

Abstract
We report a method of inducing antigen production in dendritic cells (DCs) by in vivo targeting with lentiviral vectors that specifically bind to the DC surface protein, DC-SIGN. To target the DCs, the lentivector was enveloped with a viral glycoprotein from Sindbis virus, engineered to be DC-SIGN-specific. In vitro, this lentivector specifically transduced DCs and induced DC maturation. A remarkable frequency (up to 12%) of ovalbumin (OVA)-specific CD8+ T cells and a significant antibody response were observed 2 weeks following injection of a targeted lentiviral vector encoding an OVA transgene into naïve mice. These mice were solidly protected against the growth of the OVA-expressing E.G7 tumor and this methodology could even induce regression of an established tumor. Thus, lentiviral vectors targeting DCs provide a simple method of producing effective immunity and may provide an alternative route for immunization with protein antigens.
Immunization is one of the most productive tools in modern medical practice yet it still has limitations and novel methods of immunization are likely to be needed.1 One of the great advances in our understanding of the process of immunization was the recognition of the role of dendritic cells (DCs) as specialized antigen-presenting cells (APCs).2 This has led to attempts at DC-based immunization/vaccination involving loading DCs with specific antigens.3, 4 Although significant progress has been made on various aspects of this vaccination method, many challenges still remain in order to rationally manipulate DCs to achieve protective immunity.3-5
There is growing interest in genetically modifying DCs to make them either express antigens or produce immuno-stimulatory molecules.6 Of these methods, viral vectors have been proven most effective for the delivery of genes into DCs in vitro.7 The most popular viral vectors capable of transducing and expressing transgenes in DCs are adenoviral vectors,8-10 gamma-retroviral vectors11, 12 and lentiviral vectors.13-15
Considerable effort has also gone towards direct immunization using recombinant viral vectors as vaccine carriers. In these protocols, viral vectors are injected directly into an animal with the hope that a fraction will target immune cells and stimulate the desired immunity. Direct injection of adenoviral vectors was shown to induce both innate and adaptive immune responses and is currently being evaluated as a subunit vaccine for several infectious diseases and cancer.16-18 Direct injection of lentivectors into mice does transduce DCs, leading to antigen-specific CD8+ T cell responses and anti-tumor immunity.19-22 However, these recombinant viral vectors usually have broad specificity and transduce multiple cell types. Development of viral vectors capable of transducing DCs in a cell-specific manner in vivo could potentially improve the safety and efficacy of vaccination. Whereas few attempts have been reported of targeting recombinant viral vectors to DCs,23-25 success has been reported for targeting protein antigens directly to DCs in vivo by conjugating antigens to the anti-DEC-205 antibody 26-28 and other DC-specific molecules.29,30
Here we report a new method of in vivo DC targeting through the DC-specific surface molecule called DC-SIGN (also known as CD209)31,32 by a recombinant lentivector bearing an engineered glycoprotein derived from Sindbis virus. We show here that the engineered lentivector can genetically modify DCs in vitro with remarkable efficiency and specificity. Direct administration of the targeting lentivector can induce a strong antigen-specific T cell response and an antibody response. As a model, we have explored the applicability of injecting such an engineered lentivector to achieve cancer immunotherapy in mice and found that this is a promising approach for immunization against cancer.
Results
Targeting of DC-SIGN-expressing cell lines by an engineered lentivector in vitro
Certain subsets of DCs bear on their surface the DC-SIGN protein,31, 32 a C-type lectin-like receptor capable of rapid binding and endocytosis of materials,33 an ideal targeting receptor on DCs. Sindbis virus (SV)—a member of the Alphavirus genus and the Togaviridae family—is able to infect DCs through DC-SIGN.34 However, the canonical viral receptor for the laboratory strain of SV is cell-surface heparan sulfate (HS), which is expressed by many cell types.35, 36
Taking advantage of the physical separation of the two receptor-binding sites on the SV envelope glycoprotein (designated SVG), we engineered the receptor to be blind to its canonical binding target, HS, but left intact its ability to interact with DC-SIGN (Fig. 1a). Once it is incorporated onto a lentiviral surface, this mutant glycoprotein should be able to mediate infection of DCs but not other cells. We and others have demonstrated that SVG can efficiently pseudotype lentiviruses and that alterations to SVG, including deletion of amino acids 61–64 of the SVG E3, mutations of 157KE158 into 157AA158 of the SVG E2, and an insertion of 10-amino acid tag sequence (MYPYDVPDYA) between amino acids 71 and 74 of the SVG E2, can disable its binding to HS;37, 38 we designated this modified SVG as SVGmu. Using a GFP-vpr labeling method, we found that over 70% of lentiviral particles that we produced displayed SVGmu (Fig. 1b).
Figure 1.
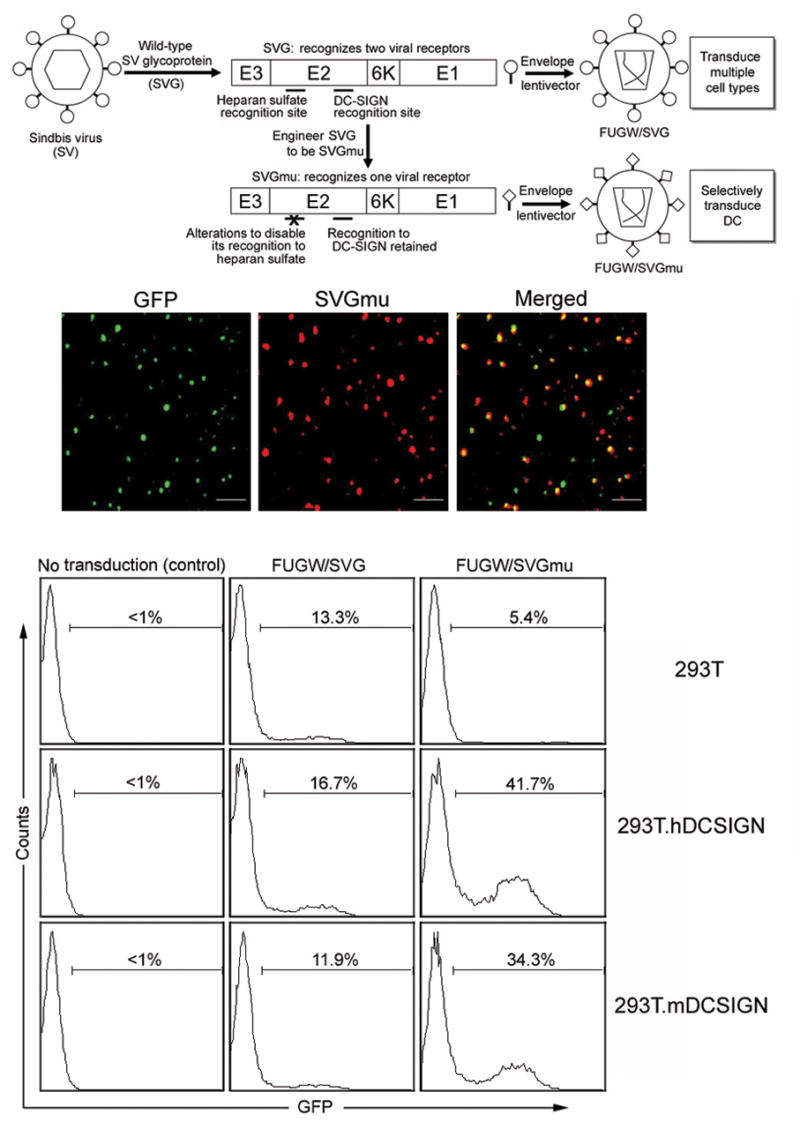
Lentivector bearing engineered Sindbis viral glycoprotein targets to DC-SIGN-expressing cells. (a) A schematic representation of the general strategy to engineer a lentivector system capable of targeting DCs. (b) Viral supernatant harvested from virus-producing cells transiently transfected with GFP-vpr, SVGmu, and other necessary packaging constructs was coated to a poly-lysine containing coverslip by centrifugation. A hemagglutinin (HA) epitope tag (YPYDVPDYA) was engineered into SVGmu to facilitate its detection by antibody. The resulting coverslip was then rinsed and immunostained with an anti-HA tag antibody (red) to label SVGmu and imaged using a laser confocal microscope. The scale bar represents 2 μm. (c) One milliliter of fresh viral supernatants of FUGW/SVG and FUGW/SVGmu were used to transduce 293T cells (2×105) expressing human DC-SIGN (293T.hDCSIGN) or murine DC-SIGN (293T.mDCSIGN). The parental 293T cells lacking the expression of DC-SIGN were included as controls. Three days later, the transduction efficiency was measured by analyzing GFP expression using flow cytometry. The specific transduction titer of FUGW/SVGmu was estimated to be ∼1×106 TU/ml for 293T.hDC-SIGN and ∼0.8×106 TU/ml for 293T.mDC-SIGN.
To facilitate our study of targeted transduction, we constructed 293T cell lines that stably expressed either human DC-SIGN (293T.hDCSIGN) or murine DC-SIGN (293T.mDCSIGN) (Supplementary Fig. 1 online). To assess transduction efficiency and specificity, we pseudotyped the lentiviral vector FUGW, which carries the GFP reporter gene under control of the human ubiquitin C promoter,39 with wild-type SVG or SVGmu to produce FUGW/SVG or FUGW/SVGmu. FUGW/SVG had similar transduction efficiency (11∼16% transduction) towards the three target cell lines (293T, 293T.hDCSIGN, and 293T.mDCSIGN) (Fig. 1c), indicating that SVG has broad specificity and the presence of DC-SIGN on the cell surface does not markedly alter the transduction ability of a SVG-pseudotyped lentiviral vector. In contrast, the FUGW/SVGmu vector could specifically transduce 293T.hDCSIGN and 293T.mDCSIGN cells, with 42% and 34% transduction efficiencies, respectively, but not the 293T cells (Fig. 1c). We confirmed the stable integration of the FUGW lentiviral vector in the transduced cells by PCR analysis of the genomic integration of the GFP reporter gene (data not shown). Adding soluble anti-human DC-SIGN antibody to the FUGW/SVGmu viral supernatant before its exposure to 293T.hDCSIGN cells reduced the transduction efficiency (data not shown). The specific titer of FUGW/SGVmu for 293T.mDCSIGN was estimated to be 1×106 TU (Transduction Units)/ml. FUGW/SVGmu could be concentrated by ultracentrifugation with a > 90% recovery, indicating that SVGmu is a stable envelope for a lentivector.
Targeting of DCs in vitro by an engineered lentivector
In a mixed mouse bone marrow culture, approximately 10% of the cells were CD11c positive DCs, of which ∼80% were DC-SIGN high (data not shown). After FUGW/SVGmu transduction, 12% of the cells were GFP+ (Fig. 2a, lower). Within the GFP+ cells, up to 95% of the transduced cells were DC-SIGN+CD11c+ DCs, indicating that FUGW/SVGmu could specifically transduce DCs expressing DC-SIGN. A blocking assay using anti-mouse DC-SIGN antibody confirmed that DC-SIGN plays an important role in the specific transduction (Fig. 2b). In contrast, although 68% of the cells were GFP-positive after exposure to lentivectors enveloped with an ecotropic murine leukemia virus glycoprotein (FUGW/Eco), only 9% of the transduced cells were DCs, within which 6.5% were DC-SIGN+ (Fig. 2a, upper). A similar non-specific transduction was observed for lentivector enveloped with vesicular stomatitis viral glycoprotein (FUGW/VSVG, Supplementary Fig. 2 online). FUGW/SVG could preferentially modify CD11c+ cells, but it was less specific for DC-SIGN+ cells (FUGW/SVG, Supplementary Fig. 2 online). Stable transduction by FUGW/SVGmu was verified by Alu PCR analysis40 of the genomic integration of the LTR of the lentivector backbone (data not shown). In addition, attempts to use FUGW/SVGmu to transduce primary T and B cells harvested from mouse spleen completely failed (Supplementary Fig. 3 online), indicating its transduction specificity.
Figure 2.
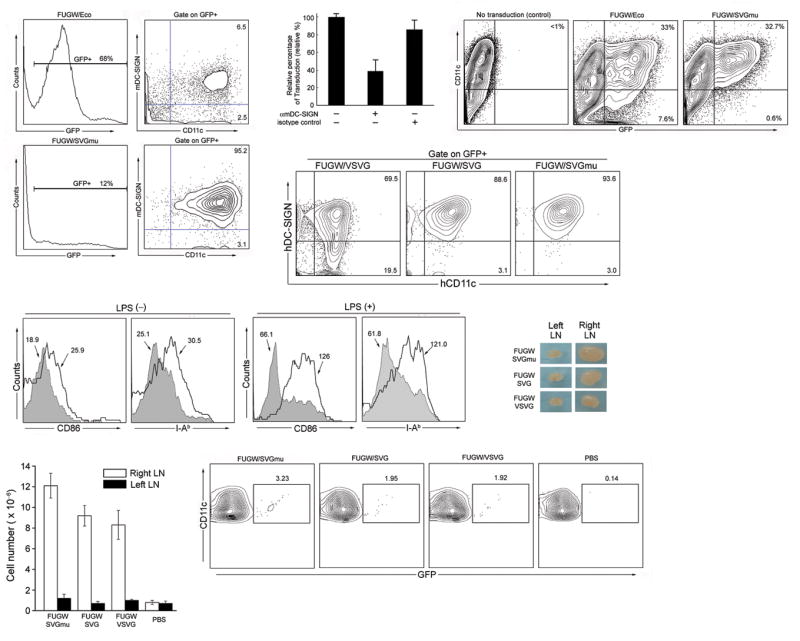
Lentivector encoding a reporter GFP gene and bearing SVGmu can selectively transduce DCs in vitro and in vivo. (a) Whole bone marrow cells isolated from B6 mice were exposed to the fresh viral supernatant of FUGW/SVGmu. The FUGW lentivector pseudotyped with the ecotropic glycoprotein (FUGW/Eco) was included as a non-targeting control. Three days post-transduction, the cells were collected for flow cytometric analysis of GFP expression. Surface antigens of the GFP-positive cells were assessed by staining with anti-CD11c and anti-DC-SIGN antibodies. (b) Anti-murine DC-SIGN antibody was added into viral supernatant during transduction of whole mouse bone marrow cells for 8 h. Then, the supernatant was replaced with fresh medium. The cells were analyzed for GFP expression after 2 days. Isotype-matched antibody was used as a control. (c) Murine bone marrow-derived DCs (mBMDCs) were generated by culturing freshly isolated bone marrow cells in the presence of cytokine GM-CSF for 6 days. The resulting cells were transduced with the fresh viral supernatant of either the targeting FUGW/SVGmu or non-targeting FUGW/Eco vector. GFP and CD11c expression were measured by flow cytometry. (d) Human monocyte-derived DCs (hMoDCs) were generated by culturing freshly purified CD14+ peripheral blood moncytes in the presence of GM-CSF and IL-4. The cells from the day 2 culture were transduced with the fresh viral supernatant of either FUGW/SVGmu, or FUGW/VSVG, or FUGW/SVG vector. GFP expression was measured by flow cytometry at day 6. (e) Upon targeted transduction of mouse BMDCs with FUGW/SVGmu, DC activation was assessed by analyzing the surface expression of CD86 and I-Ab using flow cytometry. The addition of LPS (1 μg/ml) overnight was used as a synergistic stimulator for the activation of transduced BMDCs. Shaded area, GFP negative cells (untransduced); solid line, GFP positive cells (transduced). (f-h) In vivo DC-targeting using FUGW/SVGmu lentivector. B6 mice were injected with 50×106 TU of FUGW/SVGmu, FUGW/SVG, or FUGW/VSVG, and analyzed 3 days later. Mice injected with PBS were included as a control. (f) Comparison of a representative inguinal lymph node close to the injection site (right) and the equivalent lymph node distant from the injection site (left). (g) Total cell number counts of the indicated lymph nodes. (h) Representative flow cytometric analysis of CD11c+ cells from the indicated lymph nodes that are close to the injection sites. The numbers indicate the fraction of GFP+ DC populations.
We next tested the efficiency of the lentivector bearing SVGmu to transduce in vitro-cultured, mouse bone marrow (BM)-derived DCs (mBMDCs). Flow cytometry analysis showed that FUGW/Eco transduced both CD11c+ DCs (33%) and CD11c– cells (7.6%) (Fig. 2c), which is consistent with the wide tropism of Eco. On the contrary, FUGW/SGVmu could only transduce CD11c+ DCs (32.7%) (Fig. 2c), indicating that FUGW/SGVmu can specifically modify mBMDCs. FUGW/SVGmu also had a greater specificity for transducing human monocyte-derived DCs (hMoDCs) compared to FUGW/VSVG or FUGW/SVG; the transduction is closely correlated with DC-SIGN expression (Fig. 2d).
We further examined whether the targeted transduction could activate and mature DCs. 41 Flow cytometry analysis of mBMDCs 3 days post-transduction showed that treatment with FUGW/SVGmu elevated the expression of DC activation markers, CD86 and I-Ab,41 on GFP+ DCs, as compared to GFP– DCs (Fig. 2e, left). Moreover, we found that the targeted transduction of mBMDCs could synergize with lipopolysaccharide (LPS) treatment to further mature DCs (Fig. 2e, right), suggesting that it can either work alone or in combination with other DC maturation stimuli, such as adjuvant, to induce DC activation.
Targeting of DCs in vivo by an engineered lentivector
To test whether engineered lentivectors bearing SVGmu could target DCs in vivo in mice, we injected 50×106 TU of FUGW/SVGmu subcutaneously on the right flank of a B6 mouse. On day 3, we observed a significant enlargement of the right inguinal lymph node close to the injection site (Fig. 2f), and found an approximately 10-fold increase of cell number in this lymph node compared with the equivalent lymph node at the opposite side or lymph nodes from a mouse injected with PBS (Fig. 2g), indicating that vector administration can enhance trafficking of DCS and proliferation of lymphocytes in a nearby lymph node. Flow cytometry showed that approximately 3.2% of the total CD11c+ cells in the right inguinal lymph node cells were GFP+ DCs (Fig. 2h), which had presumably migrated from the injection site.
To compare the in vivo effects of vectors pseudotyped with SVGmu, SVG and the more commonly used VSVG, we injected into the flanks of mice FUGW enveloped with each of these proteins and found that FUGW/SVGmu produced a distinctly larger node, more cellularity, and more transduced DCs than the others (Fig. 2f-h). Thus, the increased infection efficiency mediated by the mutant SVG protein compared to wild type, which we found in vitro (Fig. 1c), is paralleled by an increased effectiveness in vivo.
To examine the in vivo specificity of the DC-targeted lentivector, we constructed a lentiviral vector encoding a firefly luciferase (designated as Fluc, Supplemental Fig. 4a online), which enabled us to visualize the in vivo transduction of the tissue cells using bioluminescence imaging (BLI). We found that Fluc/VSVG-treated mice had a strong and permanent signal at the injection site, while no significant signal was detected at the injection site of Fluc/SVGmu-treated mice (Supplementary Fig. 4b online), indicating that the lentivector bearing SVGmu had a relatively stringent target specificity. At no time were we able to detect a luminescence signal in the targeted mice, probably due to the rare and sparse distribution of the DCs, which is beyond the sensitivity of the current BLI method. After a month, the mice injected with Fluc/SVGmu were subjected to biodistribution analysis by quantitative RT-PCR and no detectable copy of the lentivector was observed in all isolated organs (heart, liver, spleen, kidney, gonad, lung, skin, lymph node), verifying the lack of non-specific infection in the animals and thus the specificity of the targeted vector for DCs.
CD8+ and CD4+ T cell responses induced by lentivector-modified DCs in vitro
To determine whether the targeted transduction of DCs by a recombinant lentivector could be used to effectively deliver antigen genes to DCs for stimulation of antigen-specific CD8+ and CD4+ T cell responses, we constructed a lentivector expressing the model antigen, chicken ovalbumin (OVA), which we designated FOVA (Fig. 3a). In C57BL/6J (B6) mice, OVA is a well-characterized target antigen for the CD8+ TCR, OT1, which recognizes OVA257-269 (designated as OVAp), and for the CD4+ TCR, OT2, which recognizes OVA323-339 (designated as OVAp*).42 mBMDCs were transduced on day 6 of culture with either FOVA/SVGmu or FUGW/SVGmu encoding a non-relevant reporter gene GFP; the modified DCs were designated as DC/FOVA and DC/FUGW, respectively. The capacity of vector-transduced DCs to process and present the transgenic OVA antigen was examined by their ability to stimulate OVA-specific OT1 transgenic CD8+ T cells and OT2 transgenic CD4+ T cells. Unmodified mBMDCs pulsed with the OVAp (DC/OVAp) or OVAp* (DC/OVAp*) were included as positive controls. After a three-day coculture with varying ratios of DC/FOVA to transgenic T cells, OT1 T cells responded vigorously as measured by the release of IFN-γ (Fig. 3c) and T cell proliferation (Fig. 3d). As expected, no detectable OVA response was seen using DC/FUGW (Fig. 3cd). Flow cytometry showed that the activated OT1 T cells exhibited the typical effector cytotoxic T cell phenotype (CD25+CD69+CD62LlowCD44high) after stimulation by either DC/FOVA or DC/OVAp (Fig. 3b). When we cocultured the DCs with OT2 CD4+ T cells, we also observed T cell activation indicated by changes in the surface markers (Fig. 3e) and the production of IFN-γ (Fig. 3f). However, stimulation of CD4+ cells was not as pronounced as that of CD8+ cells, presumably due to the less efficient presentation of endogenous antigen peptides to the MHC class II molecules. By modifying the cellular localization of OVA antigen to direct it to MHC class II presentation pathway, we have achieved an enhancement of CD4 stimulation, which was even better than that of peptide-pulsed DCs (data not shown). Our results show that the method of DC targeting through lentivector infection can effectively deliver antigens to DCs and stimulate both CD8+ and CD4+ T cell responses.
Figure 3.
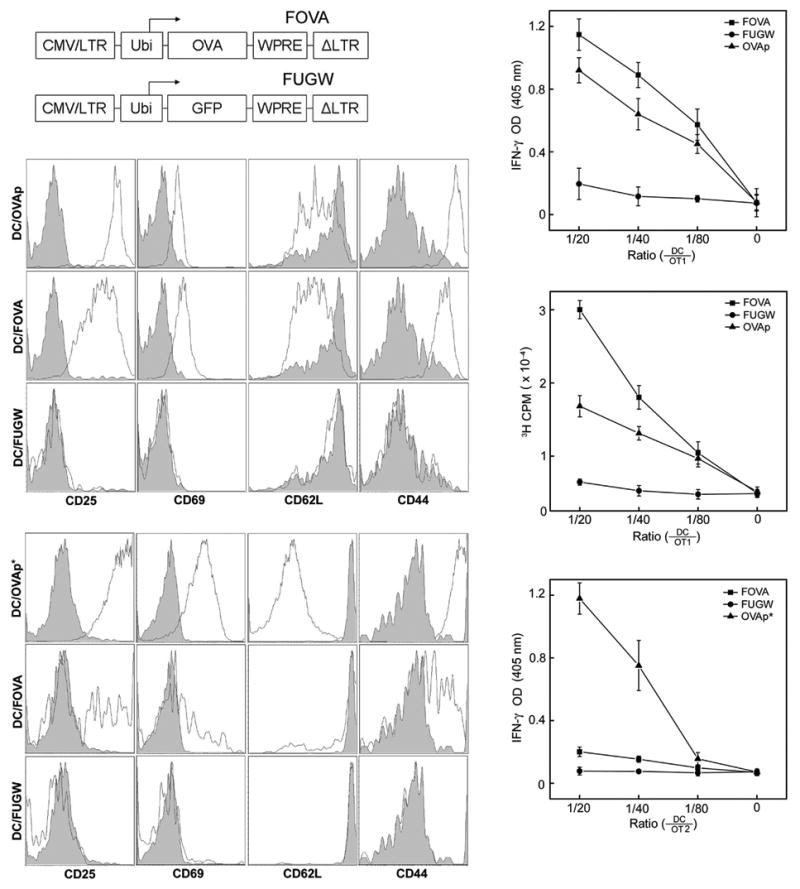
Mouse bone marrow-derived DCs (mBMDCs) transduced by a SVGmu enveloped lentivector encoding an OVA gene can stimulate OVA-specific CD8+ and CD4+ T cells in vitro. (a) A schematic representation of the lentivector encoding the OVA antigen (FOVA) or the lentivector encoding GFP (FUGW) as a control. LTR: long terminal repeat; Ubi: human ubiquitin-C promoter; WPRE: woodchuck hepatitis virus posttranscriptional regulatory element. (b-f) OVA-specific, CD8+ OT1 T cells and CD4+ OT2 T cells were harvested from the spleens of OT1 TCR- or OT2 TCR-transgenic mice (The Jackson Laboratory) and were cocultured with FOVA/SVGmu-transduced mBMDCs (DC/FOVA) in vitro for 3 days. Non-transduced BMDC pulsed with either OVAp peptide (SIINFEKL) (DC/OVAp), recognized by OT1 T cells, or OVAp* peptide (ISQAVHAAHAEINEAGR) (DC/OVAp*), recognized by OT2 T cells, were included as positive controls. mBMDCs transduced with FUGW/SVGmu (DC/FUGW) were included as a negative control. (b) Patterns of surface activation markers of OT1 T cells cocultured with various mBMDCs were assessed by antibody staining for CD25, CD69, CD62L, and CD44. Shaded area: naïve OT1 T cells harvested from transgenic animals; solid line: OT1 T cells cocultured with the indicated mBMDCs. (c) OT1 T cells were mixed with various dilutions of mBMDCs transduced with FOVA/SVGmu (■), FUGW/SVGmu (●), or pulsed with OVAp peptide (▲) and cultured for 3 days. Secretion of IFN-γ was measured by ELISA. (d) The proliferative responses of treated OT1 T cells from (c) were measured by a [3H] thymidine incorporation assay. (e) Similar to (b) except for the analysis of activated CD4+ OT2 T cells. Shaded area, naïve OT2 T cells harvested from transgenic animals; solid line, OT2 T cells cocultured with BMDCs. (f) Similar to (c) except for the responding cells being CD4+ OT2 T cells.
Induction of in vivo CTL and antibody responses by direct administration of targeting lentivector
We focused our subsequent studies on the efficacy of the in vivo DC targeting for inducing an antigen-specific CD8+ cytotoxic T lymphocyte (CTL) and antibody responses following administration of the targeting lentivector to naïve, wild-type mice. We used a single dose of 50×106 TU of FOVA/SVGmu subcutaneously and monitored the presence of OVA-specific T cells by measuring cytokine secretion and tetramer staining. At day 14 post-injection, T cells harvested from spleens were analyzed. The generated CD8+ T cells could be primed to secrete IFN-γ upon OVAp restimulation (Supplementary Fig. 5 online). Administration of the control vector FUGW/SVGmu failed to generate OVAp-specific responses (Supplementary Fig. 5 online). Using MHC class I tetramer staining., a high frequency of OVAp-specific T cells (> 6%) was observed following a single dose injection (Fig. 4a); no tetramer-positive cells were detected in the mice treated with FUGW/SVGmu (Fig. 4a), correlating well with the intracellular IFN-γ staining results (Supplementary Fig. 5 online). These cells displayed the surface characteristics of effector memory T cells (CD25lowCD69lowCD62LhighCD44high) (Fig. 4b). In addition, we also compared the level of CD8 T cell responses generated by FOVA/SVGmu with that of other vectors (FOVA/VSVG and FOVA/SVG) and found that FOVA/SVGmu provided the strongest immune response (Fig. 4c).
Figure 4.
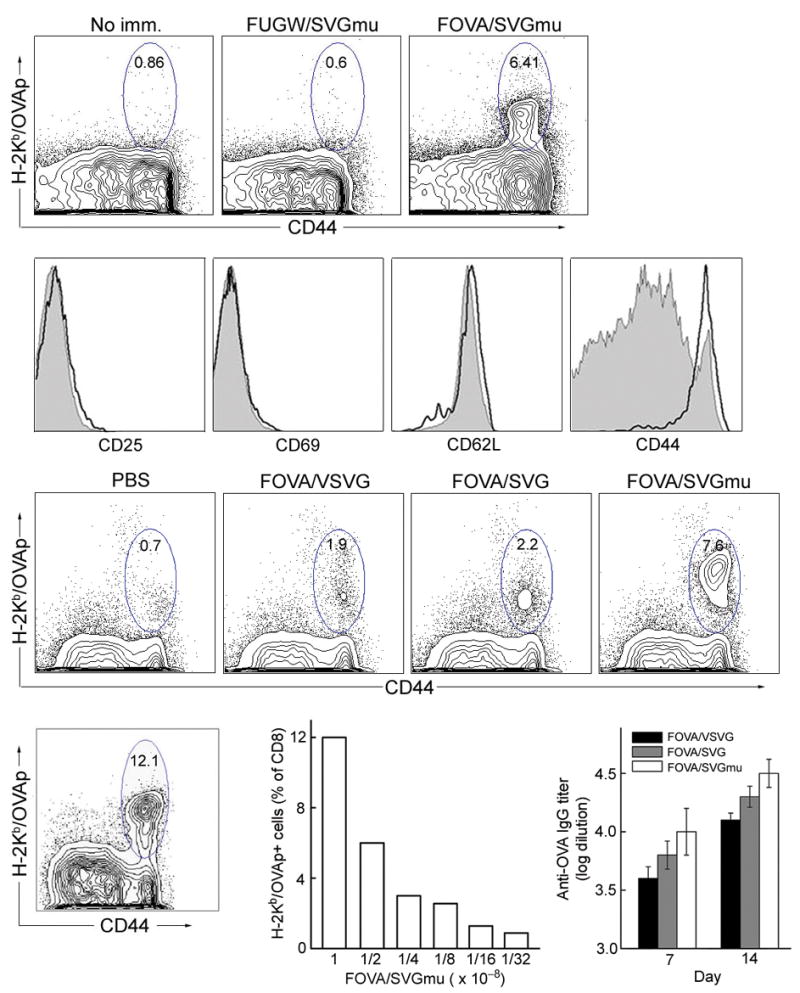
In vivo stimulation of antigen specific T cell and antibody responses in wild-type B6 mice following a subcutaneous injection of the DC-targeting lentivector FOVA/SVGmu. (a) B6 mice were immunized subcutaneously with 50×106 TU of either FOVA/SVGmu or FUGW/SVGmu (as a control). Mice without immunization (no imm.) were included as a negative control. Fourteen days post-immunization, spleen cells were harvested and analyzed for the presence of OVA-specific CD8+ T cells measured by H-2Kb-SIINFEKL-PE tetramer and CD44 staining. Indicated percentages are a percent of total CD8+ T cells. (b) Patterns of surface activation markers of OVA-specific CD8+ T cells (identified as tetramer positive cells) isolated from immunized mice 2 weeks post-injection were assessed by antibody staining for CD25, CD69, CD62L and CD44. Solid line, tetramer+CD8+ T cells from FOVA/SVGmu-immunized mice; shaded area, CD8+ T cells from non-immunized mice. (c) Naïve B6 mice were immunized by subcutaneous injection of 50×106 TU of the different lentivectors (FOVA/VSVG, FOVA/SVG, or FOVA/SVGmu). The injection of PBS was included as a control. Two weeks later, spleen cells were harvested and analyzed for the presence of OVA-specific CD8+ T cells measured by H-2Kb-SIINFEKL-PE tetramer and CD44 staining. Indicated percentages are a percent of total CD8+ T cells. (d-e) OVA-specific T cell responses seen in mice receiving different subcutaneous doses of FOVA/SVGmu. OVA-specific T cells were identified by tetramer staining as described in (a). (d) Percentage of OVA-specific CD8+ T cells following immunization with 100×106 TU of FOVA/SVGmu. (e) Dose responses of OVA-specific CD8+ T cells following injection of the indicated doses of FOVA/SVGmu. (f) OVA-specific serum IgG titer of B6 mice following immunization with 50×106 TU of the different lentivectors (FOVA/VSVG, FOVA/SVG, or FOVA/SVGmu). Sera were collected on day 7 and day 14 post-immunization and were analyzed for the titer of OVA-specific IgG using ELISA at serial 10× dilutions, starting at 1:100. The titer values were determined by the highest dilution at which the optical density was 2× standard derivations higher than that of the baseline serum at the equivalent dilution.
To investigate the dose response of lentivector administration, FOVA/SVGmu ranging from 100×106 TU to 3×106 TU were injected subcutaneously and OVAp-specific T cells in the spleen were measured at day 14 post-injection. Strikingly, an exceptionally high frequency (12%) of OVAp-specific CD8+ T cells was detected at the dose of 100×106 TU (Fig. 4d). The percentage of OVAp-specific cells correlated proportionately with the amount of recombinant vector administered (Fig. 4e). We may not have reached a plateau response with the doses we tested, suggesting that further enhancement may be possible by increasing the amount of vector injected and/or the frequency of injection.
We further examined the serum IgG levels specific for OVA in mice on the 7th and 14th days after immunization with FOVA/SVGmu (50×106 TU). The IgG serum titer was 1:10,000 on day 7 and 1:30,000 on day 14 (Fig. 4f). This is a rather impressive antibody response for a single dose injection without additional adjuvant or other stimuli, suggesting that targeted lentivector immunization can also elicit significant B cell secretion of antigen-specific antibodies. As a comparison, immunization with the same vector titer of either FOVA/VSVG or FOVA/SVG generated less serum IgG (Fig. 4f). These results show that in vivo administration of a DC-targeting lentivector can be a powerful tool to induce both cellular and humoral immune responses against the delivered antigen.
Generation of anti-tumor immunity
As a measure of the effectiveness of this mode of immunization, we evaluated the anti-tumor immunity generated after an in vivo administration of DC-targeted lentivector. We used the E.G7 tumor model,42 in which OVA serves as the tumor antigen. Mice receiving a subcutaneous administration of 50×106 TU of FOVA/SVGmu as a single dose tumor vaccination were challenged two weeks later with 5×106 E.G7 tumor cells. Vaccination with FOVA/SVGmu completely protected the mice from the tumor challenge (Fig. 5a, left), while tumors grew rapidly in mice receiving a mock vaccination with a lentivector lacking the OVA transgene (Fig. 5a, left). This protection was OVA-specific because the vaccinated mice grew control EL4 tumors that lack expression of OVA (Fig. 5a, right).
Figure 5.
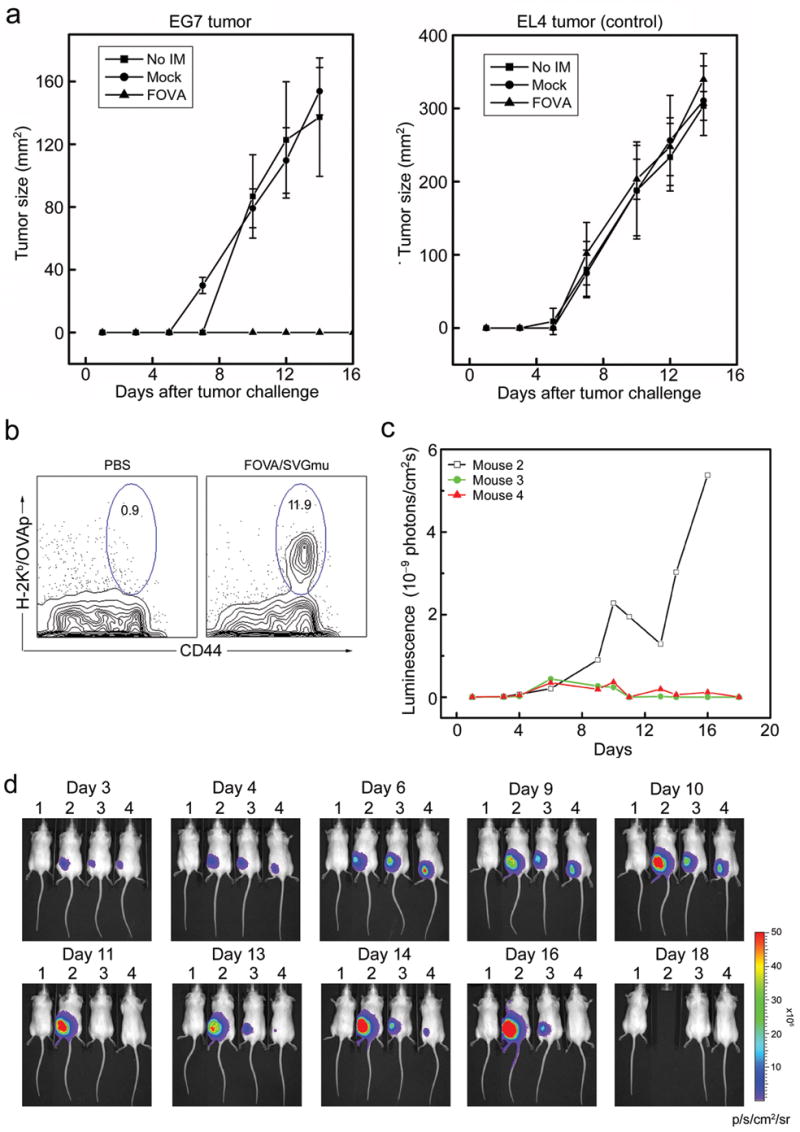
Preventive and therapeutic anti-tumor immune responses elicited through in vivo administration of the DC-targeted lentivector FOVA/SVGmu in a murine E.G7 tumor model. (a) B6 mice were immunized at day 0 by subcutaneous injection of 50×106 TU of either FOVA/SVGmu (▲) or mock vector FUW/SVGmu (●). No immunization (■) was included as a control. At day 14 post-immunization, the mice were challenged with 5×106 of either E.G7 tumor cells (expressing the OVA antigen) or EL4 tumor cells (lacking the OVA antigen, as a control) subcutaneously. Tumor growth was measured with a fine caliper and is shown as the product of the two largest perpendicular diameters (mm2). Four mice were included in each group. The experiment was repeated for 3 times and the result for one representative experiment was shown. (b) Percentage of OVA-specific T cells present following immunization with 100×106 TU of FOVA/SVGmu, or PBS (control), in the albino strain of B6 mice. The analysis was as described in Fig. 5. (c-d) B6 mice were implanted with E.G7 tumor cells stably expressing a firefly luciferase imaging gene (E.G7.luc) as described in (a). A mouse (#1) without tumor implantation was included as a control. Mice bearing tumors were treated without immunization (#2), or with immunization (#3, #4) by the injection of 50×106 TU of FOVA/SVGmu at day 3 and day 10. The kinetic growth of the tumors was monitored by live animal imaging using bioluminescence imaging (d). The p/s/cm2/sr represents photons/sec/cm2/steridian. (c) Quantitation of luminescence signals generated by the E.G7 tumors in (d). (□) for mouse #2; (
 ) for mouse #3; (
) for mouse #3; (
 ) for mouse #4.
) for mouse #4.
We then reversed the times of tumor injection and lentivector administration to test whether an established tumor could be eliminated, in a test of so-called therapeutic vaccination. To this end, we used E.G7 tumor cells expressing the firefly luciferase gene (E.G7.luc) to challenge mice, allowing us to closely monitor tumor growth kinetics in live animals using BLI. To facilitate imaging, an albino strain of B6 mice was used. Injection of these mice with FOVA/SVGmu showed a similar response to that observed in canonical B6 mice (Fig. 5b). E.G7.luc tumor cells (5×106) were implanted subcutaneously in the albino B6 mice. The mice were immunized by FOVA/SVGmu (50×106 TU per mice per time) twice on days 3 and 10 post-tumor challenge via subcutaneous injection. The experiment was repeated three times with a representative experiment shown in Fig. 5c-d. The mice receiving the DC-targeting lentivector immunization showed a decline of tumor growth starting at day 9, followed by tumor regression and a reduction of luminescence below the detection level on day 11 (Fig. 5c-d). Although minimal tumor recurrence was observed from day 12 to day 16, mice treated with FOVA/SVGmu were free of disease at the end of day 18 and thereafter; no tumor relapse was observed for as long as the experiment ran (> 60 days). In contrast, tumors grew progressively in the mice receiving no treatment and the mice had to be removed from the experiment after day 16 due to the large size of the tumors. It was an interesting observation that tumor regression started 7 days after the lentivector immunization. This timing correlates well with the kinetics of an antigen-specific immune response induced by vaccination.
Discussion
This study describes a new and highly efficient targeting method to deliver genes of antigenic proteins to DCs both in vitro and in vivo. By using a vector coated with a protein selectively targeted to a dendritic cell surface protein, we have achieved very high specificity, as measured in vitro, and high efficiency in vivo delivery of the vectored genes. One measure of the effectiveness of T-cell immunization by this route is the complete protection we achieved against a very rapidly growing tumor expressing a single protein targeted by the T cells that were stimulated by vector administration to a wild type mouse.
DC-SIGN is one of many potential receptors predominantly expressed on the surface of DCs for antigen uptake. Many viruses, are able to interact with DC-SIGN, and some can even utilize it as a receptor to mediate viral entry.43 This suggested to us that the glycoprotein derived from Sindbis virus could be engineered to target a lentiviral vector to DCs. Many alterations were introduced to the wild-type SVG, resulting in SVGmu, a protein unable to interact with cell-surface heparin sulfate but retaining the ability to interact with DC-SIGN. The lentivecotors pseudotyped with SVGmu proved to be highly specific and effective on modification of both murine and human DCs expressing DC-SIGN.
The most remarkable results were achieved by direct injection of the engineered lentivector subcutaneously into an animal. This was sufficient, in absence of any adjuvant, to produce a rapid and florid enlargement of the lymph node near the injection site with a few percent of transduced DCs detected within 3 days. Because the injection was subcutaneous, approximately 10 millimeters from the lymph node itself, it would appear that dermal DCs were transduced by the vector and the gene-modified DCs were then able to migrate to the nearby lymph node. It seems likely that it is the infection process itself, by which the DCs are transduced, that stimulates DCs to mature and migrate. The capacity of lentivectors to transduce non-dividing cells may be especially advantageous for effective delivery of antigen genes into the dermal DCs, which are considered to be poorly proliferative.44
In a mouse tumor model, we found that direct administration of a lentivector carrying the gene encoding a tumor antigen could not only protect the mice from tumor challenge, but also lead to a cession of growth and a complete regression of established solid tumor. These studies show that this in situ DC immunization method has the potential to be used in current immunotherapy protocols to improve therapeutic efficiency.
Quantitation of T cell responses after administration of a single dose of a targeted lentivector into naïve mice showed that the method could elicit high levels of antigen-specific CD8+ T cells (up to 12%). It is certainly much more efficient than conventional methods using recombinant protein antigen or adoptive transfer of antigen-loaded or viral vector-transduced DCs (Supplementary Fig. 6 online).45, 46 Although the exact mechanisms behind such an efficient immunization are still under investigation, it seems likely that the lentivector transduction through DC-SIGN can stimulate the maturation of the immature dendritic cells that are resident in the dermis. Whatever the cause, these cells must take up the vector, express its genes, process the encoded proteins, present the peptides on MHC molecules, migrate to the local lymph nodes, present their MHC/peptides to lymphocytes, and activate the lymphocytes to become effectors. The independence from an additional maturation signal represents a significant advantage of our targeting method. Unlike our approach, the protocol of anti-DEC-205-mediated delivery of protein antigens to DCs absolutely requires the co-administration of anti-CD40 antibody as a maturation stimulus for the induction of immunity.27 With our method, phenotypic analysis of the induced antigen-specific T cells showed them to be typical effector memory cells, which is important for therapeutic vaccination. Further studies are needed to quantify the efficiency of generating central memory T cells by the DC-targeting lentivector, which is essential for protective vaccination. Significant antibody responses were also detected in our experiments and we found that co-delivery of CD40L could further enhance the antibody response (data not shown).
There is much left to learn about this system. For instance, we used the highly immunogenic E.G7, OVA-expressing tumor to test immunity. How well would the method work with less immunogenic tumors, viral antigens, or antigens of other pathogens? Experiments are under way on these issues. A key issue is the specificity of the mutated Sindbis glycoprotein. There is controversy regarding the functional difference between human and murine DC-SIGNs and several studies have shown that murine DC-SIGN lacks the ability to interact with viruses, as opposed to human DC-SIGN.47 We know that DC-SIGN can be the receptor for the SVGmu-enveloped lentivector to transduce cells, but more experiments are needed to examine the possibility that other C-type lectin receptors closely related to DC-SIGN, such as the non-DC-specific L-SIGN for humans and SIGNR1-8 for mice,48 could mediate transduction. In fairly insensitive experiments, we saw no off-target effects, but that could be a property of the subcutaneous route of inoculation and there might be cells inaccessible by that route which could be transduced.
In conclusion, we demonstrate here an efficient method of targeting lentivectors to DCs in vivo by direct subcutaneous administration which elicits strong antigen-specific immune responses. This method expands the scope of genetic modification of DCs for vaccines and can significantly overcome the current limitations of the tedious and costly protocols used to induce immunity through adoptive transfer of in vitro-manipulated DCs. We have concentrated on an experimental antigen, OVA, but it has provided a proof-of-concept that this novel approach of antigen delivery for vaccination/immunization markedly improves immunity relative to conventional DC-based approaches. We are engineering and testing DC-specific lentivectors encoding clinically relevant antigens to directly evaluate the therapeutic utility of this method against cancer and infectious diseases.
Methods
Mice
C57BL/6J (designated as B6) female mice were purchased from Charles River Breeding Laboratories. The albino B6 female mice (B6(Cg)-Tyrc-2J/J), OT1 transgenic mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J) and OT2 transgenic mice (C57BL/6-Tg(TcraTcrb)425Cbn/J) were purchased from The Jackson Laboratory. All mice were housed in the animal facility in accordance with Institute regulations.
Plasmid construction
The cDNA for wild-type SVG was cloned into the pcDNA3 vector (Invitrogen) by PCR to generate plasmid SVG. SVGmu was generated by PCR mutagenesis of the E2 glycorprotein of SVG to introduce a 10-residue tag sequence (MYPYDVPDYA) between amino acids 71 and 74, and mutations of 157KE158 into 157AA158. Additional deletion was introduced to the E3 glycoprotein of SVG to remove amino acids 61-64. The cDNA of firefly luciferase was amplified from pGL4.2LucP (Promega) and cloned into FUGW39 to replace GFP, yielding the construct Fluc. FOVA was constructed from FUGW by replacing the GFP with the cDNA of chicken ovalbumin.
Lentivector production
The lentiviral transfer vectors (FUGW and its derivatives) used in this study are the third generation HIV-based lentiviral vectors, in which most of the U3 region of the 3′ LTR was deleted, resulting in a self-inactivating 3′-LTR or SIN. Lentivectors were prepared by transient transfection of 293T cells using a standard calcium phosphate precipitation protocol. 293T cells cultured in 6-cm tissue culture dishes (Corning or BD Biosciences) were transfected with the appropriate lentiviral transfer vector plasmid (5 μg), along with 2.5 μg of the envelope plasmid (SVG, SVGmu, Eco, or VSVG) and the packaging plasmids (pMDLg/pRRE and pRSV-Rev). The viral supernatants were harvested 48 and 72 hours post-transfection and filtered through a 0.45-μm filter (Corning). To prepare concentrated viral vectors for in vivo study, the viral supernatants were ultracentrifugated (Optima L-80 K preparative ultracentrifuge, Beckman Coulter) at 50,000 × g for 90 min. The pellets were then resuspended in an appropriate volume of cold PBS.
Confocal imaging of GFP-vpr labeled lentiviral vectors
GFP-vpr-labeled lentivectors were produced as described above with an additional plasmid encoding GFP-vpr (2.5 μg). Fresh viral supernatant was overlaid on polylysine-coated coverslips in a 6-well culture dish and centrifuged at 3,700× g at 4°C for 2 hours using a Sorvall Legend RT centrifuge. The coverslips were rinsed with cold PBS twice and immunostained by anti-HA-biotin antibody (Miltenyi Biotec) and Cy5-streptavidin (Invitrogen). Fluorescent images were acquired by a Zeiss LSM 510 laser scanning confocal microscope equipped with filter sets for fluorescein and Cy5. A plan-apochromat oil immersion objective (63×/1.4) was used for imaging.
Cell line construction
The 293T.hDCSIGN and 293T.mDCSIGN cell lines were generated by stable transduction of parental 293T cells with a VSVG-pseudotyped lentivector. The cDNAs for human DC-SIGN and murine DC-SIGN were amplified from plasmids pUNO-hDCSIGN1Aa and pUNO-mDCSIGN (InvivoGene) and cloned downstream of the human ubiquitin-C promoter in the lentiviral plasmid FUW to generate FUW-hDCSIGN and FUW-mDCSIGN, respectively. The lentivectors were then pseudotyped with VSVG and was used to transduce 293T cells. The resulting cells were subjected to antibody staining (anti-human DC-SIGN antibody from BD Biosciences and anti-murine DC-SIGN from eBioscience) and cell sorting to yield a uniform population of DC-SIGN+ 293T.hDCSIGN and 293T.mDCSIGN cell lines.
Targeted lentivector transduction of cell lines in vitro
Target cells (293T.hDCSIGN, 293T.mDCSIGN, or 293T cells; 0.2×106 per well) were seeded in a 24-well culture dish (Corning or BD Biosciences) and spin-infected with viral supernatants (1 ml per well) at 2,500 rpm and 30°C for 90 min using a Sorvall Legend centrifuge. Subsequently, the supernatants were replaced with fresh culture medium and incubated for 3 days at 37°C with 5% of CO2. The percentage of GFP+ cells was measured by flow cytometry. The transduction titer was determined by the dilution ranges that exhibited a linear response.
Mixed bone marrow and BMDC culture and transduction
Total bone marrow cells were harvested from B6 mouse and BMDCs were generated as previously described42. Either BM cells or BMDCs were plated in a 24-well culture dish (2×106 cells per well), and spin-infected with viral supernatant (1 ml per well) at 2,500rpm and 30°C for 90min using a Sorvall RT7 centrifuge. After the spin, the supernatant was removed and replaced with fresh RPMI medium containing 10% FBS and GM-CSF (1:20 J558L conditioned medium). The cells were cultured for 3 days and were analyzed for GFP expression using flow cytometry.
Murine T cell and B cell transduction in vitro
Spleen cells were harvested from B6 mice, and cultured in a 24-well culture dish (2×106 cells per well) in RPMI containing 10% FBS for 3 days in the presence of T cell stimuli (0.5μg/ml anti-mouse CD3 + 0.5μg/ml anti-mouse CD28, BD Biosciences), or B cell stimuli (50μg/ml LPS, Sigma). On day 1 and 2, the cells were spin-infected with viral supernatant (1 ml per well) at 2,500 rpm and 30°C for 90min. After each spin, the supernatant was removed and replaced with the T cell-specific or B cell-specific medium. On day 3 post-infection, the cells were analyzed for GFP expression using flow cytometry.
In vivo assay of targeting of DCs by lentivector
The recombinant and concentrated lentivector FUGW/SVGmu (50×106 TU resuspended in 200 μl PBS) was injected subcutaneously into the right flank of the C57BL/6 mice close to an inguinal lymph node (within 1 cm range). The right inguinal lymph node and the equivalent lymph node at the opposite site were isolated for size examination on day 3 post-injection. The cells were harvested from these nodes and their total numbers were counted. The percentage of GFP+ DCs was analyzed by flow cytometry on cells stained with anti-CD11c antibody (BD Biosciences).
In vitro DC stimulation of OT1 and OT2 T cells and functional assays
The day 6 BMDCs were spin-infected with viral supernatant, and cultured for an additional 3 days. On day 9, the non-adherent cells were collected and re-cultured in RPMI medium containing 10% FBS, GM-CSF (1:20 J558L conditional medium), and 1μg/ml LPS (Sigma). On day 10, the cells were collected and used for T cell stimulation. In parallel, non-adherent cells were collected from non-transduced day 9 BMDC culture, and were re-cultured in the same medium (RPMI containing 10% FBS, GM-CSF and LPS). On day 10, the cells were collected and loaded with either OVAp (OVA257-269, recognized by OT1 TCR) or OVAp* (OVA323-339, recognized by OT2 TCR), and used for T cell stimulation. Spleen cells were collected from the OT1 and OT2 transgenic mice and cultured with the lentivector-transduced BMDCs, or BMDCs loaded with either OVAp or OVAp*, at the indicated ratio. Three days later, the supernatant was collected and assayed for IFN-γ production using ELISA and the cells were collected and analyzed for their surface activation markers using flow cytometry. T cell proliferation was assayed using [3H] thymidine incorporation.
In vivo immunization of naïve mice
Wild-type B6 mice or albino B6 mice were given a single injection of targeting lentivector subcutaneously on the right flank at the indicated dose. On day 7 and day 14 post-immunization, blood was collected from the immunized mice through tail bleeding, and the serum anti-OVA IgG was measured using ELISA. On day 14, spleen and lymph node cells were harvested and analyzed for the presence of OVA-specific T cells and their surface activation markers using flow cytometry.
Tumor challenge study
The tumor cell lines EL4 (C57BL/6J, H-2b, thymoma) and E.G7 (EL4 cells stably expressing one copy of chicken OVA cDNA)42 were used for the tumor challenge of mice. For the tumor protection experiment, B6 mice received a single injection of 50×106 TU of the targeting lentivector on the right flank. Two weeks later, 5×106 EL4 or E.G7 cells were injected subcutaneously into the left flank of the mice. Tumor size was measured every other day using fine calipers and was shown as the product of the two largest perpendicular diameters a × b (mm2). For the tumor eradication experiment, albino B6 mice were injected subcutaneously with 5×106 E.G7.luc cells (E.G7 cells stably expressing a firefly luciferase) on the left flank. On day 3 and day 10 post-tumor-challenge, the mice received 50×106 TU of targeting lentivector through subcutaneous injection on the right flank. The tumor growth was monitored using bioluminescence imaging. In both experiments, the mice were killed when the tumors reached 400mm2.
Supplementary Material
Note: Supplementary information is available on the Nature Biotechnology website.
Acknowledgments
We are grateful to James Strauss for providing reagents and April Tai for a critical reading of the manuscript. This work was supported by a grant from the National Institute of Health (R01AI068978), a grant from the Bill and Melinda Gates Foundation through the Grand Challenges in Global Health Initiative, and by the Skirball Foundation.
Footnotes
COMPETING INTERESTS STATEMENT: The authors declare that they have no competing financial interests.
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 4.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 5.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15:138–147. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 6.Ribas A, Butterfield LH, Glaspy JA, Economou JS. Cancer immunotherapy using gene-modified dendritic cells. Curr Gene Ther. 2002;2:57–78. doi: 10.2174/1566523023348129. [DOI] [PubMed] [Google Scholar]
- 7.Kirk CJ, Mule JJ. Gene-modified dendritic cells for use in tumor vaccines. Hum Gene Ther. 2000;11:797–806. doi: 10.1089/10430340050015419. [DOI] [PubMed] [Google Scholar]
- 8.Song W, Tong Y, Carpenter H, Kong HL, Crystal RG. Persistent, antigen-specific, therapeutic antitumor immunity by dendritic cells genetically modified with an adenoviral vector to express a model tumor antigen. Gene Ther. 2000;7:2080–2086. doi: 10.1038/sj.gt.3301336. [DOI] [PubMed] [Google Scholar]
- 9.Gong J, et al. Induction of antigen-specific antitumor immunity with adenovirus-transduced dendritic cells. Gene Ther. 1997;4:1023–1028. doi: 10.1038/sj.gt.3300496. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan JM, et al. Induction of antitumor immunity with dendritic cells transduced with adenovirus vector-encoding endogenous tumor-associated antigens. J Immunol. 1999;163:699–707. [PubMed] [Google Scholar]
- 11.Meyer zum Buschenfelde C, Nicklisch N, Rose-John S, Peschel C, Bernhard H. Generation of tumor-reactive CTL against the tumor-associated antigen HER2 using retrovirally transduced dendritic cells derived from CD34+ hemopoietic progenitor cells. J Immunol. 2000;165:4133–4140. doi: 10.4049/jimmunol.165.7.4133. [DOI] [PubMed] [Google Scholar]
- 12.Temme A, et al. Efficient transduction and long-term retroviral expression of the melanoma-associated tumor antigen tyrosinase in CD34(+) cord blood-derived dendritic cells. Gene Ther. 2002;9:1551–1560. doi: 10.1038/sj.gt.3301821. [DOI] [PubMed] [Google Scholar]
- 13.Mangeot PE, et al. Development of minimal lentivirus vectors derived from simian immunodeficiency virus (SIVmac251) and their use for gene transfer into human dendritic cells. J Virol. 2000;74:8307–8315. doi: 10.1128/jvi.74.18.8307-8315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroers R, Chen SY. Lentiviral transduction of human dendritic cells. Meth Mol Biol. 2004;246:451–459. doi: 10.1385/1-59259-650-9:451. [DOI] [PubMed] [Google Scholar]
- 15.Esslinger C, Romero P, MacDonald HR. Efficient transduction of dendritic cells and induction of a T-cell response by third-generation lentivectors. Hum Gene Ther. 2002;13:1091–1100. doi: 10.1089/104303402753812494. [DOI] [PubMed] [Google Scholar]
- 16.Barouch DH, Nabel GJ. Adenovirus vector-based vaccines for human immunodeficiency virus type 1. Hum Gene Ther. 2005;16:149–156. doi: 10.1089/hum.2005.16.149. [DOI] [PubMed] [Google Scholar]
- 17.Shiver JW, Emini EA. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu Rev Med. 2004;55:355–372. doi: 10.1146/annurev.med.55.091902.104344. [DOI] [PubMed] [Google Scholar]
- 18.Tatsis N, Ertl HC. Adenoviruses as vaccine vectors. Mol Ther. 2004;10:616–629. doi: 10.1016/j.ymthe.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breckpot K, Aerts JL, Thielemans K. Lentiviral vectors for cancer immunotherapy: transforming infectious particles into therapeutics. Gene Ther. 2007;14:847–862. doi: 10.1038/sj.gt.3302947. [DOI] [PubMed] [Google Scholar]
- 20.Esslinger C, et al. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J Clin Invest. 2003;111:1673–1681. doi: 10.1172/JCI17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JH, et al. Induction of therapeutic antitumor immunity by in vivo administration of a lentiviral vaccine. Hum Gene Ther. 2005;16:1255–1266. doi: 10.1089/hum.2005.16.1255. [DOI] [PubMed] [Google Scholar]
- 22.Dullaers M, et al. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther. 2006;13:630–640. doi: 10.1038/sj.gt.3302697. [DOI] [PubMed] [Google Scholar]
- 23.Cheng C, et al. Mechanism of ad5 vaccine immunity and toxicity: fiber shaft targeting of dendritic cells. PLoS Pathog. 2007;3:e25. doi: 10.1371/journal.ppat.0030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Gruijl TD, et al. Prolonged maturation and enhanced transduction of dendritic cells migrated from human skin explants after in situ delivery of CD40-targeted adenoviral vectors. J Immunol. 2002;169:5322–5331. doi: 10.4049/jimmunol.169.9.5322. [DOI] [PubMed] [Google Scholar]
- 25.Belousova N, et al. Genetically targeted adenovirus vector directed to CD40-expressing cells. J Virol. 2003;77:11367–11377. doi: 10.1128/JVI.77.21.11367-11377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonifaz L, et al. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. 2002;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonifaz LC, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199:815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trumpfheller C, et al. Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J Exp Med. 2006;203:607–617. doi: 10.1084/jem.20052005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 30.Dakappagari N, et al. Internalizing antibodies to the C-type lectins, L-SIGN and DC-SIGN, inhibit viral glycoprotein binding and deliver antigen to human dendritic cells for the induction of T cell responses. J Immunol. 2006;176:426–440. doi: 10.4049/jimmunol.176.1.426. [DOI] [PubMed] [Google Scholar]
- 31.Geijtenbeek TB, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 32.Geijtenbeek TB, et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 2000;100:575–585. doi: 10.1016/s0092-8674(00)80693-5. [DOI] [PubMed] [Google Scholar]
- 33.Geijtenbeek TB, van Vliet SJ, Engering A, t Hart BA, van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- 34.Klimstra WB, Nangle EM, Smith MS, Yurochko AD, Ryman KD. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J Virol. 2003;77:12022–12032. doi: 10.1128/JVI.77.22.12022-12032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strauss JH, Wang KS, Schmaljohn AL, Kuhn RJ, Strauss EG. Host-cell receptors for Sindbis virus. Arch Virol. 1994;9:473–484. doi: 10.1007/978-3-7091-9326-6_46. [DOI] [PubMed] [Google Scholar]
- 36.Byrnes AP, Griffin DE. Binding of Sindbis virus to cell surface heparan sulfate. J Virol. 1998;72:7349–7356. doi: 10.1128/jvi.72.9.7349-7356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morizono K, et al. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat Med. 2005;11:346–352. doi: 10.1038/nm1192. [DOI] [PubMed] [Google Scholar]
- 38.Yang L, Bailey L, Baltimore D, Wang P. Targeting lentiviral vectors to specific cell types in vivo. Proc Natl Acad Sci USA. 2006;103:11479–11484. doi: 10.1073/pnas.0604993103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 40.Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 41.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 42.Yang L, Baltimore D. Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proc Natl Acad Sci USA. 2005;102:4518–4523. doi: 10.1073/pnas.0500600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou T, Chen Y, Hao L, Zhang Y. DC-SIGN and immunoregulation. Cell Mol Immunol. 2006;3:279–283. [PubMed] [Google Scholar]
- 44.Matsuno K, Ezaki T, Kudo S, Uehara Y. A life stage of particle-laden rat dendritic cells in vivo: their terminal division, active phagocytosis, and translocation from the liver to the draining lymph. J Exp Med. 1996;183:1865–1878. doi: 10.1084/jem.183.4.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22:1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 46.Park D, Lapteva N, Seethammagari M, Slawin KM, Spencer DM. An essential role for Akt1 in dendritic cell function and tumor immunotherapy. Nat Biotechnol. 2006;24:1581–1590. doi: 10.1038/nbt1262. [DOI] [PubMed] [Google Scholar]
- 47.Takahara K, et al. Functional comparison of the mouse DC-SIGN, SIGNR1, SIGNR3 and Langerin, C-type lectins. Int Immunol. 2004;16:819–829. doi: 10.1093/intimm/dxh084. [DOI] [PubMed] [Google Scholar]
- 48.Powlesland AS, et al. Widely divergent biochemical properties of the complete set of mouse DC-SIGN-related proteins. J Biol Chem. 2006;281:20440–20449. doi: 10.1074/jbc.M601925200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Biotechnology website.