

Abstract
Rho GTPases are key transducers of integrin/extracellular matrix and growth factor signaling. Although integrin-mediated adhesion and trophic support suppress neuronal apoptosis, the role of Rho GTPases in neuronal survival is unclear. Here, we have identified Rac as a critical pro-survival GTPase in cerebellar granule neurons (CGNs) and elucidated a death pathway triggered by its inactivation. GTP-loading of Rac1 was maintained in CGNs by integrin-mediated (RGDdependent) cell attachment and trophic support. Clostridium difficile toxin B (ToxB), a specific Rho family inhibitor, induced a selective caspase-mediated degradation of Rac1 without affecting RhoA or Cdc42 protein levels. Both ToxB and dominant-negative N17Rac1 elicited CGN apoptosis, characterized by cytochrome c release and activation of caspase-9 and -3, whereas dominant-negative N19RhoA or N17Cdc42 did not cause significant cell death. ToxB stimulated mitochondrial translocation and conformational activation of Bax, c-Jun activation, and induction of the BH3-only protein Bim. Similarly, c-Jun activation and Bim induction were observed with N17Rac1. A c-jun N-terminal protein kinase (JNK)/p38 inhibitor, SB203580, and a JNK-specific inhibitor, SP600125, significantly decreased ToxB-induced Bim expression and blunted each subsequent step of the apoptotic cascade. These results indicate that Rac acts downstream of integrins and growth factors to promote neuronal survival by repressing c-Jun/Bim-mediated mitochondrial apoptosis.
Keywords: Bax, BH3-only, c-jun N-terminal protein kinase, neuronal survival, Rho GTPase
Apoptosis is a form of programmed cell death that plays an essential role in the development of the central nervous system by mediating the removal of redundant neuronal populations (Lossi and Merighi 2003). However, aberrant neuronal apoptosis contributes to the onset and progression of several devastating neurodegenerative disorders, including Parkinson’s disease and amyotrophic lateral sclerosis (Inoue et al. 2003; Tatton et al. 2003). Consequently, identification of molecular pathways that regulate neuronal apoptosis is key to developing novel therapeutics that slow or halt these neurodegenerative diseases.
One well-studied in vitro system used to investigate neuronal apoptosis is cultured cerebellar neurons from early (day 7) post-natal rats. Cerebellar granule neurons (CGNs)require both serum-derived growth factors (e.g. insulin-like growth factor-I) and depolarization-mediated Ca2+ influx for their survival in vitro (D’Mello et al. 1993). CGNs deprived of serum growth factors and depolarizing extracellular potassium (K+ex) undergo a rapid cell death that involves induction of the BH3-only protein, Bim, and activation of an intrinsic (mitochondrial) apoptotic cascade (Gerhardt et al. 2001; Linseman et al. 2002b).
In addition to growth factors and activity-dependent Ca2+ influx, neurons also depend on survival signals originating at sites of cell attachment to the extracellular matrix (ECM). Cell-matrix adhesion is mediated via binding of heterodimeric integrin receptors to their ECM ligands. The activated (ligand-bound) integrins stimulate multiple survival signals, including AKT- and ERK-dependent pathways, by interacting with partner proteins via their cytoplasmic domains (Stupack and Cheresh 2002).
Rho family GTPases are monomeric G-proteins that act as key transducers of integrin/ECM signaling (Parise et al. 2000; Schwartz and Shattil 2000; Arthur et al. 2002) and growth factor signaling (Huang and Reichardt 2003; Burridge and Wennerberg 2004). These G-proteins are involved in a diverse array of biological functions, including cytoskeletal dynamics, membrane trafficking, cell cycle progression, gene transcription, adhesion, migration, and cell survival (Etienne-Manneville and Hall 2002; Burridge and Wennerberg 2004). The three most studied family members include RhoA, Rac1, and Cdc42.
Despite the essential roles that Rho family GTPases play in mediating survival signaling by integrins and growth factors, the potential of these G-proteins to promote neuronal survival has largely been unexplored. We have previously utilized Clostridial toxins, which act as highly specific inhibitors of Ras and Rho family GTPases (Busch and Aktories 2000), to demonstrate a critical function for these G-proteins in maintaining CGN survival (Linseman et al. 2001a). In particular, Clostridium difficile toxin B (ToxB) and Clostridium sordellii lethal toxin (LTox), are large Clostridial toxins that specifically mono-glucosylate a critical threonine residue conserved in the switch 1 region of Ras and Rho family members. The substrate specificities for these two toxins include Rho, Rac, and Cdc42 for ToxB and Rac, Cdc42 (to a lesser extent than Rac), Ras, and Rap for LTox (Just et al. 1995; Genth et al. 1996; Popoff et al. 1996; Hofmann et al. 1998; Djouder et al. 2000). Incubation of CGNs with either of these toxins induces substantial apoptosis, whereas the Rho-specific inhibitor, Clostridium botulinum C3 fusion toxin (Barth et al. 1998), does not promote CGN death. From these data, we previously concluded that Clostridial toxins with overlapping specificities for inhibiting Rac and Cdc42 GTPases are capable of inducing apoptosis of CGNs, suggesting a key role for one or both of these specific G-proteins in promoting CGN survival (Linseman et al. 2001a).
In the present study, we have utilized adenoviral dominant-negative mutants of RhoA, Rac1, and Cdc42 to explicitly identify Rac as the essential pro-survival GTPase in CGNs. In addition, we have elucidated a death pathway triggered in CGNs by antagonism of Rac and determined that it is dependent on a c-Jun-driven induction of Bim and the subsequent activation of a mitochondrial apoptotic cascade. Our findings suggest a critical role for Rac as an endogenous suppressor of c-Jun/Bim-dependent mitochondrial apoptosis in healthy neurons.
Experimental procedures
Materials
Reagents for measurement of GTP-bound Rac, including agaroseconjugated PAK-protein binding domain, were obtained from Upstate Biotechnology (Charlottesville, VA, USA) and Cytoskeleton Inc. (Denver, CO, USA). Monoclonal antibodies to Rac1 (clone 102; 1 : 1000), Rho (clone 55; 1 : 250), Cdc42 (clone 44; 1: 250), and a polyclonal to cyt C oxidase subunit IV (COXIV) were from BD Biosciences Clontech (Palo Alto, CA, USA). C. difficile toxin B was isolated and prepared as previously described (von Eichel-Streiber et al. 1987). Hoecsht dye number 33258 and 4′,6-diamidino-2-phenylindole (DAPI) were from Sigma (St Louis, MO, USA). SB203580, SP600125, the pan-caspase inhibitors, Boc-Asp(OMe)-FMK and zVAD-FMK, the caspase-9-selective inhibitor, zLEHD-FMK, the proteasome inhibitor, MG132, and RGD and RAD peptides were from Calbiochem (San Diego, CA, USA). Rabbit polyclonal antibodies to Bim (FL-198; 1 : 250) and cytochrome c (H-104; 1 : 200) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal antibodies to the active fragment of caspase-9 were purchased from Santa Cruz (H-83; 1 : 200 for western blotting) and from Cell Signaling Technology (1 : 500; Beverly, MA) for immunocytochemistry. Polyclonal antibodies to active caspase-3 were from Santa Cruz (H-227; 1 : 250 for western blotting) and Promega (rabbit pAb; 1 : 500; Madison, WI, USA) for immunocytochemistry. A plasmid encoding an amino-terminal GFP fusion protein of human Baxα was kindly provided by Dr R. J. Youle (NINDS, NIH, Bethesda, MD, USA). Monoclonal antibody to the active conformation of Bax (clone 6A7; 1 : 1000) was purchased from Alexis Biochemicals (San Diego, CA, USA). Rabbit polyclonal antibody to c-Jun (1 : 1000) was from Cell Signaling Technology. Horseradish peroxidase-linked secondary antibodies and reagents for enhanced chemiluminescence detection were obtained from Amersham Biosciences (Piscataway, NJ, USA). Cy3-conjugated secondary antibodies for immunofluo-rescence were from Jackson Immunoresearch Laboratories (West Grove, PA, USA).
CGN culture
Rat CGNs were isolated from 7-day-old Sprague-Dawley rat pups (15-19 g) as described previously (Li et al. 2001). Briefly, neurons were plated on 35-mm diameter plastic dishes coated with poly-l-lysine at a density of 2.0 × 106 cells/mL in basal modified Eagle’s medium containing 10% fetal bovine serum, 25 mm KCl, 2mm l-glutamine, and penicillin (100 units/mL)/streptomycin (100 lg/mL) (Life Technologies, Grand Island, NY, USA). Cytosinearabinoside (10 μm) was added to the culture medium 24 h after plating to limit the growth of non-neuronal cells. These cultures are highly homogeneous, consisting of approximately 95-98% cerebellar granule neurons (CGNs) with the remainder being primarily made up of Purkinje neurons (Linseman et al. 2002a). In general, experiments were performed after 6-7 days in culture.
Cell lysis and immunoblotting
Following incubation, the culture medium was aspirated, cells were washed once with 2 mL of ice-cold PBS (pH 7.4), placed on ice and scraped into lysis buffer (200 μL/35-mm well) containing 20 mm HEPES (pH 7.4), 1% Triton X-100, 50 mm NaCl, 1 mm EGTA, 5mm β-glycerophosphate, 30 mm sodium pyrophosphate, 100 lm sodium orthovanadate, 10 μg/mL leupeptin, and 10 μg/mL aprotinin. In the case of subcellular fractionation to isolate a mitochondrial pellet, CGNs were lysed and fractionated according to the manufacturer’s directions using a commercially available mitochondria/cytosol fractionation kit (Alexis Biochemicals). Cell debris was removed by centrifugation at 600 g for 3 min and the protein concentration of the supernatant was determined using a commercially available protein assay kit (Pierce, Rockford, IL, USA). Aliquots (∼80 μg) of supernatant protein were diluted to a final concentration of 1 × sodium dodecyl sulfate - polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, boiled for 5 min, and electrophoresed through 12.5 or 15% polyacrylamide gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Amersham Biosciences) and processed for immunoblot analysis.
Non-specific binding sites were blocked in phosphate-buffered saline (PBS; pH 7.4) containing 0.1% Tween 20 (PBS-T) and 1% bovine serum albumin (BSA) for 1 h at room temperature (22°C). Primary antibodies were diluted in blocking solution and incubated with the membranes for 1 h. Excess primary antibody was removed by washing the membranes three times in PBS-T. The blots were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody diluted in PBS-T for 1 h and were subsequently washed three times in PBS-T. Immunoreactive proteins were detected by enhanced chemiluminescence. Autoluminograms shown are generally representative of at least three independent experiments.
BD PowerBlot™ analysis
CGN lysates were pooled from three separate cell isolations following incubation for 24 h with either vehicle (2 μg/mL BSA, final concentration) or ToxB (40 ng/mL, final concentration). Lysates were prepared according to the manufacturer’s instructions followed by western blotting with a PowerBlot™ array of over 1000 monoclonal antibodies (BD Biosciences). Raw data in the form of image files of the actual blots and densitometric measurements of the immunoreactive proteins were obtained from the manufacturer. The blots and densitometric results shown for Rac1, RhoA, and Cdc42 are representative of a 2 × 2 comparison of duplicate control and ToxB lysates.
Adenoviral preparation and infection
Adenoviral dominant-negative Rho family GTPases were constructed as previously described (Allen et al. 2002). Briefly, Rac1, RhoA, and Cdc42 were cloned from a mouse cDNA library. Dominant-negative (T17N or T19N) mutations were generated by PCR-based site-directed mutagenesis. Adenoviral GFP, GFP/N17Rac1, GFP/N17Cdc42, and GFP/N19RhoA were generated using the AdEasy system (Qbiogene Inc., Carlsbad, CA, USA). The produced adenoviruses express both GFP and the respective dominant-negative Rho family mutant. CGN cultures were infected on day 4 in vitro with adenoviruses at a final titer of 50 infectious particles per cell. Using these conditions, the adenoviruses infected approximately 15-20% of the CGNs in the cultures. At 72 h post-infection, CGNs were fixed in paraformaldehyde and nuclei were stained with Hoechst dye for quantitation of apoptosis. Alternatively, cell lysates of infected cells were prepared for immunoblot analysis. Because of the mechanisms involved in infecting primary neurons, CGNs require at least 48 h for infection to be effective (based on visualization of GFP fluorescence). Therefore, infecting them for 72 h is essentially equivalent to exposing the cells for approximately 24 h to the dominant negative mutant.
Rac GTP-binding assays
CGNs were either deprived of serum and depolarizing K+ex for up to 4 h, detached from the culture plate by scraping for various times from 15 min to 4 h, or incubated for 8 h with one of two small peptides, H-Gly-Arg-Ala-Asp-Ser-Pro-OH (RAD; negative control, 10 mm) or H-Gly-Arg-Gly-Asp-Ser-Pro-OH (RGD; 10 mm). Cells were lysed by homogenization in a high (10 mm)Mg2+ buffer and the lysates were briefly pelleted to remove cell debris. A small aliquot of the resulting supernatant was immediately removed for detection of total Rac1 protein. To detect GTP-bound Rac1, agarose beads bound to a GST-PAK-protein binding domain were added to the remaining supernatant according to the manufacturer’s instructions (Upstate Biotechnology). The supernatants were then mixed by inversion for 1 h at 4°C and the pelleted beads were washed three times with Mg2+ lysis buffer. Aliquots of the original cell lysates and the PAK-precipitates were subjected to SDS-PAGE through 15% polyacrylamide gels, transferred to PVDF membranes, and immunoblotted for Rac1.
Quantitation of apoptosis
Treated neurons were fixed in 4% paraformaldehyde and nuclei were stained with Hoechst dye. Cells were scored as apoptotic if they contained chromatin that was condensed and/or fragmented. In general, approximately 500 cells from at least two fields of a 35-mm well were scored and each treatment was performed in duplicate in a minimum of three independent experiments. For the cultures infected with adenoviral dominant-negative Rho family GTPases, approximately 100 infected cells (GFP-positive neurons) were quantitated per coverslip, and each adenovirus was infected into triplicate coverslips in a total of four separate experiments.
Immunocytochemistry
CGNs were cultured on polyethyleneimine-coated glass cover slips at a density of approximately 5 × 105 cells per coverslip. Following adenoviral infection and/or incubation as described in Results, cells were fixed in 4% paraformaldehyde and were then permeabilized and blocked in PBS (pH 7.4) containing 0.2% Triton X-100 and 5% BSA. Cells were then incubated for approximately 16 h at 4°C with primary antibody diluted in PBS containing 0.2% Triton X-100 and 2% BSA. The primary antibody was aspirated and the cells were washed five times with PBS. The cells were then incubated with Cy3-conjugated secondary antibodies and DAPI for 1 h at room temperature. CGNs were then washed five more times with PBS and coverslips were adhered to glass slides in mounting medium (0.1% p-phenylenediamine in 75% glycerol in PBS). Fluorescent images were captured using a 63 × oil immersion objective on a Zeiss Axioplan 2 microscope equipped with a Cooke Sensicam deep-cooled CCD camera and a Slidebook software analysis program for digital deconvolution (Intelligent Imaging Innovations Inc., Denver, CO, USA).
Gene-gun transfection
CGNs were transiently transfected using the Helios Gene-Gun system (Bio-Rad, Hercules, CA, USA). Briefly, 60 μg of plasmid DNA was precipitated onto 30 mg of 0.6 μm-diameter gold beads in a CaCl2/spermidine mixture. The gold/DNA precipitates were washed three times in 100% ethanol, and re-suspended in 3 mL of ethanol containing 0.05 mg/mL polyvinylpyrrolidone. After thoroughly re-suspending the gold/DNA precipitate, it was drawn into approximately 74 cm of Tefzel tubing and the beads were allowed to settle to the bottom of the tubing. After 5 min, the ethanol was slowly drawn off while the beads adhered to the tubing. The tubing was dried under nitrogen for an additional 5 min and then cut into approximately 1.3-cm pieces. CGNs to be transfected were seeded at a density of 8 × 105 cells/well on polyethyleneimine-coated glass coverslips in 24-well plates. After 5 days in culture, the medium was removed from the wells and the 1.3-cm lengths of tubing containing the DNA-bound beads were loaded into the Gene-Gun and shot with a burst of ∼100 p.s.i. helium through a 40-μm nylon cell strainer placed over the well. The medium was immediately replaced and cells were grown an additional 48 h before exposure to ToxB and subsequent image analysis of the localization of either GFP alone or the GFP-Bax fusion protein.
Results
ToxB induces a specific caspase-mediated degradation of Rac1 in CGNs undergoing apoptosis
To define the relative pro-survival contributions of Rac and Cdc42 GTPases in CGNs, we examined the protein expression of the various Rho family members during ToxB-induced apoptosis. As previously reported (Linseman et al. 2001a), CGNs incubated for 24 h with ToxB showed a marked increase in the percentage of cells with condensed and/or fragmented nuclei, consistent with an apoptotic mechanism of cell death (Fig. 1a). After 24 h of incubation with either vehicle or ToxB, CGN lysates were prepared and electrophoresed through polyacrylamide gels for analysis using the BD PowerBlot™ screen (BD Biosciences). Representative immunoblots are shown in Fig. 1(b) that include areas of the membranes which were probed with monoclonal antibodies against either Rac1, RhoA, or Cdc42 (indicated by the arrows). Incubation with ToxB induced a striking decrease in the amount of Rac1 detected in CGN lysates (Fig. 1b, upper panels, see arrows). In contrast, neither RhoA nor Cdc42 showed any notable reduction in immunoreactivity in lysates from CGNs exposed to ToxB (Fig. 1b, middle and lower panels, see arrows). Semiquantitative densitometric analysis of the PowerBlot™ data is shown in Fig. 1(c). Incubation with ToxB for 24 h induced an approximately 15-fold reduction in the immunoreactivity of Rac1, but had no demonstrable effect on the content of either RhoA or Cdc42. Furthermore, the marked loss of Rac1 GTPase observed in CGNs exposed to ToxB was not affected by an inhibitor of the 26S proteasome, MG132, but was partially blocked by co-incubation with the pan-caspase inhibitors, Boc-Asp(OMe)-FMK or zVAD-FMK (Fig. 1d). In agreement with a recent report that identified Rac1 as a caspase substrate in lymphoma cells (Zhang et al. 2003), the current data suggest that the effects of the glucosylation of Rac by ToxB are potentially augmented by the caspase mediated degradation of this G-protein in CGNs. Thus, although ToxB glucosylates and inhibits all three Rho family members, Rho, Rac and Cdc42 (Just et al. 1995), Rac1 GTPase is the sole member degraded by caspases following its glucosylation in CGNs, likely resulting in a prolonged functional inhibition of this particular G-protein.
Fig. 1.
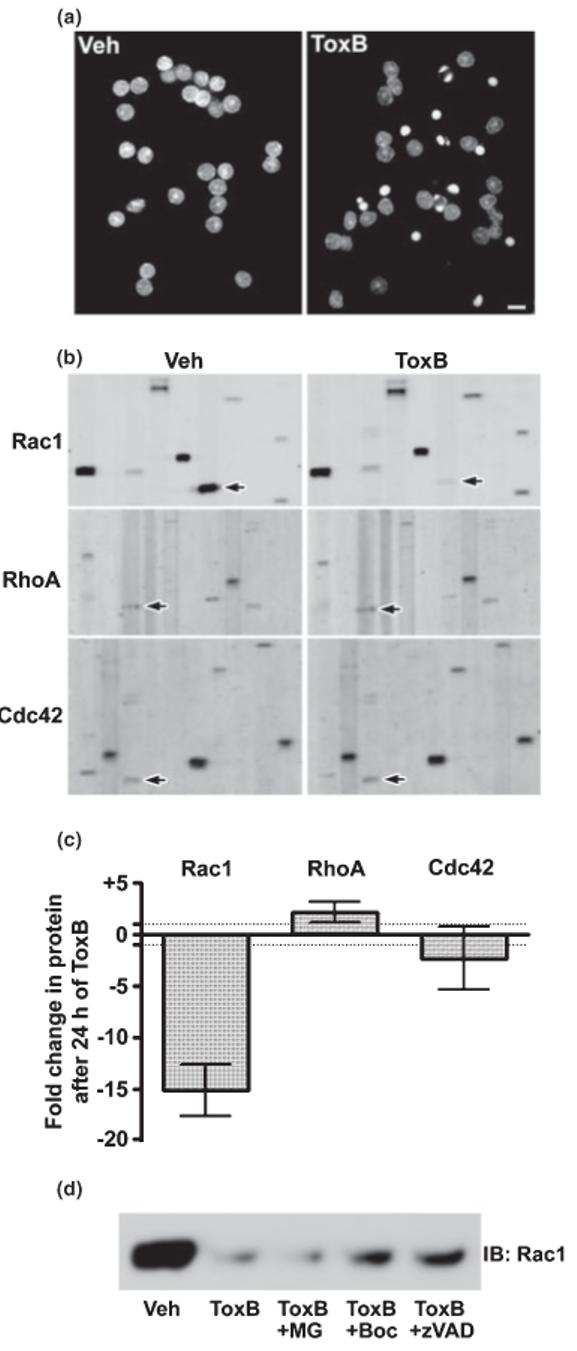
ToxB induces apoptosis and elicits a selective caspase-mediated degradation of Rac1 GTPase in CGNs. (a) CGNs were incubated for 24 h in complete medium (containing serum and 25 mm KCl) in the presence of either vehicle (Veh; 2 μg/mL BSA in PBS) or ToxB (40 ng/mL). Following incubation, cells were fixed in paraformaldehyde and nuclei were stained with Hoechst dye. CGNs exposed to ToxB showed marked nuclear condensation and fragmentation characteristic of apoptosis. Scale bar, 10 μm. (b) Representative immunoblots of lysates obtained from CGNs incubated as described in (a). The blots shown were obtained from the BD PowerBlot™ (see Experimental procedures for details) using specific monoclonal antibodies against Rac1 (upper blots), RhoA (middle blots), or Cdc42 (lower blots). The immunoreactive Rho family GTPases are indicated by the arrows. Other protein bands apparent on the membranes represent adjacent lanes of the PowerBlot™ that were probed for distinct proteins and are shown to give an indication of the overall equality of protein loading. (c) The fold change in protein content induced by 24 h of ToxB treatment for each of the Rho family members blotted for in (b) is shown. Data represent the means ± range of duplicate PowerBlot™ comparisons for each GTPase. The dotted lines indicate no change (-1 to +1 fold). (d) CGNs were exposed for 8 h to ToxB in the absence or presence of either a pan-caspase inhibitor (Boc-Asp(OMe)-FMK; 200 μm) or zVAD-FMK; 200 μm) or a proteasome inhibitor (MG132; 10 μm). Following incubation, cell lysates were immunoblotted for Rac1.
Adenoviral expression of dominant-negative Rac1 induces CGN apoptosis
Given the specific caspase-mediated degradation of Rac1 observed in ToxB-treated CGNs, and the fact that Rac1 is a common substrate for two Clostridial toxins (ToxB and LTox) that each induce apoptosis of CGNs (Linseman et al. 2001a), we postulated that Rac is the major pro-survival Rho family GTPase in these neurons. To test this hypothesis, we infected CGN cultures with adenoviruses expressing either GFP alone or in combination with dominant-negative mutants of either RhoA (N19), Rac1 (N17), or Cdc42 (N17). At 72 h post-infection, CGNs were fixed and stained with Hoechst dye to identify nuclear morphology. Infection with adenoviral GFP had essentially no effect on the survival of CGNs with less than 10% of the GFP-expressing cells undergoing apoptosis after 72 h (Fig. 2a, upper panels, and Fig. 2b). In contrast, adenoviral-mediated expression of N17Rac1 with GFP induced a significant (approximately 5.8-fold) increase in CGN apoptosis when compared with GFP alone, which was characterized by marked nuclear condensation and fragmentation (Fig. 2a, lower panels, and Fig. 2b). Although both N17Cdc42 and N19RhoA tended to enhance the apoptosis of CGNs, the effects of these dominant-negative GTPases did not reach statistical significance at 72 h post-infection (Fig. 2b). The differential effects of these dominant-negative GTPase mutants on CGN survival strongly corroborate our previous data obtained using Rho GTPase-specific inhibitory toxins (Linseman et al. 2001a), and establish Rac as a major pro-survival GTPase in CGNs.
Fig. 2.

Adenoviral expression of N17Rac1 induces apoptosis of CGNs: dominant-negative mutants of RhoA or Cdc42 do not cause significant cell death. (a) On day 4 in vitro, CGN cultures were infected with adenovirus expressing either GFP alone or in combination with dominant-negative N17Rac1. At 72 h post-infection, CGNs were fixed in paraformaldehyde and nuclei were stained with Hoechst dye. Images shown represent composites of two fields each for GFP or GFP/N17Rac1. Infected cells are indicated by the arrows. CGNs infected with adenovirus expressing GFP alone showed large intact nuclei characteristic of healthy cells (upper panels). In contrast, CGNs infected with adenovirus expressing N17Rac1 displayed marked chromatin condensation and fragmentation indicative of apoptotic cell death (lower panels). Note that non-infected cells in the N17Rac1 condition remained healthy. Scale bar, 10 μm. (b) Quantitation of CGN apoptosis in GFP-expressing cells following 72 h of infection with adenoviruses expressing either GFP alone or in combination with dominant-negative mutants of Rac1, Cdc42, or RhoA at a final titer of 50 infectious particles per cell. Using these conditions, each of the adenoviruses infected approximately 15-20% of the CGNs in the cultures. The results shown represent the means ± SEM for four experiments, each conducted on triplicate coverslips in which approximately 100 GFP-positive CGNs were counted per condition per coverslip. *Significantly different from adenoviral GFP alone (p < 0.01).
Rac activity is maintained by trophic support and integrin-mediated cell attachment in CGNs
Rho family GTPases are activated downstream of both integrin and growth factor receptor stimulation (Marcoux and Vuori 2003). Moreover, both integrin- and growth factor-dependent signals contribute to neuronal survival (Gary and Mattson 2001; Linseman et al. 2002b; Gary et al. 2003). To add further support that Rac1 activity is important for CGN survival, we assessed the relative contributions of integrins or trophic support in the activation of Rac1 by examining the effects of cell detachment or trophic factor deprivation on Rac1 GTP-binding using a PAK-based pull-down assay (Benard et al. 1999). Consistent with a growth factor receptor-dependent pathway for Rac1 activation in CGNs, removal of serum and depolarizing K+ex induced a significant loss of Rac GTP-loading in attached CGNs (Fig. 3a). Similarly, detachment from the culture dish led to a rapid loss of GTP-bound Rac1 (Fig. 3b), and this effect occurred more quickly than that observed upon trophic factor withdrawal. These data show that active (GTP-bound) Rac1 is maintained in CGNs by both trophic factor stimulation and cell attachment to the ECM.
Fig. 3.
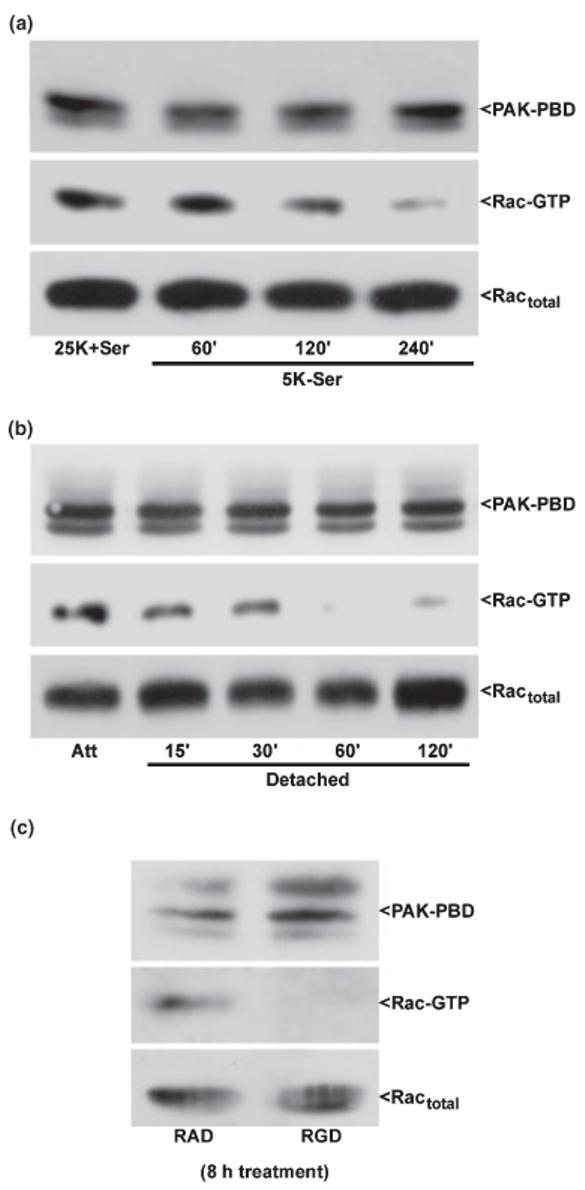
Rac1 GTP-loading is maintained in CGNs by trophic support and integrin-mediated cell attachment. CGNs were incubated under either attached conditions (a) in trophic factor-deprived medium containing 5 mm KCl and lacking serum (5K-Ser) for up to 4 h, detached conditions (b) for up to 2 h in medium containing 25 mm KCl and serum (25K + Ser), or incubated with RAD or RGD peptides (c) for 8 h. Following incubation, CGNs were immediately lysed on ice into a high Mg2+ buffer. Cell debris was removed by brief centrifugation and a small aliquot of the resulting lysate was separated for quantitation of total Rac in each sample. The remaining cell lysates were incubated for 1 h with an excess of the protein binding domain of the Rac effector, PAK (PAK-PBD), bound to agarose beads. The pellet was washed three times with lysis buffer and samples were electrophoresed through 15% polyacrylamide gels. Proteins were transferred to PVDF membranes which were immunoblotted with a monoclonal antibody against Rac1 and an HRP-conjugated secondary antibody. Immunoreactive Rac1 protein was detected using standard ECL techniques. GTP-loaded Rac was brought down in the PAK-PBD precipitation (middle blots). The PAK-PBD is detected non-specifically by the secondary antibody and is shown as a control for equal loading (upper blots). The total Rac in each lysate is also shown as a loading control as no significant loss is detected in Rac protein under these conditions (bottom blots). The blots shown are representative of results obtained in three separate experiments. Att, attached control.
Cell adhesion and subsequent signal transduction is often activated by integrins and can occur in conjunction with growth factor receptor stimulation (Gibson et al. 2005). Integrins mediate cell attachment via recognition of an Arg-Gly-Asp (RGD) motif on ECM ligands like fibronectin (Takagi 2004). To determine if the effects of cell attachment on Rac activity in CGNs are integrin-mediated, we utilized RGD-containing inhibitory peptides. The RGD peptides compete with RGD-containing ECM proteins for integrin binding, thereby reducing integrin signaling. It has previously been shown that RGD peptides inhibit integrin stimulation of Rho family GTPases (Yang et al. 1999). CGNs incubated for 8 h with RGD peptides showed a marked decrease in GTP-bound Rac1 relative to cells incubated with the negative control peptide, RAD (Fig. 3c). These results indicate that Rac1 activity is maintained in CGNs via integrin signaling downstream of cell attachment (and trophic stimulation).
ToxB and N17Rac1 each trigger cytochrome c release and activation of caspases-9 and -3 in CGNs
Previous work has shown that CGNs deprived of serum and depolarizing K+ex undergo apoptosis that involves activation of an intrinsic mitochondrial pathway (Gerhardt et al. 2001; Linseman et al. 2002b). To determine if CGN death induced by the inhibition of Rac GTPase proceeds by a similar mechanism, we analyzed the ability of ToxB or N17Rac1 to trigger mitochondrial cytochrome c (cyt C) release. Subcellular fractionation of CGN lysates showed that the vast majority of cyt C in vehicle-treated cells was localized to the mitochondrial pellet along with the integral mitochondrial membrane protein, COXIV (Fig. 4a, first and second lanes). Incubation with ToxB for 24 h resulted in a marked decrease in cyt C immunoreactivity in the mitochondrial pellet and a coincident appearance of several high molecular weight bands in the cytosolic fraction that were detected by the cyt C antibody (Fig. 4a, third and fourth lanes, see arrow-heads). We postulate that these high molecular weight forms of cyt C may represent ubiquitin conjugates of the released protein as has previously been observed for yeast cyt C (Pearce and Sherman 1997). Regardless of the precise nature of these high molecular weight forms of cyt C, it is clear from the substantial loss of immunoreactivity in the mitochondrial pellet that the majority of cyt C was indeed released from mitochondria in CGNs exposed to ToxB.
Fig. 4.
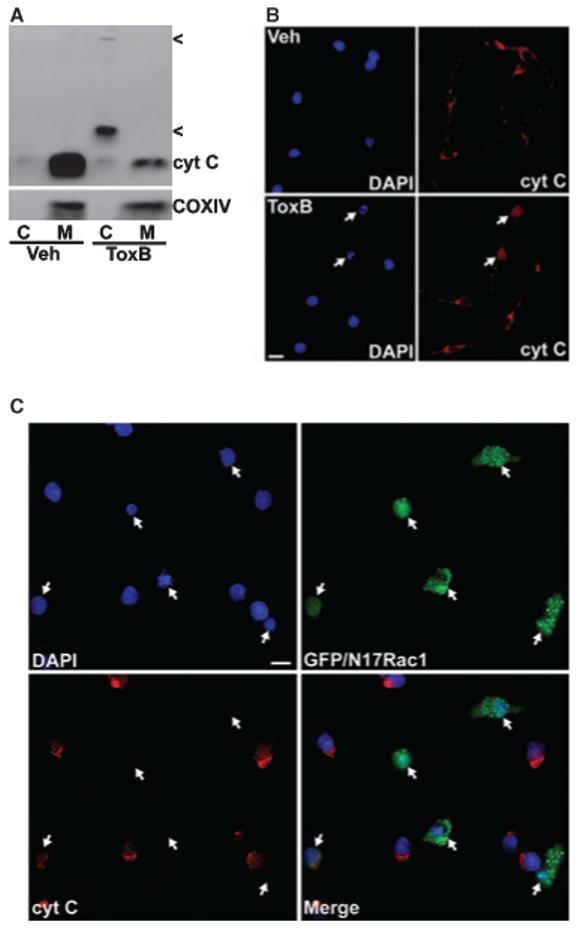
ToxB and N17Rac1 each trigger cytochrome c release from mitochondria in CGNs. (a) CGNs were incubated for 24 h in complete medium (containing serum and 25 mm KCl) in the presence of either vehicle (Veh; 2 lg/mL BSA in PBS) or ToxB (40 ng/mL). Following incubation, cell lysates were fractionated into cytosolic (C) and mitochondrial (M) subcellular fractions as described in Experimental procedures. Equivalent amounts of cytosolic (20 μg) and mitochondrial (40 μg) fractions from Veh- or ToxB-treated cells were subjected to SDS-PAGE and immunoblotted for cytochrome c (cyt C). The membranes were then stripped and re-probed for the integral mitochondrial membrane protein, cyt C oxidase subunit IV (COXIV), to verify the purity of the subcellular fractions. Note that the amount of cyt C detectable in mitochondrial fractions was significantly less in ToxB-treated CGNs, although the amount of immunoreactive COXIV was similar to Veh. The cyt C released into the cytosolic fraction in the ToxB-treated cells did not appear at the same molecular weight as that detected in the mitochondrial fractions, but instead it displayed several higher molecular weight forms (indicated by <) which may reflect ubiquitination of the cytoplasmic cyt C. The blots shown are illustrative of data from two independent experiments. (b) CGNs were exposed to Veh or ToxB as described in (a). After 24 h, cells were fixed with paraformaldehyde, blocked and permeabilized in 5% BSA and 0.2% Triton X-100, and incubated with a polyclonal antibody to cyt C. Localization of cyt C was detected using a Cy3-conjugated secondary antibody and nuclei were stained with DAPI. Veh-treated CGNs showed healthy nuclei and cyt C localization that was mostly perinuclear consistent with mitochondria (upper panels). In contrast, a significant fraction of the CGNs exposed to ToxB displayed fragmented nuclei and a diffuse distribution of cyt C over the entire cell body (lower panels, see arrows). The images shown are indicative of those from three separate experiments. Scale bar, 10 μm. (c) CGNs were incubated for 72 h after infection with adenovirus expressing GFP and N17Rac1 (at a final titer of 50 infectious particles per cell). Following infection, cells were stained for cyt C, as described in (b). The merged image shows that the majority of GFP- and N17Rac1-positive cells (indicated by the arrows) had condensed or fragmented nuclear morphology and lacked detectable cyt C staining, whereas surrounding uninfected cells were largely healthy and displayed significant perinuclear cyt C. The essentially complete loss of cyt C staining in the N17Rac1-expressing cells likely reflects the diffuse distribution of the protein over such a prolonged time post-infection. The images shown are composites of three different fields of CGNs infected with adenoviral GFP/N17Rac1. Scale bar, 10 μm.
In addition to subcellular fractionation, cyt C localization was also assessed by immunocytochemical methods. CGNs incubated with vehicle showed a cyt C distribution that was punctate and perinuclear in agreement with its localization to mitochondria (Fig. 4b, upper panels). In contrast, CGN cultures exposed to ToxB for 24 h displayed cells that had condensed and fragmented chromatin accompanied by a diffuse distribution of cyt C over the entire cell body, consistent with its release into the cytoplasm (Fig. 4b, lower panels, see arrows). In a similar manner, many CGNs which were infected with adenovirus expressing GFP and N17Rac1 showed a nearly complete loss of cyt C immunoreactivity that was coincident with condensed nuclei at 72 h postinfection, consistent with an extremely diffuse distribution of cyt C in the apoptotic cells (Fig. 4c, see arrows). Note that uninfected surrounding cells showed punctate cyt C staining localized in the perinuclear region and contained large intact nuclei. In addition, CGNs expressing GFP alone did not have condensed nuclei at 72 h, and the cyt C remained localized in the mitochondria (data not shown). These results indicate that CGN apoptosis induced by either ToxB or N17Rac1 proceeds via the release of cyt C from mitochondria.
Once cyt C is released from mitochondria, it interacts with apoptosis-activating factor-1 (Apaf-1) and pro-caspase-9 to form an oligomeric apoptosome structure that activates the intrinsic initiator caspase, caspase-9 (Zou et al. 1999). Therefore, we next examined CGNs exposed to ToxB or N17Rac1 for the appearance of active forms of the intrinsic initiator caspase-9 and the executioner caspase-3. Using antibodies that detect the cleaved (active) fragments of caspase-9 and -3, we found that CGNs incubated for 24 h with ToxB showed activation of each of these proteases (Figs 5a and b, respectively). Similarly, many CGNs expressing N17Rac1 (identified by GFP fluorescence) showed apoptotic nuclear morphology in conjunction with positive staining using antibodies that specifically detect the cleaved forms of caspase-9 and -3 (Figs 5c and d, respectively), but no active caspase staining was detected in CGNs expressing GFP alone (data not shown). Essentially every N17Rac1 GFP-positive cell that showed condensed and/or fragmented chromatin was immunoreactive for active caspase-9 or -3. Moreover, CGN apoptosis induced by a 24-h incubation with ToxB (40 ± 4% apoptosis with ToxB vs. 8 ± 2% apoptosis with vehicle, mean ± SEM, n = 3, p < 0.01 vs. vehicle) was significantly suppressed by co-incubation with either a pancaspase inhibitor [Boc-Asp(OMe)-FMK, 9 ± 1% apoptosis, p < 0.01 vs. ToxB alone] or a caspase-9 selective inhibitor (zLEHD-FMK, 13 ± 2% apoptosis, p < 0.01 vs. ToxB alone). Thus, CGN apoptosis triggered by the inhibition of Rac with either ToxB or N17Rac1 involves the cyt C-mediated activation of caspase-9 and -3.
Fig. 5.

ToxB and N17Rac1 each promote activation of caspases-9 and -3 in CGNs. (a) CGNs were incubated for 24 h in complete medium (containing serum and 25 mm KCl) in the presence of either vehicle (Veh; 2 μg/mL BSA in PBS) or ToxB (40 ng/mL). After incubation, cell lysates were resolved by SDS-PAGE on 15% polyacrylamide gels and proteins were transferred to PVDF membranes. The blots were then probed with an antibody that specifically detects the cleaved (active) form of caspase-9. (b) CGNs were treated exactly as described in (a) and lysates were western blotted with an antibody that recognizes both the pro- and cleaved (active) forms of caspase-3. The blots shown in (a) and (b) are representative of results obtained in three separate experiments. (c) CGNs were incubated for 72 h after infection with adenovirus expressing GFP and N17Rac1 (at a final titer of 50 infectious particles per cell). Following infection, cells were fixed in paraformaldehyde, blocked and permeabilized in 5% BSA and 0.2% Triton X-100, and immunostained with an antibody that specifically detects the active (cleaved) form of caspase-9 [Casp-9 (a)] and a Cy3-conjugated secondary antibody. Nuclei were stained with DAPI. The merged image shows that GFP- and N17Rac1-positive cells (indicated by the arrows) had condensed or fragmented nuclear morphology and displayed significant immunoreactivity for active caspase-9, whereas surrounding uninfected cells were largely healthy and devoid of active caspase-9. (d) CGNs were infected as described in (c) and were then immunostained for active (cleaved) caspase-3 [Casp-3 (a)]. As described above, most of the GFP- and N17Rac1-positive cells (indicated by the arrows) showed marked chromatin condensation and fragmentation and stained positively for active caspase-3. The images shown in (c) and (d) are composites of three to four different fields of CGNs infected with adenoviral GFP/N17Rac1. Ad-GFP control data not shown. Scale bars, 10 μm.
ToxB elicits Bax translocation to mitochondria and conformational activation of Bax in CGNs
Cyt C is released from mitochondria via an oligomeric channel containing the multidomain Bcl-2 family member, Bax (Antonsson et al. 2000). Bax is normally sequestered in the cytoplasm of healthy cells via binding to 14-3-3 scaffolding proteins and translocates to mitochondria in response to apoptotic stimuli (Nomura et al. 2003). In CGNs transfected via gene gun with GFP-Bax (Wolter et al. 1997), distribution of the fusion protein changed from a diffuse pattern in vehicle-treated cells (Fig. 6a, upper panels, see arrows and Fig. 6b, left panel) to a punctate mitochondrial localization following ToxB treatment (Fig. 6a, middle panels, see arrows and Fig. 6b, middle panel). The redistribution of GFP-Bax to mitochondria following ToxB exposure occurred in conjunction with substantial nuclear condensation and fragmentation. In contrast, ToxB did not cause a significant redistribution of the control protein, GFP, which maintained a relatively diffuse localization throughout the entire cell body despite the fact that significant apoptosis was induced (Fig. 6a, lower panels, see arrows and Fig. 6b, right panel).
Fig. 6.

ToxB elicits translocation of a GFP-Bax fusion protein to mitochondria and conformational activation of endogenous Bax in CGNs. (a) On day 5 in vitro, CGNs were transfected with either GFP alone or a GFP-Bax fusion protein using a helium-powered gene gun, as described in Experimental procedures. At 48 h post-transfection, cells were exposed for 24 h to either vehicle (Veh; 2 μg/mL BSA in PBS) or ToxB (40 ng/mL). CGNs were then fixed in paraformaldehyde and nuclei were stained with DAPI. The expressed GFP-Bax fusion protein had a diffuse distribution in the predominantly healthy CGNs incubated with Veh (upper panels). In contrast, cells exposed to ToxB demonstrated marked nuclear condensation and fragmentation, and GFP-Bax staining that was punctate and consistent with localization to mitochondria (middle panels). In CGNs transfected with GFP alone, ToxB still induced substantial apoptosis; however, GFP maintained a distribution that encompassed the entire cell unlike the more localized distribution of GFP-Bax under these conditions (lower panels). The images shown are representative of results from two experiments. Transfected cells in each panel are indicated by the arrows. Scale bar, 10 μm. (b) The areas demarcated by the boxes in (a) have been enlarged 300% to enhance the visualization of GFP-Bax or GFP. (c) Untransfected CGNs were incubated with either Veh or ToxB for 24 h and cells were fixed and immunostained with a monoclonal antibody (clone 6A7) that specifically detects the active conformation of Bax. Active endogenous Bax was visualized in a punctate mitochondrial distribution using a Cy3-conjugated anti-mouse secondary antibody (indicated by the arrows). Scale bar, 10 μm.
In addition to mitochondrial translocation, upon induction of apoptosis Bax also undergoes a conformational change that exposes an amino-terminal epitope detected by a specific monoclonal (6A7) antibody (Hsu and Youle 1998). CGN cultures incubated with vehicle showed little-to-no detectable immunoreactivity with the Bax 6A7 antibody (Fig. 6c, left panel). However, CGNs exposed to ToxB showed marked positive staining with the active conformation-specific Bax antibody in conjunction with chromatin condensation and fragmentation (Fig. 6c, right panel). These results show that ToxB-mediated inactivation of Rac GTPase stimulates Bax translocation to mitochondria while concurrently promoting a conformational change in Bax that is commonly associated with its cyt C-releasing activity.
ToxB and N17Rac1 each promote c-Jun activation and Bim induction in CGNs: inhibition of c-Jun and Bim activation by the pyridinyl imidazole SB203580
Conformational activation and mitochondrial translocation of Bax is stimulated by pro-apoptotic BH3-only proteins that neutralize the activities of anti-apoptotic Bcl-2 family members (Bouillet et al. 2002). Various isoforms of Bim, a BH3-only protein, play a significant role in several models of neuronal apoptosis (Putcha et al. 2001; Linseman et al. 2002b; Napankangas et al. 2003). In response to diverse apoptotic stimuli, Bim expression is up-regulated at the transcriptional level. One of the key signaling pathways that promotes Bim transcription involves the c-jun N-terminalprotein kinase (JNK)-mediated phosphorylation of c-Jun (Harris and Johnson 2001; Whitfield et al. 2001). As we previously have shown (Linseman et al. 2001a), incubation of CGNs with ToxB induced a rapid increase in the phosphorylation (detected as a decrease in electrophoretic mobility) and expression of c-Jun (Fig. 7a), effects that were largely prevented by the pyridinyl imidazole JNK/p38 MAPK inhibitor, SB203580 (Fig. 7b) (Coffey et al. 2002). Consistent with c-Jun stimulating Bim transcription, ToxB also induced a marked increase in the expression of Bims (the short isoform of Bim) which was similarly blocked by SB203580 (Fig. 7c). Finally, both an increased phosphorylation of c-Jun and induction of Bim were also observed in CGNs infected with N17Rac1 as compared with cells infected with the GFP control adenovirus (Fig. 7d). These data show that antagonism of Rac activity in CGNs promotes a robust c-Jun-dependent induction of the BH3-only protein, Bims.
Fig. 7.
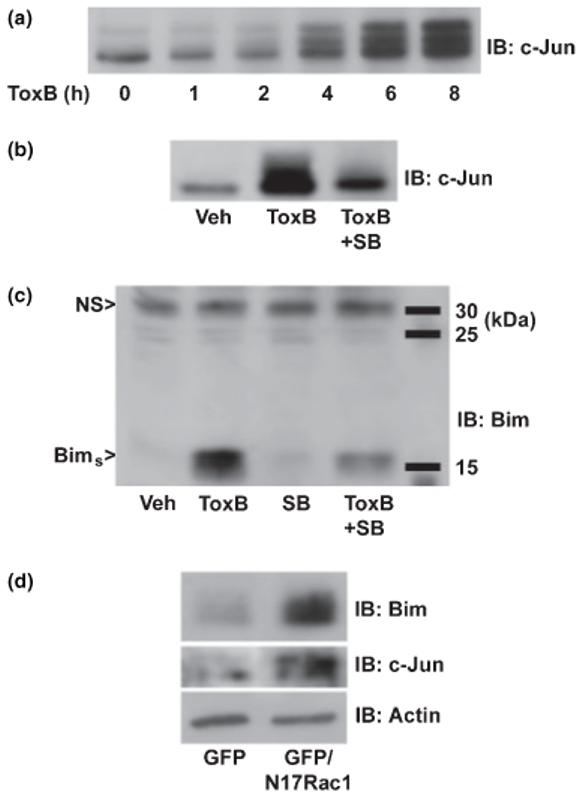
ToxB and N17Rac1 each stimulate c-Jun activation and induction of Bim in CGNs: inhibition of ToxB-induced Bim expression by the pyridinyl imidazole, SB203580. (a) CGNs were incubated for up to 8 h with ToxB (40 ng/mL) and cell lysates were immunoblotted (IB) with a polyclonal antibody to c-Jun. The decreased mobility of c-Jun observed with increasing duration of ToxB exposure is indicative of activating phosphorylation. (b) CGNs were incubated for 6 h with either BSA/PBS vehicle (Veh) or ToxB ± SB203580 (SB; 20 μm). Following treatment, CGN lysates were immunoblotted for c-Jun. (c) CGNs were incubated for 6 h as described in (b). Cell lysates were then immunoblotted with a polyclonal antibody that detects an approximately 15-kDa isoform of Bim, Bim short (Bims). (d) CGNs were infected with either adenoviral GFP alone or in combination with N17Rac1. At 96 h post-infection, cell lysates were obtained and immunoblotted for c-Jun, Bims, and actin (as a loading control). NS, non-specific protein bands that are detected by the antibodies and are shown as loading controls.
SB203580 significantly inhibits ToxB-induced Bax activation, cytochrome c release, and caspase activation in CGNs
To determine if the c-Jun-dependent induction of Bim is necessary for initiation of the mitochondrial apoptotic pathway elicited in CGNs by ToxB, we examined the effects of SB203580 on Bax conformational change, cyt C release, and caspase activation. The enhanced immunoreactivity for conformationally active Bax (i.e. positive 6A7 antibody staining) induced by ToxB was significantly suppressed by co-incubation with SB203580 (Fig. 8a). Concurrently, inclusion of SB203580 substantially decreased the number of condensed and/or fragmented nuclei observed in ToxB-treated CGN cultures. Moreover, co-incubation with SB203580 prevented the ToxB-induced loss of cyt C from the mitochondrial pellet and inhibited the cleavage of caspase-9 and -3 to the active fragments (Fig. 8b). Thus, in CGNs in which Rac has been inactivated, a c-Jun-driven induction of Bim lies upstream of a Bax-dependent mitochondrial death pathway.
Fig. 8.
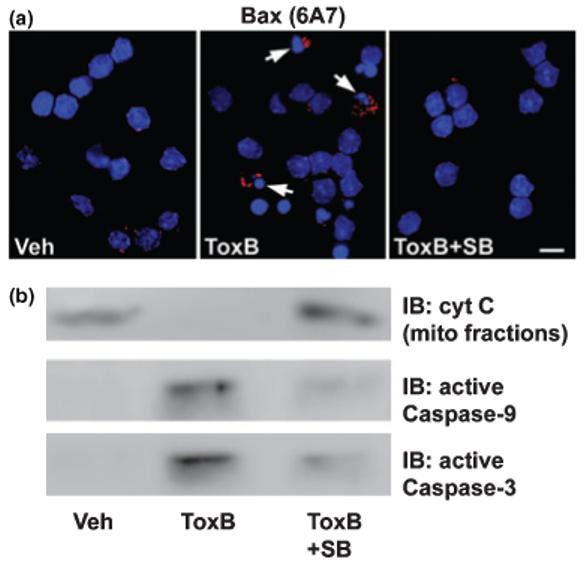
The pyridinyl imidazole, SB203580, inhibits mitochondrial apoptotic signaling induced by ToxB in CGNs. (a) CGNs were incubated for 24 h with either vehicle (Veh; 2 μg/mL BSA in PBS) or ToxB (40 ng/mL) ± SB203580 (SB; 20 μm). Following incubation, cells were immunostained with a monoclonal antibody to the active conformation of Bax (clone 6A7) and nuclei were stained with DAPI. The arrows indicate cells that stained positively for active Bax. Scale bar, 10 μm. (b) CGNs were incubated exactly as described in (a) and either mitochondrial fractions were immunoblotted (IB) for cyt C or whole cell lysates were probed for active caspase-9 or -3. The blots shown are each indicative of data obtained in three separate experiments.
The anthrapyrazol, SP600125, a JNK-specific inhibitor, significantly blocks ToxB-induced c-Jun phosphorylation, Bim induction, and CGN apoptosis
As SB203580 inhibits both JNK and p38 MAP kinase activities, we evaluated the effects of a more selective JNK inhibitor, SP600125 (Bennett et al. 2001), on c-Jun phosphorylation and the subsequent apoptotic cascade elicited by ToxB in CGNs. In agreement with the results obtained for SB203580, SP600125 significantly suppressed the ToxB-induced phosphorylation and increased expression of c-Jun (Fig. 9a). Furthermore, the induction of Bim was also significantly blocked by inclusion of SP600125 (Fig. 9b), providing further evidence that the Bim-initiated apoptotic cascade is mediated by c-Jun. Finally, quantitation of CGN apoptosis revealed that co-incubation of ToxB with SP600125 significantly decreased the number of neurons with condensed and/or fragmented nuclei (Fig. 9c). Thus, the JNK-specific inhibitor, SP600125, protects CGNs from apoptosis induced by the Rac-inhibitory ToxB.
Fig. 9.
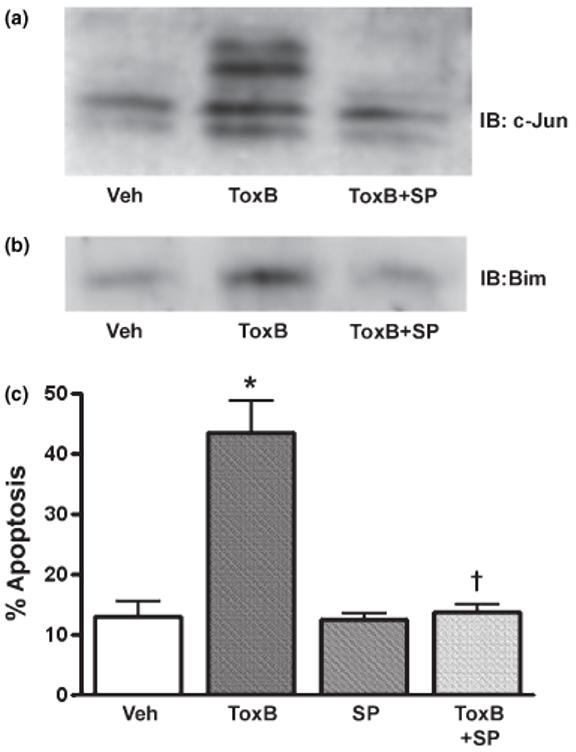
The JNK-specific inhibitor, SP600125, significantly blocks c-Jun phosphorylation, Bim induction, and apoptosis in ToxB-treated neurons. (a) CGNs were incubated for 8 h with ToxB (40 ng/mL) ± SP600125 (20 μm). Cell lysates were then immunoblotted with a polyclonal antibody for c-Jun. Activation of phosphorylation is seen by the decreased mobility of c-Jun in a time-dependent manner upon incubation with ToxB. (b) CGNs were incubated as above in (a) for 8 h. The cell lysates were then immunoblotted with a polyclonal antibody that detects Bims, approximately 15 kDa. (c) CGNs were incubated as described above for 24 h. The cells were then fixed with paraformal-dehyde and stained with Hoechst to visualize nuclei. The cells incubated with SP600125 showed a significant decrease in apoptosis. The results shown represent the means ± SEM of three experiments with approximately 400 cells counted per condition in each experiment. *Significantly different from Veh-treated cells (p< 0.01). †Significantly different from ToxB-treated cells (p< 0.01).
Discussion
Several previous studies have demonstrated a significant role for integrin/ECM and growth factor signaling in promoting neuronal survival. Integrin-mediated signaling was recently shown to protect cultured hippocampal neurons from glutamate- or staurosporine-induced toxicity (Gary and Mattson 2001; Gary et al. 2003). Moreover, disruption of ECM proteins, either by gene deletion or infusion of ECMneutralizing proteins or antibodies, exacerbated neuronal death in vivo in response to focal ischemia or excitotoxicity (Sakai et al. 2001; Chen et al. 2003). Growth factors also play a key role in neuronal survival. For example, we have shown that IGF-I promotes neuronal survival by blocking FKHRL1-dependent transcription of Bim, thereby suppressing apoptosis (Linseman et al. 2002b). Collectively, these findings illustrate a critical function for integrin/ECM and growth factor signaling in maintaining neuronal survival both in vitro and in vivo.
Yet despite the fact that Rho family GTPases are key downstream effectors of integrin activation and growth factor signaling, little is known about the role of these G-proteins in regulating neuronal survival. Several elegant studies by Bazenet and colleagues have shown that Rac/Cdc42 GTPases promote the apoptotic death of NGF-deprived sympathetic neurons by stimulating two distinct MAP kinase kinase kinases, apoptosis signal-regulating kinase 1 (ASK1) and mixed lineage kinase 3 (MLK3), upstream of JNK/c-Jun activation (Bazenet et al. 1998; Kanamoto et al. 2000; Mota et al. 2001). Furthermore, it was shown that p75-regulated activation of Rac induced JNK after withdrawal of NGF in rat oligodendrocytes, supporting a pro-apoptotic role of Rac (Harrington et al. 2002). Other studies, however, suggest that the pro-apoptotic activity of Rho family GTPases observed in sympathetic neurons and oligodendrocytes is not consistently seen in all types of neurons. For example, previous work by several groups correlated a loss of plasma membrane-associated Rho family GTPases with induction of apoptosis in primary cortical neurons and brain neuroblasts (Tanaka et al. 2000; Garcia-Roman et al. 2001; Meske et al. 2003). Each of these studies utilized inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase (statins) to block the isopr-enylation and membrane localization of Ras and Rho GTPases. More recently, Kobayashi et al. (2004) demonstrated that targeted dominant-negative mutants of either Rho or Rho kinase significantly enhance the apoptosis of spinal motor neurons in vivo during embryonic development. In addition, it was recently shown that Rac1 also plays a primary role in the survival of motor neurons; the juvenile-onset form of amyotrophic lateral sclerosis is caused by truncation and loss of function of the Rac1 GEF, alsinLF (Kanekura et al. 2005). The above studies indicate that, as has been previously described for non-neuronal cells (Murga et al. 2002), the relative pro-survival or pro-apoptotic effects of Rho family GTPases in neurons are cell-type specific.
Consistent with several prior studies which showed a pro-survival role for Rho family GTPases in specific neuronal populations, we have previously reported that CGNs undergo apoptosis following incubation with Clostridial toxins that inhibit Rac/Cdc42 GTPases (Linseman et al. 2001a). Here, we utilized adenoviral dominant-negative Rho family GTPases to identify Rac as the major pro-survival GTPase in CGNs, as significant apoptosis was induced by N17Rac1 but not dominant-negative mutants of RhoA or Cdc42. Although we cannot conclude irrefutably that Cdc42 does not play at least a minor role in CGN survival, our data provide strong evidence that Rac is the major pro-survival Rho family GTPase in these neurons. First, although dominant-negative mutants of Rac and Cdc42 can compete for the same guanine nucleotide exchange factors (GEFs), and therefore may have inhibitory effects that overlap on pathways downstream of one another, this lack of selectivity was not observed in CGNs where apoptosis was significantly elevated following infection with adenoviral N17Rac1 but not N17Cdc42 (see Fig. 2). The differential sensitivity of CGN survival to dominant-negative Rac versus dominant- negative Cdc42 was observed despite the fact that both adenoviruses appeared to infect the neurons equally based on GFP fluorescence.
Second, although Rac was a substrate for caspases in CGNs incubated with ToxB, Cdc42 was not degraded by caspases in this model (see Fig. 1). Therefore, while de novo Cdc42 synthesis could potentially overcome the effects of ToxB glucosylation over time (i.e. as the catalytic activity of ToxB diminishes), newly synthesized Rac would continually be degraded by caspases. The specificity of the caspase-mediated degradation for Rac versus Cdc42 further supports a major pro-survival role for Rac in CGNs.
Third, we previously showed that incubation with C. sordellii LTox mimicks the effects of ToxB on CGN apoptosis (Linseman et al. 2001a). While LTox definitely overlaps with ToxB in the glucosylation of Rac, it has been reported to inhibit Cdc42 in some cell-free assays (Genth et al. 1996; Hofmann et al. 1998) but not in others (Popoff et al. 1996). Moreover, LTox is generally regarded as a more potent inhibitor of Rac than Cdc42 within cells, further suggesting that it is the inhibition of Rac rather than Cdc42 that ultimately triggers CGN apoptosis.
Finally, we show that Rac1 activity (GTP-loading) is significantly decreased in CGNs deprived of serum and depolarizing K+ex (see Fig. 3a). In contrast, Cdc42 activity as measured by increased tyrosine phosphorylation of the specific Cdc42 effector, ACK-1 (Linseman et al. 2001b), is sustained under these conditions (data not shown). These results indicate that, while loss of Rac activity is coincident with induction of apoptosis, Cdc42 activity is regulated independently of apoptotic signals. Collectively, these findings indicate that Rac is the major Rho family GTPase involved in promoting CGN survival and any role for Cdc42 is likely secondary.
Given that ToxB or N17Rac1 each induce CGN apoptosis in the presence of serum and depolarizing K+ex, our data also suggest that loss of Rac function is not compensated for by trophic support. Moreover, we have previously shown that CGN apoptosis induced by Rac inactivation with either ToxB or LTox is not mimicked by direct disruption of F-actin using the actin ADP-ribosylation factor, C. botulinum C2 toxin (Linseman et al. 2001a), indicating that the pro-survival action of Rac is independent of its effects on the actin cytoskeleton. Our current research is focused on identifying the effectors downstream of Rac that promote CGN survival.
Antagonism of Rac in CGNs using either ToxB or N17Rac1 triggered the release of cyt C from mitochondria and activation of the intrinsic initiator, caspase-9, and the executioner, caspase-3. Moreover, CGNs incubated with ToxB showed a c-Jun-dependent induction of the BH3-only protein, Bim, and mitochondrial translocation and conformational activation of Bax. c-Jun activation and Bim induction were similarly observed in CGNs infected with adenoviral N17Rac1. The pyridinyl imidazole JNK/p38 inhibitor, SB203580, and the JNK-specific inhibitor, SP600125, suppressed c-Jun activation, Bim induction and mitochondrial apoptosis in CGNs exposed to ToxB. These results show that inactivation of Rac GTPase is sufficient to induce a c-Jun/Bim-dependent mitochondrial apoptotic pathway in CGNs that are maintained in full trophic support (i.e. serum and depolarizing K+ex). Thus, serum-derived growth factors and depolarization-mediated calcium influx do not provide a buffer against c-Jun activation and Bim induction in the face of Rac inactivation. These data suggest that Rho family GTPases, and specifically Rac in CGNs, regulate the transcriptional control of Bim at the level of c-Jun phosphorylation. Although many studies have indicated that Rac/Cdc42 GTPases act as upstream activators of JNK/c-Jun signaling (Bazenet et al. 1998), others have recently reported that these G-proteins can also suppress the JNK pathway via PAK-mediated phosphorylation of the MAP kinase kinase kinase, MEKK1 (Gallagher et al. 2002). Therefore, whether Rac GTPase positively or negatively regulates the JNK/c-Jun pathway in a given cell type likely depends on the specific MAP kinase kinase kinases present. The ability of PAK to antagonize c-Jun activation and, as a result, to inhibit Bim induction, complements its direct phosphorylation and inhibition of another BH3-only protein, Bad (Schurmann et al. 2000). Thus, Rac signaling through PAK may act as a key pro-survival pathway in some types of neurons by suppressing BH3-only protein-dependent mitochondrial apoptosis.
Finally, a recent study by Reginato et al. (2003) demonstrated that Bim is strongly induced in epithelial cells by cell detachment, and down-regulation of Bim using RNA interference prevents detachment-induced apoptosis or anoikis. These authors further showed that the up-regulation of Bim during cell detachment was because of loss of b1 integrin ligation to the ECM and down-regulation of EGF receptor signaling to the ERK pathway. In this context, several groups have recently shown that Bim is targeted for degradation following ERK-mediated phosphorylation (Ley et al. 2003; Marani et al. 2004). Given that Rac1 has previously been shown to transduce signals from the ECM-ligated β1 integrin to the ERK MAP kinase pathway (Hirsch et al. 2002), it will be interesting to determine if inactivation of Rac in CGNs leads to suppression of the ERK pathway and if this effect contributes to the consequential increase in Bim expression that ultimately triggers neuronal apoptosis.
In conclusion, we have shown that inactivation of Rac GTPase, using either C. difficile toxin B or dominant negative N17Rac1, provokes apoptosis of primary cultured CGNs. The apoptosis induced by antagonism of Rac function occurs via a Bim-dependent mitochondrial death pathway that is significantly inhibited by blockade of c-Jun activation. Our findings that Rac is a key pro-survival molecule for CGNs complement those of Kobayashi et al. (2004) and Kanekura et al. (2005) who recently showed a critical role for Rho and Rac, respectively, in motor neuron survival. Elucidation of the pro-survival signals generated by Rho family GTPases in neurons is key to understanding how integrin-mediated adhesion and growth factors suppress neuronal apoptosis.
Acknowledgements
This work was supported by a Department of Veterans Affairs Merit Review Entry Program (MREP) Award (to DAL) and a Department of Defense Grant USAMRMC no. 03281009 (to DAL and KAH). The authors thank Tracey Laessig and Brent Butts for technical assistance.
Abbreviations used
- BSA
bovine serum albumin
- CGNs
cerebellar granule neurons
- COXIV
cyt C oxidase subunit IV
- cyt C
cytochrome c
- ECM
extracellular matrix
- JNK
c-jun N-terminal protein kinase
- K+ex
extracellular potassium
- LTox
Clostridium sordellii lethal toxin
- PBS
phosphate-buffered saline
- SDS-PAGE
sodium dodecyl sulfate - polyacrylamide gel electrophoresis
- ToxB
Clostridium difficile toxin B
References
- Allen MP, Linseman DA, Udo H, Xu M, Schaack JB, Varnum B, Kandel ER, Heidenreich KA, Wierman ME. Novel mechanism for gonadotropin-releasing hormone neuronal migration involving Gas6/Ark signaling to p38 mitogen-activated protein kinase. Mol. Cell Biol. 2002;22:599–613. doi: 10.1128/MCB.22.2.599-613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- Arthur WT, Noren NK, Burridge K. Regulation of Rho family GTPases by cell-cell and cell-matrix adhesion. Biol. Res. 2002;35:239–246. doi: 10.4067/s0716-97602002000200016. [DOI] [PubMed] [Google Scholar]
- Barth H, Hofmann F, Olenik C, Just I, Aktories K. The N-terminal part of the enzyme component (C2I) of the binary Clostridium botulinum C2 toxin interacts with the binding component C2II and functions as a carrier system for a Rho ADP-ribosylating C3-like fusion toxin. Infect. Immun. 1998;66:1364–1369. doi: 10.1128/iai.66.4.1364-1369.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazenet CE, Mota MA, Rubin LL. The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc. Natl Acad. Sci. USA. 1998;95:3984–3989. doi: 10.1073/pnas.95.7.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 1999;274:13 198–13 204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl Acad. Sci. USA. 2001;98:13 681–13 686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P, Strasser A, Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. BH3-only proteins - evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death: degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. J. Cell Sci. 2002;115:1567–1574. doi: 10.1242/jcs.115.8.1567. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Busch C, Aktories K. Microbial toxins and the glycosylation of rho family. GTPases. Curr. Opin. Struct. Biol. 2000;10:528–535. doi: 10.1016/s0959-440x(00)00126-3. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Indyk JA, Strickland S. The hippocampal laminin matrix is dynamic and critical for neuronal survival. Mol. Biol. Cell. 2003;14:2665–2676. doi: 10.1091/mbc.E02-12-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET, Smiciene G, Hongisto V, Cao J, Brecht S, Herdegen T, Courtney MJ. c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J. Neurosci. 2002;22:4335–4345. doi: 10.1523/JNEUROSCI.22-11-04335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl Acad. Sci. USA. 1993;90:10 989–10 993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouder N, Prepens U, Aktories K, Cavalie A. Inhibition of calcium release-activated calcium current by Rac/Cdc42-inactivating clostridial cytotoxins in RBL cells. J. Biol. Chem. 2000;275:18 732–18 738. doi: 10.1074/jbc.M001425200. [DOI] [PubMed] [Google Scholar]
- von Eichel-Streiber C, Harperath U, Bosse D, Hadding U. Purification of two high molecular weight toxins of Clostridium difficile which are antigenically related. Microb. Pathog. 1987;2:307–318. doi: 10.1016/0882-4010(87)90073-8. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Gallagher ED, Xu S, Moomaw C, Slaughter CA, Cobb MH. Binding of. JNK/SAPK to MEKK1 is regulated by phosphorylation. J. Biol. Chem. 2002;277:45 785–45 792. doi: 10.1074/jbc.M207702200. [DOI] [PubMed] [Google Scholar]
- Garcia-Roman N, Alvarez AM, Toro MJ, Montes A, Lorenzo MJ. Lovastatin induces apoptosis of spontaneously immortalized rat brain neuroblasts: involvement of nonsterol isoprenoid biosynthesis inhibition. Mol. Cell Neurosci. 2001;17:329–341. doi: 10.1006/mcne.2000.0904. [DOI] [PubMed] [Google Scholar]
- Gary DS, Mattson MP. Integrin signaling via the PI3-kinase-Akt pathway increases neuronal resistance to glutamate-induced apoptosis. J. Neurochem. 2001;76:1485–1496. doi: 10.1046/j.1471-4159.2001.00173.x. 332001. [DOI] [PubMed] [Google Scholar]
- Gary DS, Milhavet O, Camandola S, Mattson MP. Essential role for integrin linked kinase in Akt-mediated integrin survival signaling in hippocampal neurons. J. Neurochem. 2003;84:878–890. doi: 10.1046/j.1471-4159.2003.01579.x. [DOI] [PubMed] [Google Scholar]
- Genth H, Hofmann F, Selzer J, Rex G, Aktories K, Just I. Difference in protein substrate specificity between hemorrhagic toxin and lethal toxin from Clostridium sordellii. Biochem. Biophys. Res. Commun. 1996;229:370–374. doi: 10.1006/bbrc.1996.1812. [DOI] [PubMed] [Google Scholar]
- Gerhardt E, Kugler S, Leist M, Beier C, Berliocchi L, Volbracht C, Weller M, Bahr M, Nicotera P, Schulz JB. Cascade of caspase activation in potassium-deprived cerebellar granule neurons: targets for treatment with peptide and protein inhibitors of apoptosis. Mol. Cell Neurosci. 2001;17:717–731. doi: 10.1006/mcne.2001.0962. [DOI] [PubMed] [Google Scholar]
- Gibson RM, Craig SE, Heenan L, Tournier C, Humphries MJ. Activation of integrin α5β1 delays apoptosis of Ntera2 neuronal cells. Mol. Cell Neurosci. 2005;28:588–598. doi: 10.1016/j.mcn.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO. Activation of. Rac GTPase by p75 is necessary for c-Jun N-terminal kinase-mediated apoptosis. J. Neurosci. 2002;22:156–166. doi: 10.1523/JNEUROSCI.22-01-00156.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Johnson EM., Jr BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J. Biol. Chem. 2001;276:37 754–37 760. doi: 10.1074/jbc.M104073200. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Barberis L, Brancaccio M, et al. Defective Rac-mediated proliferation and survival after targeted mutation of the β1 integrin cytodomain. J. Cell Biol. 2002;157:481–492. doi: 10.1083/jcb.200111065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Busch C, Aktories K. Chimeric clostridial cytotoxins: identification of the N-terminal region involved in protein substrate recognition. Infect. Immun. 1998;66:1076–1081. doi: 10.1128/iai.66.3.1076-1081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 1998;273:10 777–10 783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Inoue H, Tsukita K, Iwasato T, et al. The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 2003;22:6665–6674. doi: 10.1093/emboj/cdg634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- Kanamoto T, Mota M, Takeda K, Rubin LL, Miyazono K, Ichijo H, Bazenet CE. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol. Cell Biol. 2000;20:196–204. doi: 10.1128/mcb.20.1.196-204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura K, Hashimoto Y, Kita Y, Sasabe J, Aiso S, Nishimoto I, Matsuoka M. A. Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J. Biol. Chem. 2005;280:4532–4543. doi: 10.1074/jbc.M410508200. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi M, Matsushita N, Miyazaki J, Koike M, Yaginuma H, Osumi N, Kaibuchi K, Kobayashi K. Survival of developing motor neurons mediated by Rho GTPase signaling pathway through Rho-kinase. J. Neurosci. 2004;24:3480–3488. doi: 10.1523/JNEUROSCI.0295-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the. ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J. Biol. Chem. 2003;278:18 811–18 816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Li M, Linseman DA, Allen MP, Meintzer MK, Wang X, Laessig T, Wierman ME, Heidenreich KA. Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. J. Neurosci. 2001;21:6544–6552. doi: 10.1523/JNEUROSCI.21-17-06544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Laessig T, Meintzer MK, McClure M, Barth H, Aktories K, Heidenreich KA. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J. Biol. Chem. 2001a;276:39 123–39 131. doi: 10.1074/jbc.M103959200. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Heidenreich KA, Fisher SK. Stimulation of M3 muscarinic receptors induces phosphorylation of the Cdc42 effector activated Cdc42Hs-associated kinase-1 via a Fyn tyrosine kinase signaling pathway. J. Biol. Chem. 2001b;276:5622–5628. doi: 10.1074/jbc.M006812200. [DOI] [PubMed] [Google Scholar]
- Linseman DA, McClure ML, Bouchard RJ, Laessig TA, Ahmadi FA, Heidenreich KA. Suppression of death receptor signaling in cerebellar Purkinje neurons protects neighboring granule neurons from apoptosis via an insulin-like growth factor I-dependent mechanism. J. Biol. Chem. 2002a;277:24 546–24 553. doi: 10.1074/jbc.M201098200. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Phelps RA, Bouchard RJ, Le SS, Laessig TA, McClure ML, Heidenreich KA. Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J. Neurosci. 2002b;22:9287–9297. doi: 10.1523/JNEUROSCI.22-21-09287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossi L, Merighi A. In vivo cellular and molecular mechanisms of neuronal apoptosis in the mammalian CNS. Prog. Neurobiol. 2003;69:287–312. doi: 10.1016/s0301-0082(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Marani M, Hancock D, Lopes R, Tenev T, Downward J, Lemoine NR. Role of Bim in the survival pathway induced by Raf in epithelial cells. Oncogene. 2004;23:2431–2441. doi: 10.1038/sj.onc.1207364. [DOI] [PubMed] [Google Scholar]
- Marcoux N, Vuori K. EGF receptor mediates adhesion-dependent activation of the Rac GTPase: a role for phosphatidylinositol 3-kinase and Vav2. Oncogene. 2003;22:6100–6106. doi: 10.1038/sj.onc.1206712. [DOI] [PubMed] [Google Scholar]
- Meske V, Albert F, Richter D, Schwarze J, Ohm TG. Blockade of HMG-CoA reductase activity causes changes in microtubule-stabilizing protein tau via suppression of geranylgeranylpyrophosphate formation: implications for Alzheimer’s disease. Eur. J. Neurosci. 2003;17:93–102. doi: 10.1046/j.1460-9568.2003.02433.x. [DOI] [PubMed] [Google Scholar]
- Mota M, Reeder M, Chernoff J, Bazenet CE. Evidence for a role of mixed lineage kinases in neuronal apoptosis. J. Neurosci. 2001;21:4949–4957. doi: 10.1523/JNEUROSCI.21-14-04949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga C, Zohar M, Teramoto H, Gutkind JS. Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-κB. Oncogene. 2002;21:207–216. doi: 10.1038/sj.onc.1205036. [DOI] [PubMed] [Google Scholar]
- Napankangas U, Lindqvist N, Lindholm D, Hallbook F. Rat retinal ganglion cells upregulate the pro-apoptotic BH3-only protein Bim after optic nerve transection. Brain Res. Mol. Brain Res. 2003;120:30–37. doi: 10.1016/j.molbrainres.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, Tsujimoto Y. 14-3-3 interacts directly with and negatively regulates pro-apoptotic Bax. J. Biol Chem. 2003;278:2058–2065. doi: 10.1074/jbc.M207880200. [DOI] [PubMed] [Google Scholar]
- Parise LV, Lee J, Juliano RL. New aspects of integrin signaling in cancer. Semin. Cancer Biol. 2000;10:407–414. doi: 10.1006/scbi.2000.0337. [DOI] [PubMed] [Google Scholar]
- Pearce DA, Sherman F. Differential ubiquitin-dependent degradation of the yeast apo-cytochrome c isozymes. J. Biol. Chem. 1997;272:31 829–31 836. doi: 10.1074/jbc.272.50.31829. [DOI] [PubMed] [Google Scholar]
- Popoff MR, Chaves-Olarte E, Lemichez E, et al. Ras, Rap, and Rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J. Biol. Chem. 1996;271:10 217–10 224. doi: 10.1074/jbc.271.17.10217. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotoic protein Bim to prevent anoikis. Nat. Cell Biol. 2003;5:733–740. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- Sakai T, Johnson KJ, Murozono M, Sakai K, Magnuson MA, Wieloch T, Cronberg T, Isshiki A, Erickson HP, Fassler R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat. Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC, Bokoch GM. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol. Cell Biol. 2000;20:453–461. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Shattil SJ. Signaling networks linking integrins and rho family GTPases. Trends Biochem. Sci. 2000;25:388–391. doi: 10.1016/s0968-0004(00)01605-4. [DOI] [PubMed] [Google Scholar]
- Stupack DG, Cheresh DA. Get a ligand, get a life: integrins, signaling and cell survival. J. Cell Sci. 2002;115:3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- Takagi J. Structural basis for ligand recognition by RGD (Arg-Gly-Asp)-dependent integrins. Biochem. Soc. Trans. 2004;32:403–406. doi: 10.1042/BST0320403. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tatsuno I, Uchida D, et al. Geranylgeranyl-pyrophosphate, an isoprenoid of mevalonate cascade, is a critical compound for rat primary cultured cortical neurons to protect the cell death induced by 3-hydroxy-3-methylglutaryl-CoA reductase inhibition. J. Neurosci. 2000;20:2852–2859. doi: 10.1523/JNEUROSCI.20-08-02852.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton WG, Chalmers-Redman R, Brown D, Tatton N. Apoptosis in Parkinson’s disease: signals for neuronal degradation. Ann. Neurol. 2003;53:S61–S70. doi: 10.1002/ana.10489. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ, Hsu YT, Wolter KG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis: cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. J. Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Lin Q, Guan JL, Cerione RA. Activation of the Cdc42-associated tyrosine kinase-2 (ACK-2) by cell adhesion via integrin \g=b1. J. Biol. Chem. 1999;274:8524–8530. doi: 10.1074/jbc.274.13.8524. [DOI] [PubMed] [Google Scholar]
- Zhang B, Zhang Y, Shacter E. Caspase 3-mediated inactivation of rac. GTPases promotes drug-induced apoptosis in human lymphoma cells. Mol. Cell Biol. 2003;23:5716–5725. doi: 10.1128/MCB.23.16.5716-5725.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999;274:11 549–11 556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]