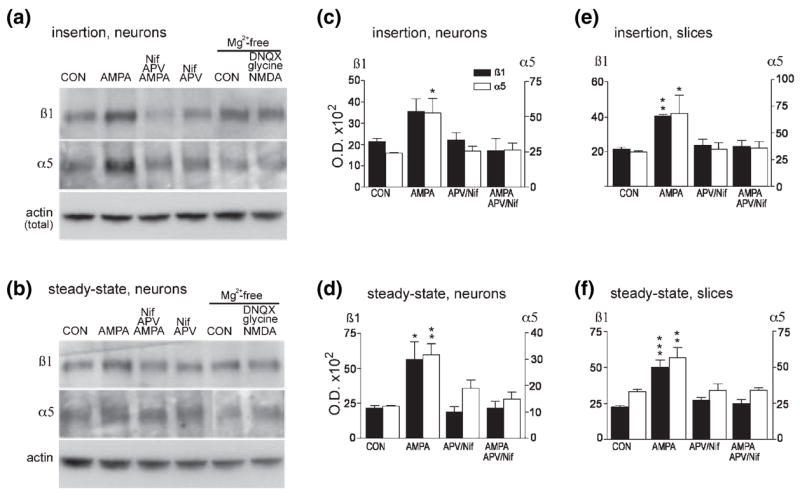
Activity of AMPA receptors, NMDA receptors and L-type VSCCs is required for full AMPA-induced increases in neuronal β1 and α5 integrin surface expression. Dissociated cortical/hippocampal neurons were treated for 10 min with normal medium (CON), 100 μM AMPA, AMPA plus 50 μM APV and 10 μM Nif, or APV plus Nif only. α5 and β1 immunoreactivities in the newly inserted and steady-state surface protein pools were evaluated. Western blots from a representative dissociated cell experiment show that, relative to control levels, AMPA increased α5 and β1 integrin protein levels in both new membrane insertion (a) and steady-state (b) surface protein pools and that these increases were blocked by APV and Nif co-treatment. APV/Nif treatment alone did not clearly influence surface α5 and β1 levels. Right two lanes in (a) and (b) show samples from parallel sets of cells placed in Mg2+-free medium and then treated with normal medium (CON) or with a cocktail of 50 μM NMDA, 200 μM glycine and 40 μM DNQX for 10 min, and processed for new membrane insertion and steady-state surface protein assays. NMDA treatment did not increase integrin protein levels in either new insertion or steady-state surface pools. In (a) and (b) lower panels show that levels of actin immunoreactivity were comparable in aliquots removed from total cell lysates before execution of insertion and steady-state assays, (c–f) Quantification of AMPA effects on α5 and β1 new membrane insertion (c,e) and steady-state surface levels (d,f) in primary neuronal cultures (c,d) and cultured hippocampal slices (e,f). Values were obtained by den-sitometric analysis of western blots and are mean ± SEM of n = 3 per group. *p < 0.05, **p < 0.01 and ***p < 0.001 versus CON (SNK test).
To test whether NMDA receptor stimulation alone is sufficient to increase integrin trafficking to the membrane, NMDA (50 μm, 10 min) was applied in the presence of the AMPA receptor antagonist DNQX. These experiments were conducted in magnesium-free medium and with 200 μM glycine added to the NMDA treatment cocktail to alleviate the voltage-dependent magnesium block and to facilitate activation of the NMDA receptors respectively. As shown in the two righthand lanes of Figs 8(a) and (b), under these conditions NMDA application did not increase either new membrane insertion or steady-state membrane levels of the α5 or β1 integrin subunits. Quantification of western blots found no significant effects of NMDA on either measure of α5 or β1 surface expression (p-values ranged from 0.24 to 0.93 across comparisons, paired two-tailed t-test, n = 3 per group).
To determine whether AMPA treatment effects on integrin trafficking are dependent on NMDA receptor and VSCC activities in more mature cells embedded in an organotypic neuropil, the above studies were repeated using cultured hippocampal slices. Measures of western blots, evaluating AMPA treatment effects on new membrane insertion and steady-state membrane levels of α5 and β1 integrin, are presented in Figs 8(e) and (f) respectively (values are mean ± SEM for three independent experiments). As with cultured neurons, in hippocampal slices AMPA treatment increased membrane insertion of α5 and β1 (Fig. 8e) and these increases were blocked by APV/Nif co-treatment (for β1 p = 0.001 for AMPA treatment and interactions between AMPA and APV/Nif treatments, for α5 interactions between AMPA and APV/Nif were not significant, two-way ANOVA; p < 0.05 and p < 0.01 for α5 and β1 respectively for post hoc comparisons of control vs. AMPA and for AMPA vs. AMPA + APV/Nif, SNK test). Treatment effects were also robust for effects on steady-state membrane expression: AMPA treatment significantly increased total α5 and β1 surface expression and these effects were completely blocked by APV/Nif (for α5 and β1 respectively p = 0.028 and p = 0.008 for effects of AMPA treatment, and p = 0.03 and p = 0.0043 for interactions between AMPA and APV/Nif treatment, two-way ANOVA; for α5 and β1 p < 0.01 and p < 0.001 respectively for comparison of control vs. AMPA and of AMPA vs. AMPA + APV/Nif, SNK test) (Fig. 8f). As found with experiments using dissociated neurons, in cultured hippocampal slices NMDA treatment in the presence of DNQX did not increase new insertion or steady-state membrane levels of α5 or β1 integrin (p-values ranged from 0.56 to 0.94 across comparisons, paired two-tailed t-test).
Discussion
Studies of synaptic plasticity in hippocampus indicate that integrins (Staubli et al. 1990, 1998; Bahr et al. 1997; LeBaron et al. 2003), particularly the α5β1 integrin (Chun et al. 2001; Chan et al. 2003), are critical for stabilization of LTP at glutamatergic synapses. Although the mechanisms of integrin involvement in LTP are still unclear, recent work has shown that integrin ligands activate kinases (Kramár et al. 2003; Bernard et al. 2005) implicated in LTP (Ali and Salter 2001; Huang et al. 2001; Poncer et al. 2002; Yang et al. 2003) and trigger phosphorylation of both AMPA and NMDA receptors (Kramár et al. 2003; Bernard et al. 2005), including phosphorylation of a GluR1 site (Kramár et al. 2003) that becomes phosphorylated with LTP (Barria et al. 1997). These phosphorylation events are accompanied by increases in AMPA and NMDA receptor-gated synaptic currents (Kramár et al. 2003; Lin et al. 2003; Bernard et al. 2005). Although these results provide evidence for an influence of synaptic integrins on neighboring glutamate receptors, they leave open the question of how intense stimulation of these neurotransmitter receptors (the induction event for LTP) might engage integrin activities. The present study examined one possibility, namely that AMPA receptor stimulation causes the insertion of additional LTP-related integrins into the cell membrane, resulting in new ligation and signaling. The results demonstrate that stimulation of GluR1 homomeric AMPA receptors in GluR1/COS7 cells increased (1) α5 and β1 integrin membrane insertion, (2) adhesion to fibronectin- and anti-α5-coated substrates, and (3) kinase signaling upon attachment to a fibronectin-coated substrate. Similar AMPA-induced increases in new membrane insertion and total surface expression of the α5 and β1 integrin subunits were obtained in studies with dissociated neurons and cultured hippocampal slices. Other work in our laboratory and elsewhere has shown that in differentiated neurons integrin proteins are localized, and in the instance of α5 concentrated, at synapses (Nishimura et al. 1998; Rodriquez et al. 2000). Together these results provide evidence that glutamate receptor activation drives new integrins into the membrane and suggest that synaptic glutamate receptor activation is likely to elicit a similar increase in integrin exposure and function with the induction of LTP.
It should be noted that these stimulatory effects on integrin trafficking probably occur amidst other changes in synaptic adhesive chemistries that play a role in LTP (Benson et al. 2000). In addition to integrins, cadherin and immunoglobulin class adhesion receptors have been localized to the synaptic or parasynaptic region and implicated in LTP (Tang et al. 1998; Murase and Schuman 1999; Benson et al. 2000; Bukalo et al. 2004), and there is evidence that the functional representation of these receptors is modulated by intense glutamatergic and/or synaptic activity. The cadherins increase their adhesivity with cis-dimerization and the assumption of a rigid, rod-like structure (Tanaka et al. 2000). In neurons and hippocampal slices depolarization and the induction of LTP rapidly (within 1 min) increases cadherin dimerization, protease resistance and adhesive strength, suggesting the recruitment of pre-existing receptors to an activated state (Bozdagi et al. 2000; Tanaka et al. 2000; Huntley et al. 2002). Studies of the immunoglobulin class neural cell adhesion molecule (NCAM) have shown that LTP-inducing stimulation activates proteases that cleave the extracellular domain of the receptor (Hoffman et al. 1998a, 1998b; Endo et al. 1999). This may be linked with increased receptor turnover; depolarization of cultured neurons reportedly increases trafficking of polysialylated-NCAM to the membrane (Kiss et al. 1994), although studies of more mature neurons in cultured hypothalamic slices described constitutive polysialylated NCAM trafficking that was not modulated by glutamate receptor or calcium channel activities (Pierre et al. 2001).
Time courses of LTP stabilization and increases in surface integrins are comparable
The idea that AMPA receptor-induced increases in integrin surface expression are involved in LTP stabilization is reinforced by the good alignment between the temporal parameters of these effects (Chun et al. 2001; Gall and Lynch 2004). After induction by afferent stimulation, LTP can be reversed by various manipulations (e.g. transient anoxia, adenosine, low-frequency stimulation) for about 30 min, with the degree of LTP reversal declining with greater poststimulus latencies during this interval (Arai et al. 1990; Staubli and Chun 1996; Huang et al. 1999). Studies in our laboratories using ligand peptides, neutralizing antibodies and potent disintegrins antagonists to manipulate integrin function (Bahr et al. 1997; Staubli et al. 1998; Chun et al. 2001; Kramár et al. 2002) have shown that integrins are important for LTP stabilization for about 20–30 min after induction, with the magnitude of effects on potentiation declining over this interval. Applying integrin antagonists before induction does not increase the level of disruption and applying them 30 min or more after induction does not disturb potentiation. However, using lower doses of integrin ligands, LeBaron et al. (2003) described integrin involvement in LTP extending only a few minutes after stabilization. Our results lead to the suggestion that integrins become involved in the potentiation phenomenon after the initial induction phase, and are engaged, or initiate processes that are engaged, in LTP stabilization through a period of about 30 min. In GluR1/COS7 cells, AMPA-induced increases in integrin surface expression became evident within 3–5 min; with pulse treatment increases were maximal at 5–8 min and declined slowly thereafter. Thus, increases in surface expression are maximal at latencies of predicted maximal integrin involvement in LTP stabilization and decline slowly over a time period in which there is clear but diminishing requirement for integrins in the consolidation of synaptic plasticity.
In our studies, continuous AMPA treatment led to protracted increases in integrin surface expression, whereas pulse treatment led to a more transient response particularly for the α5 subunit. The fact that increases in insertion and steady-state levels followed each other quite closely suggests that AMPA treatment did not stimulate comparable increases in integrin endocytosis which, had they been present, could have maintained surface integrin immunoreactivities closer to control levels despite new insertion. It is noteworthy that the two subunits showed somewhat different time courses of response, most particularly with pulse treatment. This is not unexpected given that β1 integrin is predicted to be included in several different integrin dimers (β1 pairs with α1–8 and v) that might have different responses to AMPA treatment. In contrast, the α5 subunit pairs with β1 alone (Hynes 1992, 1999). Integrin heterodimers form in the ER before export to the plasma membrane (Cheresh and Spiro 1987); thus trafficking of α5 to the membrane provides an index for movements of the α5β1 dimer specifically.
AMPA pretreatment increased integrin adhesive and signaling functions
Ligands for α5β1 include fibronectin, fibrinogen and LI (Ruppert et al. 1995; Plow et al. 2000; Milner and Campbell 2002). Tests of AMPA treatment effects on α5β1 integrin function focused on responses to fibronectin- or anti-α5-coated substrates. AMPA stimulation of GluR1/COS7 cells increased their adhesion to both substrates; this result is consistent with there being greater numbers of functional (i.e. activated) α5β1 integrins at the cell surface of AMPA pretreated compared with control cells. AMPA pretreatment effects were greater for adhesion to anti-α5 than to fibronectin. This difference may be due to the involvement of integrins other than α5β1 in adhesion to fibronectin. Although α5β1 is known as the ‘fibronectin receptor’, this matrix protein is a ligand for several other integrins including α3β1, α4β1, α8β1 and αvβ1 (Plow et al. 2000; Milner and Campbell 2002). It is possible that one or more of these other integrin dimers contributed to fibronectin adhesion of GluR1/ COS7 cells but were unaffected by AMPA treatment. Alternatively, differences in the magnitude of AMPA effects on adhesion to the different substrates may be related to the strength of attachment to anti-α5 compared with fibronectin and hence to the efficiency of the assay with use of different attachment substrates.
Measures of AMPA treatment effects on levels of phosphoproteins known to be influenced by integrin signaling reinforced the conclusion that AMPA treatment increased numbers of functional integrins at the cell surface. Although integrins lack kinase activity, upon ligand binding or clustering these receptors stimulate increases in tyrosine and serine kinase signaling (Zhu and Assoian 1995; Vuori 1998; Giancotti and Ruoslahti 1999; Yamada and Even-Ram 2002). These cascades typically include increases in phosphorylation of FAK and paxillin that bind integrin proteins directly (Schaller et al. 1995), Src family kinases associated with activated FAK and the FAK homologue Pyk2 (Parsons et al. 2000) and, further downstream, p44/42MAPK (Zhu and Assoian 1995). Therefore, to test whether AMPA-induced increases in α5β1 integrin surface expression subserve increased integrin signaling function, the effects of fibronectin attachment on levels of p-FAK, p-Src, p-paxillin and p-p44/42MAPK were evaluated using phospho-specific antisera to the activation sites for FAK and Src, to a tyrosine within the SH2 binding domain for paxillin, and to the Thr-Glu-Tyr motif for p44/42MAPK (Hanks and Hunter 1995). The results demonstrated that adhesion to fibronectin elicited significantly larger increases in relative levels of each of these phosphoproteins in AMPA-pretreated compared with naive cells; similar AMPA pretreatment effects were not evident in cells remaining in suspension. These results support the conclusion that integrins, driven into the membrane by AMPA treatment, are active and mediate increased kinase signaling upon contact with ligand (i.e. the fibronectin substrate). The generally low phosphoprotein levels in suspended cells, either pretreated with AMPA or not, indicates that these phosphorylation events are adhesion dependent and not induced by AMPA treatment alone.
AMPA treatment effects on surface integrins depend on exocytotic and PKC activities
The most likely explanation for the present results is that AMPA stimulation resulted in translocation of integrins from the ER and/or endocytotic vesicles into the plasma membrane. Although relatively little is known of the precise mechanisms regulating integrin membrane insertion, results from other systems suggest that conventional BFA-sensitive exocytotic processes and PKC are likely to be involved. For example, studies of blastocyst implantation have shown that ligand-induced up-regulation of integrin-mediated adhesion to fibronectin is blocked by both BEA and antagonists of PKC but is unaffected by the protein synthesis inhibitor cycloheximide (Schultz and Armant 1995; Wang et al. 2002); in this system increased adhesion is also associated with a redistribution of integrins to the membrane and a BFA-sensitive clearance of vesicles from below the plasma membrane surface (Schultz and Armant 1995). These combined results suggest that rapid increases in integrin adhesion can be mediated by PKC- and BFA-sensitive mobilization of receptors from ER-derived transport vesicle fusion. Studies of migrating cells indicate that integrins are also trafficked to and from the surface membrane via an endocytotic recycling pathway (Maxfield 1995; Pierini et al. 2000); these studies have implicated PKC in both membrane insertion and internalization (Ng et al. 1999; Ivaska et al. 2002) although, in some cases, this endocytotic integrin cycling was found to be insensitive to BFA (Ng et al. 1999). To test whether similar mechanisms mediated increased integrin surface expression and signaling in the present studies, the effects of the PKC antagonist CCl and BFA were examined. CCl blocked both AMPA-induced increases in new α5 and β1 integrin membrane insertion, as determined by the surface protein bio tiny lation assay, and enhancement of fibronectin adhesion-induced signaling by AMPA pretreatment. The first of these observations provides the most direct evidence for PKC involvement in α5 and β1 integrin trafficking whereas the latter probably reflects increased membrane insertion as well as potential PKC-mediated increases in integrin ligand affinity (Kolanus and Seed 1997; Hughes and Pfaff 1998).
BFA also blocked AMPA effects on new membrane insertion of the α5 and β1 integrin proteins, indicating that conventional mechanisms of exocytosis of products from the Golgi-ER network, which are blocked by BFA (Klausner et al. 1992; Shah and Klausner 1993), are required for the AMPA-induced mobilization of α5β1 integrin to the cell surface.
The observation that AMPA-induced increases in integrin surface expression depend on extracellular calcium in GluR1/COS7 cells accords with evidence from studies of integrin ligand effects on adhesion, i.e. that increases in intracellular calcium provide the effective trigger for PKC-and BEA-dependent increases in integrin trafficking to the membrane (Maxfield 1995; Pierini et al. 2000). Indeed, in trophoblasts treatment with the calcium ionophore ionomycin mimics the effects of fibronectin contact in stimulating integrin trafficking and increased adhesive function (Wang et al. 2002). Similar calcium-dependent exocytotic processes may have mediated increased integrin surface expression and function in both GluR1/COS7 and neuronal preparations in the present studies. The GluR1/COS7 cells used here expressed GluR1 homomeric receptors (Verdoorn et al. 1991) that flux calcium upon activation (Hollmann et al. 1991; Jonas and Burnashev 1995). Therefore, AMPA treatment of the GluR1/COS7 cells would be expected to have increased calcium influx and intracellular calcium content, and the latter might have been sufficient to trigger integrin trafficking to the membrane. Mechanisms subserving the observed AMPA-induced increases in integrin surface expression in neurons and in cultured hippocampal slices were undoubtedly somewhat different because the great majority of native, neuronal AMPA receptors contain a GluR2 subunit that prevents calcium influx (Hollmann et al. 1991; Jonas and Burnashev 1995). However, in neurons AMPA receptor-mediated depolarization would be expected to alleviate the voltage block from NMDA receptors and VSCCs that, in turn, could support calcium influx and mobilization of integrins to the synaptic membrane. Tests of this possibility verified that in both neuronal preparations AMPA-induced increases in α5 and β1 surface expression are indeed dependent on NMDA receptor and/or L-type VSCC activities. The fact that NMDA receptor activation alone did not increase membrane insertion or surface expression suggests that AMPA effects on trafficking depend (1) primarily on VSCC-mediated calcium influx or (2) on a coordination of events including calcium influx and other, yet to be identified, consequences of AMPA receptor activation. Future studies will test these possibilities.
Regardless of precise mechanism, the effects of AMPA stimulation on integrin trafficking described here suggest that intense, activity-driven AMPA receptor stimulation will similarly mobilize integrins to the synaptic membrane. Considered more broadly, these results suggest that with high-frequency or repeated-burst afferent stimulation, as used to elicit LTP, AMPA receptor activation will lead to local increases in intracellular calcium that may, in turn, drive synapse-specific insertion of integrins into the synaptic membrane and recruitment of new integrin signaling into processes mediating the consolidation of LTP.
Acknowledgments
The authors thank Dr Peter Seeburg (University of Heidelberg, Germany) and Dr Kathy Partin (Colorado State University) for providing the GluR1/pRK5 plasmid used in these studies; and Annie Liu, Yilu Xie and Thien Ngo for technical assistance. This research was supported by National Institute of Neurological Disorders and stroke grant NS377099 to CMG and National Institute of Mental Health MH61007 to GL.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-proprionate
- APV
DL-2-Amino-5-phosphonovaleric acid
- BFA
brefeldin A
- CCl
chelerythrine chloride
- DMEM
Dulbecco’s modified Eagle’s medium
- DNQX
6,7 dinitroquinoxaline-2,3-dione
- ER
endoplasmic reticulum
- FAK
focal adhesion kinase
- FBS
fetal bovine serum
- LTP
long-term potentiation
- MAPK
mitogen-activated protein kinase p42/44
- NCAM
neural cell adhesion molecule
- NHS
N-hydroxy-succinimide
- Nif
nifedipine
- NMDA
N-methyl-D-aspartate
- PBS
phosphate-buffered saline
- PKC
protein kinase C
- Pyk2
proline-rich tyrosine kinase 2
- SNK
Student-Newman-Keuls
- VSCC
voltage-sensitive calcium channel
References
- Ali DW, Salter MW. NMDA receptor regulation by Src kinase signalling in excitatory synaptic transmission and plasticity. Curr Opin Neurobiol. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- Alpin A, Howe A, Alahari S, Juliano R. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev. 1998;50:197–263. [PubMed] [Google Scholar]
- Arai A, Larson J, Lynch G. Anoxia reveals a vulnerable period in the development of long-term potentiation. Brain Res. 1990;511:353–357. doi: 10.1016/0006-8993(90)90184-d. [DOI] [PubMed] [Google Scholar]
- Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 2000;12:123–133. doi: 10.1016/s0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Staubli U, Xiao P, Chun D, Ji Z-X, Esteban ET, Lynch G. Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: Pharmacological studies and the characterization of a candidate matrix receptor. J Neurosci. 1997;17:1320–1329. doi: 10.1523/JNEUROSCI.17-04-01320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Benson DL, Schnapp LM, Shapiro L, Huntley GW. Making memories stick: cell-adhesion molecules in synaptic plasticity. Trends Cell Biol. 2000;10:473–182. doi: 10.1016/s0962-8924(00)01838-9. [DOI] [PubMed] [Google Scholar]
- Bernard JA, Kramár EA, Torp R, Lin C-Y, Pineda EA, Lynch G, Gall CM. Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J Neurochem. 2005;93:834–849. doi: 10.1111/j.1471-4159.2005.03062.x. [DOI] [PubMed] [Google Scholar]
- Bongiorno-Borbone L, Onofri F, Giovedi S, Ferrari R, Girault JA, Benfenati F. The translocation of focal adhesion kinase in brain synaptosomes is regulated by phosphorylation and actin assembly. J Neurochem. 2002;81:1212–1222. doi: 10.1046/j.1471-4159.2002.00906.x. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Bukalo O, Fentrop N, Lee A, Aalmen B, Law JW, Wotjak CT, Schweizer M, Dityatev A, Schachner M. Conditional ablation of the neural cell adhesion molecule reduces precision of spatial learning, long-term potentiation, and depression in the CA1 subfield of mouse hippocampus. J Neurosci. 2004;24:1565–1577. doi: 10.1523/JNEUROSCI.3298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C-S, Weeber EJ, Kurup S, Sweatt JD, Davis RL. Integrin requirement for hippocampal synaptic plasticity and spatial memory. J Neurosci. 2003;23:7107–7116. doi: 10.1523/JNEUROSCI.23-18-07107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheresh DA, Spiro RC. Biosynthetic and functional properties of an Arg-Gly-Asp-directed receptor involved in human melanoma cell attachment to vitronectin, fibrinogen, and von Willebrand factor. J Biol Chem. 1987;262:17 703–17 711. [PubMed] [Google Scholar]
- Chun D, Gall CM, Bi X, Lynch G. Evidence that integrins contribute to multiple stages in the consolidation of long term potentiation. Neuroscience. 2001;105:815–829. doi: 10.1016/s0306-4522(01)00173-7. [DOI] [PubMed] [Google Scholar]
- Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matri-cryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–1498. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedhar S, Hannigan GE. Integrin cytoplasmic interactions and bidirectional transmembrane signalling. Curr Opin Cell Biol. 1996;8:657–669. doi: 10.1016/s0955-0674(96)80107-4. [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B. Activity and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol. 2000;150:1423–1433. doi: 10.1083/jcb.150.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo A, Nagai N, Urano T, Takada Y, Hashimoto K, Takada A. Proteolysis of neuronal cell adhesion molecule by the tissue plasminogen activator–plasmin system after kainate injection in the mouse hippocampus. Neurosci Res. 1999;33:1–8. doi: 10.1016/s0168-0102(98)00105-9. [DOI] [PubMed] [Google Scholar]
- Gall CM, Lynch G. Consolidation: a view from the synapse. In: Stanton PK, Bramhan C, Scharfman H, editors. Synaptic Plasticity and Transsynaptic Signaling. Kluwer/Academic Press; New York: 2005. In press. [Google Scholar]
- Gall CM, Lynch G. Integrins, synaptic plasticity, and epileptogenesis. In: Scharfman H, Binder D, editors. Recent Advances in Epilepsy Research: Molecular Mechanisms of Epileptogenesis. Vol. 548. Kluver Academic; Georgetown, TX: 2004. pp. 12–33. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix – cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Grooms S, Jones L. RODS tetrapeptide and hippocampal in vitro kindling in rats: evidence for integrin-mediated physiological stability. Neurosci Lett. 1997;231:139–142. doi: 10.1016/s0304-3940(97)00524-7. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Hoffman KB, Larson J, Bahr BA, Lynch G. Activation of NMDA receptors stimulates extracellular proteolysis of cell adhesion molecules in hippocampus. Brain Res. 1998a;811:152–155. doi: 10.1016/s0006-8993(98)00907-x. [DOI] [PubMed] [Google Scholar]
- Hoffman KB, Martinez J, Lynch G. Proteolysis of cell adhesion molecules by serine proteases: a role in long term potentiation? Brain Res. 1998b;811:29–33. doi: 10.1016/s0006-8993(98)00906-8. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca 2+ permeability of KA-AMPA-gated receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Howe A, Aplin AE, Alahari SK, Juliano RL. Integrin signaling and cell growth control. Curr Opin Cell Biol. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- Huang CC, Liang YC, Hsu KS. A role for extracellular adenosine in time-dependent reversal of long-term potentiation by low-frequency stimulation at hippocampal CA1 synapses. J Neurosci. 1999;19:9728–9738. doi: 10.1523/JNEUROSCI.19-22-09728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-Q, Lu WY, Ali DW, et al. CAKp/PYK2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001;29:485–196. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- Hughes PE, Pfaff M. Integrin affinity modulation. Trends Cell Biol. 1998;8:359–364. doi: 10.1016/s0962-8924(98)01339-7. [DOI] [PubMed] [Google Scholar]
- Huntley GW, Gil O, Bozdagi O. The cadherin family of cell adhesion molecules: multiple roles in synaptic plasticity. Neuroscientist. 2002;8:221–233. doi: 10.1177/1073858402008003008. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Cell adhesion: old and new questions. Trends Cell Biol. 1999;9:M33–M37. [PubMed] [Google Scholar]
- Ivaska J, Whelan RDH, Watson R, Parker PJ. PKC-epsilon controls the traffic of β1 intgrins in motile cells. EMBO J. 2002;21:3608–3619. doi: 10.1093/emboj/cdf371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Burnashev N. Molecular mechanisms controlling calcium entry through AMPA-type glutamate receptor channels. Neuron. 1995;15:987–990. doi: 10.1016/0896-6273(95)90087-x. [DOI] [PubMed] [Google Scholar]
- Kiss JZ, Wang C, Olive S, Rougon G, Lang J, Baetens D, Harrey D, Pralong WF. Activity-dependent mobilization of the adhesion molecule polysialic NCAM to the cell surface of neurons and endocrine cells. EMBO J. 1994;13:5284–5292. doi: 10.1002/j.1460-2075.1994.tb06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Sippincott-Schwartz J. Brefeldin a: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolanus W, Seed B. Integrins and inside-out signal transduction: converging signals from PKC and PIP3. Curr Opin Cell Biol. 1997;9:725–731. doi: 10.1016/s0955-0674(97)80127-5. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Bernard JA, Gall CM, Lynch G. Alpha3 integrin receptors contribute to the consolidation of long-term potentiation. Neuroscience. 2002;110:29–39. doi: 10.1016/s0306-4522(01)00540-1. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Bernard JA, Gall CM, Lynch G. Integrins modulate fast excitatory transmission at hippocampal synapses. J Biol Chem. 2003;278:10 722–10 730. doi: 10.1074/jbc.M210225200. [DOI] [PubMed] [Google Scholar]
- Lafrenie R, Yamada K. Integrin-dependent signal transduction. J Cell Biochem. 1996;61:543–553. doi: 10.1002/(sici)1097-4644(19960616)61:4<543::aid-jcb7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Lynch G, Vanderklish P, Arai A, Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J Neurosci. 2000;20:8–21. doi: 10.1523/JNEUROSCI.20-01-00008.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBaron RG, Hernandez RV, Orfila JE, MartineZ JLJ. An integrin binding peptide reduces rat CA1 hippocampal long-term potentiation during the first few minutes following theta burst stimulation. Neurosci Lett. 2003;339:199–202. doi: 10.1016/s0304-3940(03)00037-5. [DOI] [PubMed] [Google Scholar]
- Lee H-K, Takamiya K, Han JS, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Li A, Massingill JL, O’Dowd DK, Smith MA. Agrin gene expression in mouse somatosensory cortical neurons during development in vivo and in cell culture. Neuroscience. 1997;79:191–201. doi: 10.1016/s0306-4522(96)00654-9. [DOI] [PubMed] [Google Scholar]
- Liang F, Huganir RL. Coupling of agonist-induced AMPA receptor internalization with receptor recycling. J Neurochem. 2001;77:1626–1631. doi: 10.1046/j.1471-4159.2001.00377.x. [DOI] [PubMed] [Google Scholar]
- Lin B, Arai AC, Lynch G, Gall CM. Integrins regulate NMDA receptor-mediated synaptic currents. J Neurophysiol. 2003;89:2874–2878. doi: 10.1152/jn.00783.2002. [DOI] [PubMed] [Google Scholar]
- Liu S, Calderwood DA, Ginsberg MH. Integrin cytoplasmic domain-binding proteins. J Cell Sci. 2000;113:3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- Loeser RF, Forsyth CB, Samarel AM, Im HJ. Fibronectin-fragment activation of proline-rich tyrosine kinase mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem. 2003;278:24 577–24 585. doi: 10.1074/jbc.M304530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation – a decade of progress. Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Maxfield FR. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Zengping J. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39:163–176. doi: 10.1016/s0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- Milner R, Campbell IL. The integrin family of cell adhesion molecules has multiple functions within the CNS. J Neurosci Res. 2002;69:286–291. doi: 10.1002/jnr.10321. [DOI] [PubMed] [Google Scholar]
- Mould AP. Getting integrins into shape: recent insights into how integrin activity is regulated by conformational changes. J Cell Sci. 1996;109:2613–2618. doi: 10.1242/jcs.109.11.2613. [DOI] [PubMed] [Google Scholar]
- Mould AP, Akiyama SK, Humphries MJ. Regulation of integrin α5β1 fibronectin interactions by divalent cations. J Bol Chem. 1995;270:26 270–26 277. doi: 10.1074/jbc.270.44.26270. [DOI] [PubMed] [Google Scholar]
- Murase S, Schuman EM. The role of cell adhesion molecules in synaptic plasticity and memory. Curr Opin Cell Biol. 1999;11:549–553. doi: 10.1016/s0955-0674(99)00019-8. [DOI] [PubMed] [Google Scholar]
- Ng T, Shima D, Squire A, Bastiaens PIH, Gschmeissner S, Humphries MJ, Parker PJ. PKCalpha regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 1999;18:3909–3923. doi: 10.1093/emboj/18.14.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura SL, Boylen KP, Einheber S, Milner TA, Ramos DM, Pytela R. Synaptic and glial localization of the integrin cxvBS in mouse and rat brain. Brain Res. 1998;791:271–282. doi: 10.1016/s0006-8993(98)00118-8. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase; a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–5613. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Piech V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4:917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- Pierini LM, Lawson MA, Eddy RJ, Hendey B, Maxfield FR. Oriented endocytic recycling of alpha5beta1 in motile neutrophils. Blood. 2000;95:2471–2480. [PubMed] [Google Scholar]
- Pierre K, Bonhomme R, Dupouy B, Poulain DA, Theodosis DT. The polysialylated neural cell adhesion molecule reaches cell surfaces by hypothalamic neurons and astrocytes via the constitutive pathway. Neuroscience. 2001;103:133–142. doi: 10.1016/s0306-4522(00)00536-4. [DOI] [PubMed] [Google Scholar]
- Pinkstaff JK, Detterich J, Lynch G, Gall C. Integrin subunit gene expression is regionally differentiated in adult brain. J Neurosci. 1999;19:1541–1556. doi: 10.1523/JNEUROSCI.19-05-01541.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275:21 785–21 788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Esteban JA, Malinow R. Multiple mechanisms for the potentiation of AMPA receptor-mediated transmission by alpha-Ca 2+/calmodulin-dependent protein kinase II. J Neurosci. 2002;22:4406–4411. doi: 10.1523/JNEUROSCI.22-11-04406.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previtali SC, Feltri ML, Archelos JJ, Quattrini A, Wrabet ZL, Hartung HP. Role of integrins in the peripheral nervous system. Prog Neurobiol. 2001;64:35–19. doi: 10.1016/s0301-0082(00)00045-9. [DOI] [PubMed] [Google Scholar]
- Rodriquez MA, Pesold C, Liu WS, Kriho V, Guidotti A, Pappas GD, Costa E. Colocalization of integrin receptors and reelin in dendritic spine postsynaptic densities of adult nonhuman primate cortex. Proc Natl Acad Sci USA. 2000;97:3550–3555. doi: 10.1073/pnas.050589797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J, Grotewiel MS, Davis RL, Broadie K. Integrin-mediated regulation of synaptic morphology, transmission and plasticity. J Neurosci. 2000;20:6868–6878. doi: 10.1523/JNEUROSCI.20-18-06868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppert M, Aigner S, Hubbe M, Yagita H, Altevogt P. The LI adhesion molecule is a cellular ligand for VLA-5. J Cell Biol. 1995;131:1881–1891. doi: 10.1083/jcb.131.6.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MA, Basson MD. Collagen IV-dependent ERK activation in human Caco-2 intestinal epithelial cells requires focal adhesion kinase. J Biol Chem. 2000;275:38 040–38 047. doi: 10.1074/jbc.M003871200. [DOI] [PubMed] [Google Scholar]
- Sanna P, Berton F, Cammalleri M, Tallent MK, Siggins GR, Blom FE, Francesconi W. A role for Src kinase in spontaneous epileptiform activity in the CA3 region of the hippocampus. Proc Natl Acad Sci USA. 2000;97:8653–8657. doi: 10.1073/pnas.140219097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Otey CA, Hildebrand JD, Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking β integrin cytoplasmic domains. J Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Parsons JT. Complex formation with focal adhesion kinase: a mechanism to regulate activity and subcellular localization of Src kinases. Mol Biol Cell. 1999;10:3489–3505. doi: 10.1091/mbc.10.10.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D, Hunter T. Integrin signalling and tyrosine phosphorylation: just the FAKs? Trends Cell Biol. 1998;8:151–157. doi: 10.1016/s0962-8924(97)01172-0. [DOI] [PubMed] [Google Scholar]
- Schmitt WB, Sprengel R, Mack V, Draft RW, Seeburg PH, Deacon RMJ, Rawlins JNP, Bannerman DM. Restoration of spatial working memory by genetic rescue of GluR-A-deficient mice. Nat Neurosci. 2005;8:270–272. doi: 10.1038/nn1412. [DOI] [PubMed] [Google Scholar]
- Schultz JF, Armant DR. β1- and β3-class integrins mediate fibronectin binding activity at the surface of developing mouse peri-implantation blastocysts. J Biol Chem. 1995;270:1522–11531. doi: 10.1074/jbc.270.19.11522. [DOI] [PubMed] [Google Scholar]
- Schuster T, Krug M, Stalder M, Hackel N, Gerardy-Schahn R, Schachner M. Immunoelectron microscopic localization of the neural recognition molecules LI, NCAM, and its isoform NCAM180, the NCAM-associated polysialic acid, β1 integrin and the extracellular matrix molecule tenascin-R in synapses of the adult rat hippocampus. J Neurobiol. 2001;49:142–158. doi: 10.1002/neu.1071. [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Ginsberg MH. Network and crosstalk: integrin signalling spreads. Nat Cell Biol. 2002;4:E65–E68. doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- Shah N, Klausner RD. Brefeldin A reversibly inhibits secretion in Saccharomyces cerevisiae. J Biol Chem. 1993;268:5345–5348. [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Staubli U, Chun D. Factors regulating the reversibility of long term potentiation. J Neurosci. 1996;16:853–860. doi: 10.1523/JNEUROSCI.16-02-00853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Vanderklish PW, Lynch G. An inhibitor of integrin receptors blocks LTP. Behav Neural Biol. 1990;53:1–5. doi: 10.1016/0163-1047(90)90712-f. [DOI] [PubMed] [Google Scholar]
- Staubli U, Chun D, Lynch G. Time-dependent reversal of long-term potentiation by an integrin antagonist. J Neurosci. 1998;18:3460–3469. doi: 10.1523/JNEUROSCI.18-09-03460.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR. Molecular modification of N-cadherin in response to synaptic activity. Neuron. 2000;25:93–107. doi: 10.1016/s0896-6273(00)80874-0. [DOI] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long term potentiation. Neuron. 1998;20:1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- Tomita S, Nicoll RA, Bredt DS. PDZ protein interactions regulating glutamate receptor function and plasticity. J Cell Biol. 2001;153:F19–F23. doi: 10.1083/jcb.153.5.f19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderklish P, Neve R, Bahr BA, Arai A, Hennegriff M, Larson J, Lynch G. Translational suppression of a glutamate receptor subunit impairs long-term potentiation. Synapse. 1992;12:333–337. doi: 10.1002/syn.890120410. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg P, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- Vuori K. Integrin signaling: tyrosine phosphorylation events in focal adhesions. J Membr Biol. 1998;165:191–199. doi: 10.1007/s002329900433. [DOI] [PubMed] [Google Scholar]
- Wang J, Mayernick L, Armant DR. Integrin signaling regulates blastocyst adhesion to fibronectin at implantation: intracellular calcium transients and vesicle trafficking in primary trophoblast cells. Dev Biol. 2002;245:270–279. doi: 10.1006/dbio.2002.0644. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Even-Ram S. Integrin regulation of growth factor receptors. Nat Cell Biol. 2002;4:E75–E76. doi: 10.1038/ncb0402-e75. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Miyamoto S. Integrin transmembrane signaling and cytoskeletal control. Curr Opin Cell Biol. 1995;7:681–689. doi: 10.1016/0955-0674(95)80110-3. [DOI] [PubMed] [Google Scholar]
- Yang YC, Ma YL, Chen SK, Wang CW, Lee EHY. Focal adhesion kinase is required, but not sufficient, for the induction of long-term potentiation in dentate gyrus neurons in vivo. J Neurosci. 2003;23:4072–1080. doi: 10.1523/JNEUROSCI.23-10-04072.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Assoian RK. Integrin-dependent activation of MAP kinase: a link to shape-dependent cell proliferation. Mol Biol Cell. 1995;6:273–282. doi: 10.1091/mbc.6.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]