

Abstract
The individual steps in single-nucleotide base excision repair (SN-BER) are coordinated to enable efficient repair without accumulation of cytotoxic DNA intermediates. The DNA transactions and various proteins involved in SN-BER of abasic sites are well known in mammalian systems. Yet, despite a wealth of information on SN-BER, the mechanism of step-by-step coordination is poorly understood. In this study we conducted experiments toward understanding step-by-step coordination during BER by comparing DNA binding specificities of two major human SN-BER enzymes, apurinic/aprymidinic endonuclease 1 (APE) and DNA polymerase β (Pol β). It is known that these enzymes do not form a stable complex in solution. For each enzyme, we found that DNA binding specificity appeared sufficient to explain the sequential processing of BER intermediates. In addition, however, we identified at higher enzyme concentrations a ternary complex of APE·Pol β·DNA that formed specifically at BER intermediates containing a 5′-deoxyribose phosphate group. Formation of this ternary complex was associated with slightly stronger Pol β gap-filling and much stronger 5′-deoxyribose phosphate lyase activities than was observed with the Pol β·DNA binary complex. These results indicate that step-by-step coordination in SN-BER can rely on DNA binding specificity inherent in APE and Pol β, although coordination also may be facilitated by APE·Pol β·DNA ternary complex formation with appropriate enzyme expression levels or enzyme recruitment to sites of repair.
The single base lesion is the most common form of DNA damage occurring in the human genome. A DNA base can be lost through spontaneous hydrolysis (1) or can be oxidized (2, 3) and/or alkylated (4, 5) during physiologic metabolism and can be modified by exogenous DNA damaging agents (6). Cumulatively, endogenous sources of DNA damage lead to a high frequency of base lesions in the cell (5). Accumulation of base damage, especially oxidized bases, in the human genome has been implicated in a variety of common disorders, including cancer (7-10), neurodegenerative disease (11), atherosclerosis (12), and aging (13). In addition, an increase in oxidized bases has been observed as a function of aging in the promoter regions of genes involved in memory and learning (14).
To combat adverse effects of DNA base damage and preserve the integrity of their genome, cells maintain a complex base excision repair (BER)2 system. Two major BER subpathways have been identified thus far based on the total number of nucleotides that are replaced in the damaged strand during the repair process. One subpathway involving replacement of just one nucleotide is called single-nucleotide BER (SN-BER), whereas the subpathway involving replacement of two or more nucleotides in the damaged strand is termed long-patch BER (LP-BER). Typically, SN-BER is initiated by spontaneous base loss or a DNA glycosylase that cleaves the N-glycosidic bond of the damaged base resulting in an abasic site in double-stranded DNA (5, 15). Subsequently, apurinic/apyrimidinic endonuclease 1 (APE) incises the damaged strand at the 5′-side of the abasic site, generating a 1-nt gap with 3′-hydroxyl and 5′-deoxyribose phosphate (dRP) groups at the margins (16, 17). DNA polymerase β (Pol β) then fills the gap and removes the dRP group with its intrinsic dRP lyase activity (18-20). The strand integrity of the resulting nicked DNA intermediate is finally restored by a DNA ligase. Alternatively, if the dRP group is altered by oxidation or reduction and cannot be removed by the lyase, repair will be shunted into the LP-BER subpathway involving the replacement of 2-12 nucleotides of the damaged strand (21, 22). In this subpathway, Pol β performs strand-displacement synthesis to create a multinucleotide flap containing the 5′-dRP group that is subsequently cleaved by flap endonuclease 1. Alternately, flap endonuclease 1 removes a single-nucleotide with the 5′-dRP group via a “hit and run” mechanism that involves Pol β-dependent single-nucleotide gap-filling reactions (23).
Studies in vivo and in vitro have suggested that SN-BER accounts for most of the BER activity in mammalian cells (24-28). Efficient SN-BER is accomplished by the coordination of a series of enzymatic reactions allowing hand-off of the various BER intermediates from one enzyme to another (29). It seems likely that the efficiency of SN-BER could be enhanced through specific protein interactions with other proteins and/or sub-strates, and a large number of such interactions have been reported for BER proteins in solution. These include interactions of various BER enzymes with accessory proteins, such as x-ray repair cross-complementing 1 (30-33), p53 (34), and proliferating cell nuclear antigen (35). Because of the diversity of the proteins and DNA intermediates occurring during the multiple sequential steps of SN-BER and the macromolecular interactions involved, the mechanistic aspects of how these steps are coordinated are poorly understood.
The enzymes and accessory factors involved in the BER subpathways in mammalian cells have received considerable attention. As summarized above, five distinct enzymatic reaction are involved during SN-BER. These are 1) removal of a modified base by a lesion-specific monofunctional DNA N-glycosylase, 2) 5′-incision of the abasic site by a hydrolytic strand incision enzyme, 3) DNA synthesis by a nucleotidyltransferase, 4) removal of the 5′-dRP group by a β-elimination reaction, and 5) nick sealing by DNA ligase (36, 37). Specific enzymes conducting each reaction were discovered through biochemical and genetic approaches along with comparative analysis in a number of biological systems. These DNA enzymes share a common feature in that they must all recognize a DNA substrate and release the product DNA after the enzymatic reaction to provide the substrate for the next step. Therefore, the ability to conduct a DNA binding step that differentiates a substrate molecule from a product molecule or normal DNA, i.e. substrate binding specificity, is an important task for all of these enzymes. Because BER intermediates are toxic (28, 38), a situation where an intermediate is inefficiently processed or allowed to accumulate can trigger the cellular DNA damage recognition system leading to cell cycle arrest and/or cell death. Hence, masking of BER intermediates from the damage surveillance machinery must be a fundamental property of BER. Information on the mechanisms masking BER intermediates and the coordination of the steps of BER is limited. Yet, it has been proposed that protein-protein interactions between the repair enzymes may facilitate the coordination among sequential steps thereby enhancing the efficiency of the overall SN-BER process and masking the intermediates from the DNA damage surveillance system (29).
Bennett et al. (39) reported an interaction between APE and Pol β on a single-nucleotide gapped BER intermediate. They found that this interaction stimulated the dRP lyase activity of Pol β, and Wong and Demple (40) found that the polymerase activity of Pol β was decreased through the ability of APE to bind its strand incision product. These results suggest that protein complex-DNA interactions during BER may occur on certain DNA intermediates, and this feature may enhance substrate binding specificity to enable a more efficient coordination of SN-BER steps. Sokhansanj et al. (41) conducted a mathematical simulation of BER using reported values for enzyme reaction rates and Km for substrate DNA. The analysis predicted that SN-BER dominates total BER, composed of SN-BER and LP-BER, and Pol β regulates overall BER throughput. Additionally, it suggested that BER is more efficient than expected from the reported kinetic constants for the known BER enzymes. These kinetic values were taken from available steady-state measurements rather than actual binding constants for the individual reactions. This is an important consideration since Michaelis constants of DNA enzymes often reflect the contribution from multiple reaction steps. Clearly, comparative DNA binding analysis of BER enzymes conducted in a dedicated fashion should further inform modeling efforts to simulate cellular repair pathways.
The aim of the present study was to initiate systematic efforts toward understanding substrate dependent step-by-step coordination in BER as well as protein-protein interactions and their effects on the sequential coordination of the SN-BER subpathway. We used oligonucleotides representing various intermediates in BER of the abasic site lesion with purified human APE and Pol β. The relative binding affinity of the enzymes for various BER intermediates was measured, and formation of a multiprotein-DNA complex was identified with a specific DNA substrate by native gel electrophoresis. Our results indicate that DNA binding specificity of APE and Pol β can determine the orchestrated coordination of the sequential steps of SN-BER, whereas a multiprotein-DNA complex may further facilitate coordination. A model on the roles of DNA binding specificity and DNA-specific protein-protein interactions in step-by-step coordination of SN-BER is discussed.
EXPERIMENTAL PROCEDURES
Materials
DNA oligonucleotides were from Integrated DNA Technologies (IDT; Coralville, IA). The [γ-32P]ATP (7000 mCi/mmol) used in labeling was from MP Biomedicals (Irvine, CA), and [α-32P]dideoxy-ATP (3000 mCi/mmol) was from Amersham Biosciences-GE Healthcare. Optikinase and terminal deoxynucleotidyltransferase were from USB Corp. (Cleveland, OH) and Fermentas Inc. (Hanover, MD), respectively. Deoxynucleotides were from Roche Applied Science. SuperSignal West Pico Chemiluminescent substrate was from Pierce. All other reagents were from Sigma-Aldrich.
Enzymes and Antibodies
The expression and purification of human APE and Pol β were as described previously (42, 43). The concentration of each BER enzyme was measured by absorbance at 280 nm (44). The extinction coefficients used for APE and Pol β were 55,550 and 21,170 m-1 cm-1, respectively. The rabbit polyclonal antibodies against APE and Pol β were IgG preparations and were subjected to affinity purification.
Oligonucleotide Substrates
DNA oligonucleotides were designed to mimic the structures of SN-BER intermediates. These included a double-stranded DNA with an internal tetrahydrofuran (THF), 1-nucleotide (nt) gap with a 5′-THF group, nicked DNA with a 5′-THF group, 1-nt-gapped DNA with a 5′-phosphate, nicked DNA, and double-stranded DNA as a control. The sequences of these oligonucleotides are given in Table 1. A double-stranded DNA substrate with an abasic site was created by annealing an oligonucleotide with a THF residue to a 31-mer template. For the other substrates, each was constructed by annealing an upstream and downstream oligonucleotide with a template. The substrate was radiolabeled at the 3′-end of the damaged strand or downstream oligonucleotide with [α-32P]dideoxy-ATP and terminal deoxynucleotidyltransferase. In some cases, the 5′-end of the upstream oligonucleotide was labeled with [γ-32P]ATP and Optikinase. The unincorporated [α-32P]dideoxy-ATP or [γ-32P]ATP was removed with a Bio-Rad P6 spin column. The radiolabeled oligonucleotides for native gel electrophoresis assay were purified by 12% polyacrylamide, 7 m urea denaturing gel electrophoresis. A radiolabeled oligonucleotide was then annealed to a template at a molar ratio of 1.5:1 to create a double-stranded DNA substrate with an intact abasic site or was annealed to a template with a corresponding upstream oligonucleotide at a molar ratio of 1.5:1.5:1.
TABLE 1.
Oligonucleotide sequences
| Oligonucleotide | Length, nt | Sequencea |
|---|---|---|
| Double-stranded (DS) | ||
| DS1 | 31 | 5′-CTGCAGCTGATGCGCFGTGCGGATCCGGTGC-3′ |
| DS2 | 31 | 5′-CTGCAGCTGATGCGCCGTGCGGATCCGGTGC-3′ |
| Downstream (D) | ||
| DdRPb | 16 | 5′-pUGTGCGGATCCGGTGC-3′ |
| Dgap/nick | 15 | 5′-pGTGCGGATCCGGTGC-3′ |
| DTHF flap | 15 | 5′-pFGTGCGGATCCGGTGC-3′ |
| Template (T) | ||
| T | 31 | 3′-GACGTCGACTACGCGGCACGCCTAGGCCACG-5′ |
| Upstream (U)c | ||
| Ugap | 15 | 5′-CTGCAGCTGATGCGC-3′ |
| Unick | 16 | 5′-CTGCAGCTGATGCGCC-3′ |
| U3′-mismatched-nick | 16 | 5′-CTGCAGCTGATGCGCT-3′ |
Unannealed residues are in boldface. F denotes THF, and U denotes deoxyuridine. p stands for a phosphate group.
The subscript designations refer to the final annealed BER intermediates. For DdRP, a downstream 5′-terminal deoxyuridine residue, is employed as a substrate for uracil DNA glycosylase, which will ultimately generate a 3′-dRP residue. For example, Dgap/nick generates a 1-nt-gapped or nicked DNA.
The subscripts describe the annealed BER intermediates. Ugap is the upstream oligonucleotide for generating a 1-nt-gapped DNA, whereas Unick generates nicked DNA.
Native Gel Electrophoresis Assay
Apparent substrate and product binding affinities for APE and Pol β were measured by a gel mobility shift assay. The enzymes and DNA were mixed in a buffer containing 50 mm Tris-HCl, pH 7.5, 50 mm KCl, 0.1 mg/ml bovine serum albumin, 0.5 mm EDTA, and 0.01% Non-idet P-40 in the presence or absence of magnesium. After incubation for 8 min at 37 °C, the enzyme-DNA complexes were separated from the free DNA by 1% agarose, 0.1% polyacrylamide gel electrophoresis at 4 °C as described previously (23). The apparent Kd for DNA binding was determined as described previously (23).
Identification of Ternary Complex Containing APE·Pol β·DNA
Experiments to identify the APE·Pol β·DNA ternary complex were performed by incubating various concentrations of APE (0.5-5 nm) and 1nm Pol β with 5 nm DNA with a 5′-THF flap. The conditions for the enzyme-DNA incubation were the same as described above for native gel electrophoresis mobility shift assay. Affinity-purified IgG antibodies against APE (∼0.03 μg/μl) and Pol β (∼0.05 μg/μl) were employed to identify any requirement for APE or Pol β in ternary complex formation. After incubation with the antibodies, the protein-DNA mixture was subject to native agarose-polyacrylamide gel electrophoresis at 4 °C for 2 h, permitting separation of the antibody-enzyme-DNA complex from the enzyme-DNA complex and free DNA. The DNA-protein complexes were measured by determining the amount of labeled DNA with phosphorimaging, as described previously (23). The APE and Pol β in the DNA-protein ternary complex was also probed by immunoblotting. This experiment was performed in two separate steps. In the first step, the protein-DNA complex was separated in a native agarose-polyacrylamide gel at 4 °C for 2 h. Then the gel was laid on the top of a methanol-activated PVDF membrane under which DEAE paper was placed. The gel was immediately vacuum-dried at 80 °C for 1 h. The DNA-protein ternary complex was then detected by phosphor-imagery. In the second step, the PVDF membrane (attached to the vacuum-dried gel) was soaked in washing buffer (20 mm Tris-HCl, pH 7.5, 50 mm NaCl, and 5% Tween 20) with shaking for 1 h. The gel was then carefully separated from the PVDF membrane. The membrane was hydrated again with methanol and subsequently subjected to overnight immunoblotting at 4 °C with anti-APE or anti-Pol β polyclonal antibody (0.3 μg/ml). After washing, the membrane was incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG for 1 h. After several washes, APE and Pol β were detected by enhanced chemiluminescence with Kodak x-ray film.
Enzymatic Activity Assays
The dRP lyase activity of Pol β was determined by measuring the removal of a 5′-dRP group. The 5′-dRP residue was enzymatically produced by Escherichia coli uracil-DNA glycosylase excision of a 5′-uracil-containing oligonucleotide that had been annealed as part of a downstream oligonucleotide. This generates a 1-nt-gapped, nicked, or 3′-upstream mismatched, nicked DNA molecule. Each of these substrates was radiolabeled at the 3′-end of the downstream oligonucleotide and incubated with 20 nm uracil-DNA glycosylase in a reaction buffer containing 50 mm HEPES, pH 7.5, 20 mm KCl, 0.5 mm EDTA and 1 mm dithiothreitol at 37 °C for 15 min. The resulting DNA with a 5′-dRP group was then incubated with 5 nm Pol β in the absence or presence of increasing concentrations of APE (10, 25, 50 nm) at 30 °C for 15 min. The uncleaved dRP group was trapped by the addition of 200 nm NaBH4 and incubated on ice for 30 min. The substrate and product were then ethanol-precipitated, dissolved in gel loading buffer, separated by electrophoresis in a denaturing 15% polyacrylamide gel, and detected by phosphor-imagery as described previously (45).
APE 3′-exonuclease activity was examined by measuring the removal of a 3′-mismatch (T/G; primer/template) in a 5′-radiolabeled upstream oligonucleotide substrate. APE (5 nm) was incubated with 10 nm substrate in the absence or presence of various concentrations of Pol β (10, 25, 50 nm) at 37 °C for 10 min. The reactions were performed in a buffer containing 50 mm Tris HCl, pH 7.5, 50 mm KCl, 5 mm MgCl2, 0.1 mg/ml bovine serum albumin, and 0.1 mm EDTA. The substrate and product were then quantified as described above after denaturing 15% polyacrylamide gel electrophoresis.
Pol β DNA synthesis activity was measured as described previously (23). Briefly, 1 nm Pol β was incubated with a 1-nt-gapped substrate with a 5′-THF flap in the absence and presence of 25 nm APE with the buffer conditions described above. The reaction was initiated with 5 mm MgCl2 and incubated at 37 °C. Aliquots (10 μl) were removed at timed intervals (30-150 s), and the reaction was stopped by the addition of 250 mm EDTA. The substrates and products were separated by 15% denaturing polyacrylamide gel electrophoresis as described above.
RESULTS
Relative DNA Binding Affinities of APE and Pol β
The apparent dissociation constant (Kd,app) of each enzyme for binding to its canonical substrate and product was measured under native conditions using agarose-acrylamide gel electrophoresis. Fig. 1 illustrates typical results of APE and Pol β binding to the abasic site containing DNA and to a 1-nt-gapped BER intermediate with a 5′-dRP group at the margin of the gap. These two substrates mimic BER intermediates resulting from spontaneous base loss or removal of a base by a monofunctional DNA glycosylase and 5′-incision of the abasic site by APE, respectively. The results indicate that the concentration dependence of both APE and Pol β for interaction with these BER intermediates yielded hyperbolic binding curves (Fig. 1).
FIGURE 1. Substrate DNA binding by APE and Pol β.
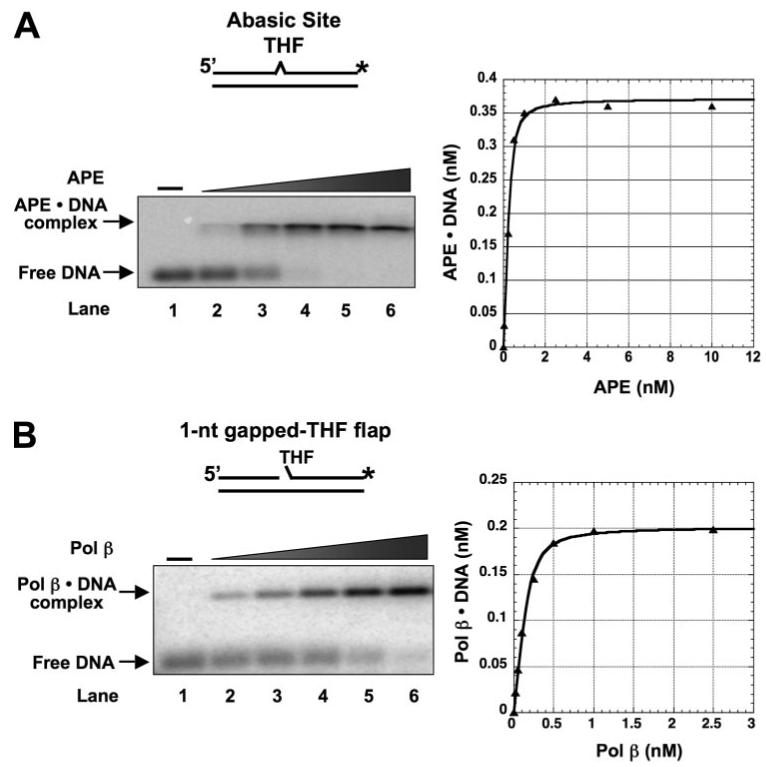
A, a double-stranded DNA with an internal THF residue opposite G in the template strand was employed to measure the binding affinity of APE. The substrate was made by annealing a DNA strand with THF (DS1) to a template strand (T) (see Table 1). The substrate was radiolabeled at the 3′-end of the THF-containing strand (asterisk). Various concentrations of APE (0.05, 0.1, 0.5, 1, and 2.5 nm) were incubated with 0.5 nm substrate at 37 °C in the absence of Mg2+. B, a 1-nt-gapped THF flap substrate was used to examine Pol β substrate binding. The substrate was constructed by annealing an upstream primer (Ugap), a downstream oligomer (DTHF-flap) to the template strand (T). The substrate was radiolabeled at the 3′-end of the downstream oligonucleotide. Pol β (0.05, 0.1, 0.2, 0.25, and 0.5 nm) was incubated with 0.5 nm substrate at 37 °C for 8 min. After incubation, the protein-DNA complex was separated from free DNA by agarose-acrylamide gel electrophoresis as described under “Experimental Procedures.” The binding curves of the enzymes are shown in the right panels. The DNA substrates are schematically illustrated above each gel photograph.
With the DNA containing an intact abasic site, increasing amounts of DNA·APE complex were observed as a function of increasing APE concentration; DNA at 0.5 nm was almost completely bound at an equal concentration of APE (Fig. 1A, lane 4). Likewise, Pol β formed a complex with the 1-nt-gapped DNA (Fig. 1B), and complex formation followed a hyperbolic binding isotherm. Table 2 summarizes the apparent dissociation constants for APE and Pol β binding to these and other related DNA ligands. Among the various DNAs tested, APE bound with highest affinity to the abasic site DNA followed by a 1-nt-gapped THF flap, nicked-THF flap, 1-nt-gapped DNA, and 3′-mismatched nicked THF flap, whereas APE binding to nicked DNA and the double-stranded DNA control was the weakest. Thus, APE bound its initial BER substrate, i.e. the abasic site DNA, with highest affinity but also bound with relatively high affinity to other BER intermediates with a 5′-dRP group. In contrast, Pol β bound the abasic site containing DNA weakly but bound the 1-nt-gapped dRP-flap DNA and 1-nt-gapped DNA with highest affinity. The plot in Fig. 2 summarizes the results of this comparative substrate and product binding of APE and Pol β to various BER intermediates. Both enzymes bound the dRP-containing BER intermediates with similar affinities. Pol β binding to the DNAs representing a gap-filling reaction product, the nicked-THF flap and nicked DNA, was weaker than for the two gapped DNAs (Table 2). These results on relative substrate binding affinity for APE and Pol β are consistent with their proposed roles in SN-BER, where APE binds to the abasic site and incises the strand creating a 1-nt-gapped dRP flap intermediate that is a preferred substrate for Pol β binding. After Pol β fills the gap and releases the reaction product, it again binds to the dRP group-containing intermediate and removes the dRP-flap to generate nicked DNA. Interestingly, these results suggest that enzyme coordination during these early steps of the BER pathway could be explained by the intrinsic enzyme-substrate/product binding affinities at each step. Nevertheless, APE and Pol β exhibited similar binding affinities for the two intermediates with a dRP-flap, as illustrated by the highlighted (i.e. circled) data points in Fig. 2. This suggested that APE and Pol β either compete with each other for these intermediates or bind to the intermediates simultaneously, perhaps through a protein-protein interaction.
TABLE 2.
Affinities of APE and Pol β for BER Intermediate DNA Substrates
| BER Steps | BER Substrates |
Kd,app(nM) |
APE·Pol β·DNA Ternary Complex | |
|---|---|---|---|---|
| APE | Pol β | |||
| 2.9 (0.3)a | >50 | |||
| 1 | 0.04 (0.02) | 3.3 (0.7) | - | |
| 2 | 0.2 (0.04) | 0.1 (0.03) | + | |
| 3 | 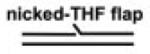 |
0.3 (0.08) | 0.5 (0.01) | + |
| 4 |  |
4.3 (1.1) | 0.4 (0.08) | - |
| 0.5 (0.02) | 0.05 (0.01) | - | ||
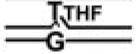 |
0.6 (0.1) | 0.4 (0.03) | + | |
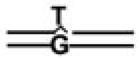 |
0.7 (0.03) | 0.1 (0.01) | - | |
The S.D. are shown in the parentheses
FIGURE 2. Binding affinity of APE and Pol β to various SN-BER intermediates.
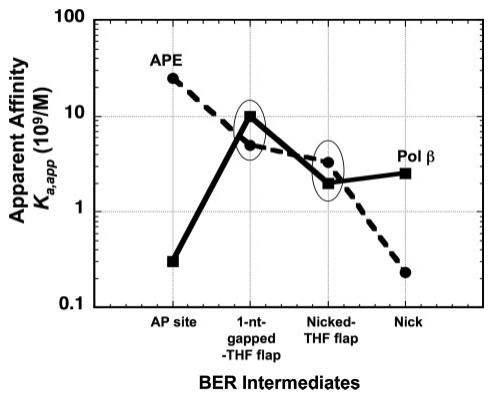
The apparent binding affinities (Ka,app; 1/Kd,app) of APE and Pol β to a double-strand DNA with an abasic site, a 1-nt-gapped THF flap, a nicked-THF flap, and a nicked DNA (Table 2) were plotted as a function of the sequential BER intermediate steps during simple SN-BER. The oval circles highlight the intermediates to which Pol β and APE exhibit similar apparent binding affinities.
To further explore the implications of the similar binding affinities for the BER intermediate(s) with a 5′-dRP group, we examined binding using the nicked-dRP flap. In these experiments low concentrations (<1nm) of APE and Pol β were used. The concentration of each enzyme was varied in the presence of a constant concentration of the other enzyme. Typical results are illustrated in Fig. 3. Increasing concentrations of APE reduced the amount of Pol β·DNA complex, and a reciprocal titration with increasing concentrations of Pol β had a similar effect on APE·DNA complex formation. However, when equal concentrations of APE and Pol β were titrated, slightly more APE·DNA complex was formed relative to Pol β·DNA complex. Thus, the enzymes competed with each other for binding to this BER intermediate, and the results were in agreement with the slightly stronger APE binding affinity noted in Table 2. These results may explain the APE inhibition of Pol β gap-filling observed by Wong and Demple (40) on a substrate with a dRP group.
FIGURE 3. APE and Pol β compete for a nicked-THF flap BER intermediate.
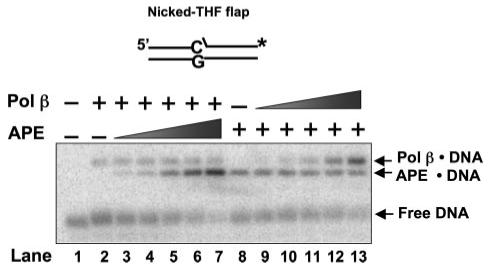
Increasing concentrations of APE (lanes 3-7) and Pol β (lanes 9-13) were incubated with 0.25 nm Pol β or 0.25 nm APE and with 0.5 nm substrate DNA. The binding assay was performed with the conditions described under “Experimental Procedures.” Lanes 3-7 include 0.05, 0.1, 0.25, 0.5, and 1 nm APE along with 0.25 nm Pol β, whereas lanes 9-13 include the same concentrations of Pol β along with 0.25 nm APE. Lane 2 was the mixture that contained only Pol β and substrate DNA, whereas lane 8 was the mixture that contained APE and substrate DNA. Lane 1 is substrate DNA alone. The substrate is schematically illustrated above the gel.
Pol β and APE Protein-Protein Interaction on BER Intermediates
It had been reported in mammalian cell systems that the concentration of APE was higher than that of Pol β (46). Our direct measurements in the present study using a quantitative immunoblotting assay supported these observations and indicated that the cellular level of APE was about 3-fold higher than Pol β (data not shown). Yet, components of the mammalian BER machinery are recruited to sites of DNA damage (47), and this will result in higher local concentrations of APE and Pol β at a lesion than the total absolute level in the nucleoplasm. Therefore, we chose to further examine binding of APE and Pol β to various BER intermediates using higher enzyme concentrations. The results in Fig. 4 illustrate enzyme-DNA complex formation with the nicked-dRP flap DNA and various concentrations of Pol β and APE. Under these conditions (1 nm APE and 2.5 nm Pol β), a novel complex migrating slower than the Pol β·DNA binary complex was observed (Fig. 4A, lane 8). This complex was not detected with incubations containing DNA, Pol β, or APE alone (Fig. 4A, lanes 1, 2, and 5). In the presence of 1 nm Pol β and increasing concentrations of APE, the slower migrating complex again was detected when APE concentration reached 5 nm (Fig. 4B, lane 10); this complex was not observed with APE concentrations lower than 5 nm (Fig. 4B, lanes 7-9) or with DNA or APE alone (Fig. 4B, lanes 1 and 6). Thus, both APE and Pol β were required for detection of this slower migrating complex. We suspected that this complex represented a ternary complex containing APE and Pol β along with the labeled DNA (noted in Fig. 4), and this observation is consistent with an APE·Pol β complex reported by Bennett et al. (39).
FIGURE 4. Formation of APE·Pol β·DNA ternary complex.
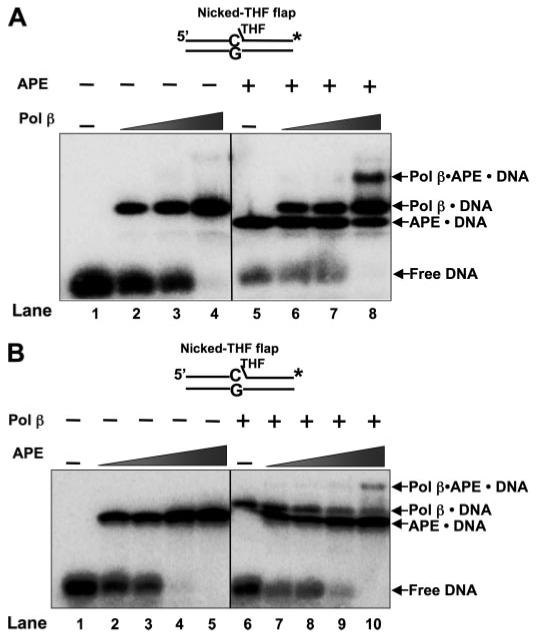
A, increasing concentrations of Pol β were incubated with 5 nm nicked-THF flap substrate DNA in the absence (lanes 2-4) and presence of 1 nm APE (lanes 6-8). Lane 1 represents the binding mixture without enzyme. Lanes 2-4 correspond to mixtures containing increasing concentrations of Pol β (0.5, 1, and 2.5 nm, respectively) and DNA. Lanes 6-8 indicate mixtures containing the same concentrations of Pol β as lanes 2-4 along with 1 nm APE. Lane 5 indicates a binding mixture with APE and the DNA substrate. B, increasing concentrations of APE (0.5, 1, 2.5, and 5 nm, respectively) were incubated with 5 nm nicked-THF flap substrate DNA in the absence (lanes 2-5) and presence of 1 nm Pol β (lanes 7-10). Lane 1 represents the reaction mixture without enzymes, whereas lane 6 corresponds to the mixture containing Pol β and DNA substrate. The enzymes were incubated with the DNA substrates with the conditions described under “Experimental Procedures,” and the protein-DNA complexes were analyzed. The substrate DNA is schematically illustrated above the gel.
To probe components of the putative ternary complex, we used two immunological approaches (Fig. 5). In the first, specific antibodies against APE or Pol β were included in the incubation mixture. This resulted in elimination of the ternary complex in both cases (Fig. 5A, lanes 5 and 6). The Pol β antibody disrupted the ternary complex, but the APE·DNA binary complex persisted (Fig. 5A, lane 5); the APE antibody also disrupted the ternary complex, and the Pol β·DNA binary complex remained (Fig. 5A, lane 6). In the second approach, we conducted native gel electrophoresis and then transferred the DNA-protein complexes to a PVDF membrane. Immunoblotting was then conducted with anti-APE and anti-Pol β antibodies (Fig. 5B). The putative ternary complex was found immunoreactive with both antibodies (Fig. 5B, lanes 6 and 7). Taken together, these results confirm that both APE and Pol β were present in the complex. Next, we evaluated DNA structural requirements for ternary complex formation. In addition to the DNA used in Fig. 5, the ternary complex was formed with the 1-nt-gapped dRP flap DNA and the 3′-mismatched-nicked dRP flap DNA but not detected with the DNAs lacking a 5′-dRP group (Table 2). These results suggest a requirement of a dRP-flap at the margin of the gap for ternary complex formation. Finally, because we wished to study functional implications of the complex in the presence of MgCl2, we examined the effect of including 5 mm MgCl2 in the incubation mixture. No effect on complex formation in the presence of magnesium was found.
FIGURE 5. Identification of components of the APE·Pol β·DNA ternary complex.
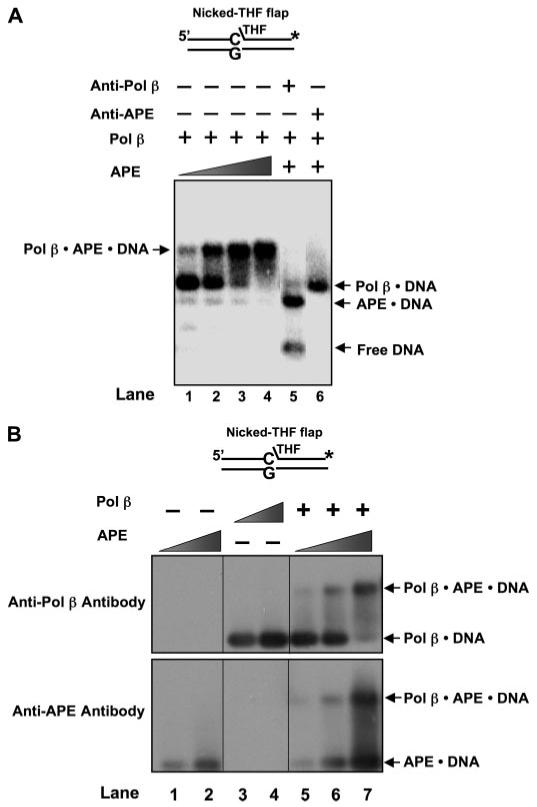
The co-existence of APE and Pol β in the ternary complex was probed by using specific polyclonal antibody against either APE or Pol β and immunoblotting. A, each antibody was incubated with the enzymes and 5 nm nicked-THF flap substrate with the conditions described under “Experimental Procedures.” Lanes 1-4 represent mixtures containing various concentrations of APE (1, 2.5, 10, 25 nm, respectively), 1 nm Pol β, and DNA. Lane 5 corresponds to the mixture containing Pol β, APE, the specific antibody against Pol β (∼0.3 μg/μl), and the DNA substrate, whereas lane 6 represents the mixture with APE, Pol β, specific antibody against APE (∼0.3 μg/μl), and the DNA substrate. The substrate is schematically illustrated above the gel. B, the protein-DNA complexes were initially separated from free DNA by electrophoresis and transferred to PVDF membrane by the vacuum-dry procedure, and the membrane was immunoblotted as described under “Experimental Procedures.” The upper panel illustrates immunoblotting results with anti-Pol β antibody, and the lower panel represents the blot with anti-APE antibody. Lanes 1 and 2 are reactions with 10 and 20 nm APE, respectively, whereas lanes 3 and 4 represent mixtures with 10 and 20 nm Pol β, respectively. Lanes 5-7 represent mixtures of 20 nm Pol β and 10, 20, or 30 nm APE, respectively. The substrate is schematically illustrated above the gel.
APE·Pol β·DNA Ternary Complex Enhances Pol β but Not APE Activities
To explore any functional significance of the ternary complex during SN-BER, we first examined the dRP lyase activity of Pol β and the 3′-exonuclease activity of APE under the conditions where most of the respective enzyme was in the ternary complex. The dRP lyase activity of Pol β was much higher (>5-fold) in the ternary complex than in the Pol β·DNA complex (Fig. 6A); under the conditions used in Fig. 6A, virtually all of the Pol β in the incubation mixture was in the APE·Pol β·DNA ternary complex (Fig. 5A, lane 4). The low DNA concentration chosen for this assay provides an estimate for the change in catalytic efficiency for this reaction. The excessive product formation, therefore, underestimates the true impact APE has on the efficiency of the dRP removal. A similar increase in dRP lyase activity was observed with the 3′-mismatched-nicked-dRP flap (Fig. 6B) and 1-nt-gapped dRP flap DNA substrates (data not shown). These results indicate that formation of the APE·Pol β·DNA ternary complex stimulated the Pol β dRP lyase activity. We also examined Pol β gap-filling activity as a function of the presence of the enzyme in the ternary complex. The rate of Pol β1-nt gap filling was ∼2-fold higher for the ternary complex than for the Pol β·DNA binary complex (Fig. 7). Pol β catalytic cycling is partially limited by product release (48). This 2-fold increase in activity appears to be due to a change in the product dissociation rate constant since a singleturnover analysis of the effect of APE on Pol β nucleotide insertion indicated that insertion (without catalytic cycling) is not affected (data not shown).
FIGURE 6. APE·Pol β·DNA ternary complex stimulates Pol β lyase activity.
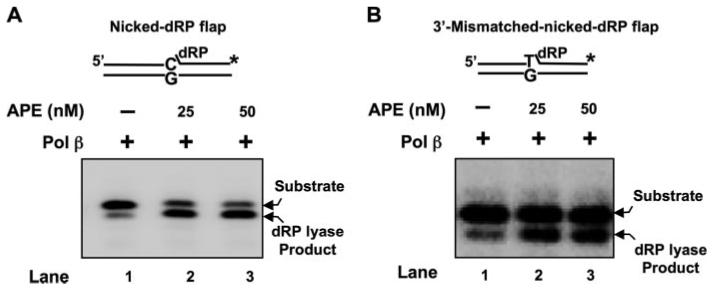
Pol β (2 or 5 nm) was incubated with 10 nm 3′-matched (A) or mismatched (B) nicked-dRP flap substrate in the absence (lane 1) and presence of 25 and 50 nm APE (lanes 2 and 3) at 30 °C for 10 min. The reaction mixtures were incubated and quenched as described under “Experimental Procedures.” The product and substrate were then separated by denaturing polyacrylamide gel electrophoresis and measured as described under “Experimental Procedures.” The nickeddeoxyuridine flap substrate was constructed by annealing the 3′-end radiolabeled (asterisk) downstream oligonucleotide and the upstream primer to the template strand. The dRP lyase substrates were prepared by treating with uracil DNA glycosylase just before performing the lyase assay. The substrate is schematically illustrated above the gel.
FIGURE 7. Stimulation of Pol β 1-nt gap-filling activity by APE·Pol β·DNA ternary complex.
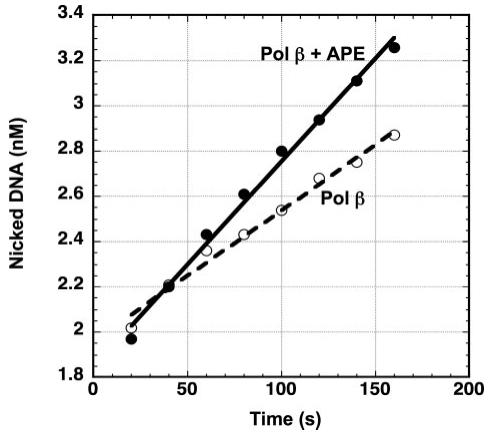
The rate of Pol β 1-nt gap-filling activity was measured with the conditions of APE·Pol β·DNA complex formation as described under “Experimental Procedures.” The reaction was performed in the presence of 1 nm Pol β or 1 nm Pol β and 25 nm APE, 5 mm MgCl2,50 μm dCTP, and 10 nm DNA substrate. Ten-μl aliquots were collected at timed intervals (0.5-5 min) and quenched with 250 mm EDTA. The data were fitted to a linear equation, and the rate was obtained from the slope.
Because formation of the ternary complex was observed with the 3′-mismatched, nicked-dRP flap DNA intermediate of BER (Table 2), we examined the possibility that the ternary complex could modulate the 3′-5′-exonuclease activity of APE. We compared the 3′-exonuclease activity of the ternary complex with that of the APE·DNA binary complex. No significant change in activity was observed (Fig. 8).
FIGURE 8. APE·Pol β·DNA ternary complex does not affect the APE 3′-5′ exonuclease activity.
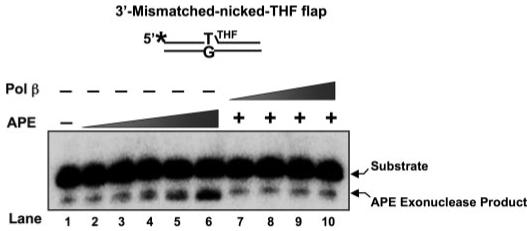
Mismatched-nicked-THF flap substrate (10 nm) was incubated either with increasing concentrations of APE (5, 25, 50 nm) (lanes 2-6, respectively) or with 2.5 nm APE and increasing concentrations of Pol β (5-50 nm) (lanes 7-10, respectively) at 37 °C for 15 min as described under “Experimental Procedures.” Lane 1 was the reaction with only substrate DNA. The product and substrate were separated by denaturing polyacrylamide gel electrophoresis as described under “Experimental Procedures.” The substrate was made by annealing the 5′-end radiolabeled (asterisk) upstream primer and the downstream oligomer to the template. The substrate is schematically illustrated above the gel.
DISCUSSION
To understand the step-by-step coordination of SN-BER, we characterized the binding of APE and Pol β to oligonucleotides representing various BER intermediates. The strongest binding interactions were observed for the enzyme and DNA combinations corresponding to the canonical sequential steps in SN-BER (Table 2, values in bold). This was consistent with the idea that a cell could make use of intrinsic substrate binding properties of these enzymes as a means of coordinating the steps of BER provided the enzymes are expressed in appropriate levels. In this scenario, an enzyme would bind to the appropriate BER intermediate, conduct the enzymatic reaction, and then release the product of the reaction. The affinities measured here appear to be consistent with this type of mechanism of BER coordination, and it is generally considered that the levels of Pol β and APE in the nucleus are sufficient to support such a mechanism.
Interestingly, both APE and Pol β displayed similar binding affinities for BER intermediates with a 5′-dRP group at the margin in the gap, suggesting that the enzymes may compete with each other or, at higher enzyme concentrations, cooperate for interaction with these intermediates. Because the in vivo nuclear concentration of APE can be elevated by transport from the cytoplasm (49, 50), we examined APE and Pol β DNA binding simultaneously under conditions where the APE concentration was greater than that of Pol β. A ternary complex of APE·Pol β·DNA was observed under these conditions. The complex also was observed with other BER intermediates containing the 5′-dRP group, including the product of the APE strand incision reaction at the abasic site (Table 2). Understanding the biological significance of this ternary complex will require further study. Yet, when bound as a component of the ternary complex, Pol β was able to conduct the dRP lyase step of BER considerably more efficiently than was the Pol β·DNA binary complex. The ternary complex also was associated with a slight enhancement in the rate of the Pol β gap-filling activity. Thus, the results suggest that ternary complex formation could have implications for SN-BER since the dRP lyase activity is thought to be a rate-limiting step during BER (20, 28).
Methods for Measuring Kd for Enzyme Binding to DNA Substrates
The robust native gel electrophoresis method used here yielded apparent binding constants for enzyme and substrate DNA interactions that are somewhat lower than values obtained by others each employing a different assay methodology with different salt concentrations (42, 48, 51). Nevertheless, the values reported here are considered as good indicators of relative binding affinities for these BER enzymes since they are performed (i.e. assayed) and determined (i.e. approach) under identical conditions with the same model BER intermediates. Other than different assay conditions and techniques, the explanation for the stronger binding observed here is unclear, but could be due to concentration effects as the macromolecules enter the native gel.
APE·Pol β·DNA Ternary Complex Could Alter the Sequence of Steps in SN-BER
Previous studies demonstrated that Pol β activity for gap-filling was faster than its activity for dRP removal (20, 40, 52). In considering the order of the steps in BER, these observations suggested that Pol β is more likely to conduct the gap-filling step before removal of the dRP group. In light of the stronger dRP lyase activity observed here and by Wong and Demple (40) when Pol β was in the ternary complex, the dRP removal step in this case would occur with similar frequency as gap-filling, thereby potentially altering the order of the BER steps (as described below). With dRP group removal before gap-filling, Pol β would then bind to the single-nucleotide-gapped substrate molecule in the next step of BER, and this is consistent with the strong affinity observed here for Pol β binding to such a structure (Table 2).
To consider the question of the order of these reactions under conditions of ternary complex formation, we compared the rate of dRP group removal with that of 1-nt gap-filling by Pol β; the 3′-exonuclease activity of APE was examined also. With essentially all of the Pol β in the reaction mixture in the ternary complex form, we found that gap-filling synthesis was ∼2-fold faster than dRP removal, but APE 3′-exonuclease activity was ∼1000-fold slower than dRP removal. This information may have implications for BER. First, in the presence of concentrations of APE and Pol β that allow formation of the ternary complex, the Pol β gap-filling and dRP removal steps may be random rather than ordered, and the dRP removal step may not be completely rate-limiting for overall SN-BER, and second, the faster removal of the dRP group from a BER intermediate with a 3′ mismatch would enhance proofreading by the APE 3′-exonuclease, since this activity is much stronger on the 3′ mismatched intermediate without the dRP group than on the same intermediate with the dRP group (Refs. 40 and 53 and data not shown).
The results allow us to propose further models to describe coordination of the sequential BER steps that are governed by DNA binding specificity and interactions of APE and Pol β (Fig. 9). After loss of a base and generation of an abasic site, APE binds to this intermediate and incises the damaged strand leaving a gap with a 5′-dRP group onto which the APE·Pol β·DNA ternary complex can assemble. Three different alternate subpathways of SN-BER may occur depending upon the order of the dRP lyase and gap-filling activities of Pol β. If the dRP lyase removes the dRP group before gap-filling synthesis occurs, as shown in subpathway 1, this will generate 1-nt-gapped DNA with a 5′-phosphate at the margin. APE does not bind strongly to this intermediate, allowing Pol β to bind without APE involvement. On the other hand, if Pol β gap-filling occurs before lyase activity, SN-BER proceeds through subpathway 2 or 3. In subpathway 2, the APE·Pol β·DNA ternary complex supports dRP group removal. Subpathway 3 represents the rare situation where Pol β misinserts a nucleotide after the dRP group is removed. Because APE 3′-exonuclease activity is inhibited by the presence of the dRP group (Refs 40 and 53 and data not shown), the proofreading 3′-exonuclease will not function effectively. Thus, stimulation of the dRP lyase activity afforded by the ternary complex will allow APE 3′-exonuclease to more efficiently remove a misinserted nucleotide, regenerating a gap for Pol β to fill with the correct nucleotide.
FIGURE 9. Mechanism of SN-BER repair as function of substrate binding specificity and protein-protein interactions.
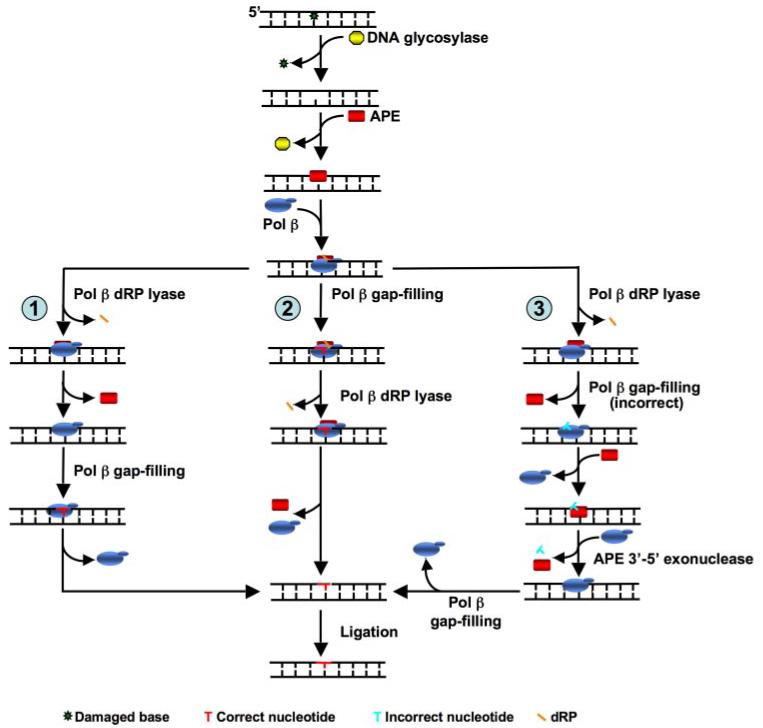
DNA glycosylase removes a damaged base and leaves an abasic site. APE binds and makes a 5′-incision adjacent to the AP site leaving a 1-nt gap with a 5′-dRP residue. With a high local enzyme concentration, APE and Pol β can form a protein-DNA ternary complex on this intermediate that stimulates Pol β activities (1-nt gap-filling and dRP lyase). Three subpathway choices may occur at this point; in one scenario, as indicated in subpathway 1, Pol β dRP lyase activity stimulated by APE·Pol β·DNA ternary complex removes the dRP group, generating a 1-nt-gapped DNA intermediate. The lower binding affinity of APE for this intermediate may promote dissociation of APE, whereas Pol β effectively binds to this intermediate and fills the gap producing the nicked DNA substrate necessary for ligation. In another scenario, the protein-DNA ternary complex with Pol β performs 1-nt gap-filling DNA synthesis before it removes the dRP group, thereby creating a nicked-dRP flap intermediate (subpathway 2). Under high local enzyme concentrations, the ternary complex may dissociate before the next step in the pathway. However, when a dRP-containing intermediate is present, APE and Pol β can rapidly reform the ternary complex (Table 2). These dissociation and re-association steps are not explicitly illustrated to simplify the overall scheme. The Pol β dRP lyase then removes the sugar-phosphate residue, creating nicked DNA for ligation. In subpathway 3, Pol β removes the dRP group before misinserting a nucleotide, generating a nicked intermediate with a 3′-mismatch. APE 3′-exonuclease activity can then remove the 3′-misinserted nucleotide. This results in 1-nt-gapped DNA that Pol β will efficiently insert the correct nucleotide, creating a nicked DNA that can then be ligated.
Faster removal of the dRP group by the ternary complex also could have implications for DNA damage signaling as well. PARP-1 is involved in the DNA damage response (54, 55) and has a preference for recognizing dRP-containing gapped DNA intermediates (54-56). Activation of PARP-1 poly(ADP-ribose) synthesis activity by virtue of such binding inhibits signaling pathways that lead to cell cycle arrest, apoptosis, and necrosis (54, 55). More efficient removal of the dRP-group by virtue of ternary complex formation could bypass such PARP-1-mediated regulation. Another implication of these results is that the level of expression or recruitment of APE and Pol β to sites of damage would be expected to alter the levels of alternate BER subpathways. In the presence of excess APE and formation of the ternary complex, a faster dRP lyase step would favor SN-BER provided the DNA ligase concentration is high enough to drive ligation once the nicked intermediate is generated.
Binding of APE to its incision product is well known (42, 53, 56), and the functional significance of this binding has been a topic of investigation. It was suggested that interaction between APE and dRP-containing intermediates could assist in SN-BER versus LP-BER subpathway choice, and a large excess of APE was found to stimulate Pol β strand-displacement synthesis during LP-BER (46). This stimulation was inhibited by the addition of PARP-1 (46). Additionally, APE was able to compete with and block PARP-1 binding to the dRP-containing intermediate and, hence, may interfere with the role of PARP-1 as a stimulatory factor in LP-BER (53). Our finding that the APE·Pol β·DNA ternary complex can form solely on BER intermediates containing the dRP group was consistent with the APE binding preference for dRP group-containing molecules. We found that formation of the APE·Pol β·DNA complex was dependent on DNA structure rather than a preformed APE·Pol β complex in solution. This result was consistent with the fact that in the absence of DNA substrate, no APE·Pol β complex in the solution could be identified (57). It is conceivable that APE binds the dRP group-containing intermediate and then recruits Pol β to the complex thereby stimulating the dRP-lyase and gap-filling activities of Pol β (39). Thus, these studies point to a critical role of APE in mediating coordination during SN-BER.
Finally, we would like to emphasize that the pathways outlined in Fig. 9 represent a small number of alternatives that depend highly on “local” enzyme concentrations as well as the presence of other BER proteins that are not considered in this study. Changes in individual rate constants do not impact the pathways outlined in Fig. 9 but would be expected to alter the magnitude of the respective pathways. These pathways represent initial models in an attempt to visualize the complexity and versatility of cellular BER and will certainly need to be refined as more details emerge.
Acknowledgment
We thank Jennifer Myers for editorial assistance.
Footnotes
This research was supported by the Intramural Research Program of the NIEHS, National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- BER
- base excision repair
- APE
- apurinic/apyrimidinic endonuclease
- dRP
- deoxyribose phosphate
- LP
- long-patch
- nt
- nucleotide
- Pol β
- DNA polymerase β
- SN
- single-nucleotide
- THF
- tetrahydrofuran
- PVDF
- polyvinylidene difluoride
- PARP
- poly(ADP-ribose) polymerase
REFERENCES
- 1.Lindahl T, Ljungquist S. Basic Life Sci. 1975;5:31–38. doi: 10.1007/978-1-4684-2895-7_5. [DOI] [PubMed] [Google Scholar]
- 2.Ames BN. Free Radic. Res. Commun. 1989;7:121–128. doi: 10.3109/10715768909087933. [DOI] [PubMed] [Google Scholar]
- 3.Ames BN, Gold LS. Mutat. Res. 1991;250:3–16. doi: 10.1016/0027-5107(91)90157-j. [DOI] [PubMed] [Google Scholar]
- 4.Lutz WK. Mutat. Res. 1990;238:287–295. doi: 10.1016/0165-1110(90)90020-c. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl T. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 6.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. American Society for Microbiology; Washington, D. C.: 1995. pp. 169–171. [Google Scholar]
- 7.Ames B, Shigenaga M, Hagen T. Proc. Natl. Acad. Sci. U. S. A. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ames BN. Science. 1983;221:1256–1264. doi: 10.1126/science.6351251. [DOI] [PubMed] [Google Scholar]
- 9.Poulsen HE, Loft S, Prieme H, Vistisen K, Lykkesfeldt J, Nyyssonen K, Salonen JT. Free Radic. Res. 1998;29:565–571. doi: 10.1080/10715769800300601. [DOI] [PubMed] [Google Scholar]
- 10.Waris G, Ahsan H. J. Carcinog. 2006;5:14. doi: 10.1186/1477-3163-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pratico D. Neurobiol. Aging. 2005;26:581–583. doi: 10.1016/j.neurobiolaging.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Circulation. 2002;106:927–932. doi: 10.1161/01.cir.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- 13.Harman D. Mutat. Res. 1992;275:257–266. doi: 10.1016/0921-8734(92)90030-s. [DOI] [PubMed] [Google Scholar]
- 14.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 15.Lindahl T. Annu. Rev. Biochem. 1982;51:61–87. doi: 10.1146/annurev.bi.51.070182.000425. [DOI] [PubMed] [Google Scholar]
- 16.Doetsch PW, Cunningham RP. Mutat. Res. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]
- 17.Mosbaugh DW, Bennett SE. Prog. Nucleic Acid Res. Mol. Biol. 1994;48:315–370. doi: 10.1016/s0079-6603(08)60859-4. [DOI] [PubMed] [Google Scholar]
- 18.Matsumoto Y, Kim K. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 19.Piersen CE, Prasad R, Wilson SH, Lloyd RS. J. Biol. Chem. 1996;271:17811–17815. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- 20.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. J. Biol. Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 21.Dianov G, Price A, Lindahl T. Mol. Cell. Biol. 1992;12:1605–1612. doi: 10.1128/mcb.12.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumoto Y, Kim K, Bogenhagen DF. Mol. Cell. Biol. 1994;14:6187–6197. doi: 10.1128/mcb.14.9.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. J. Biol. Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- 24.Dianov G, Bischoff C, Piotrowski J, Bohr VA. J. Biol. Chem. 1998;273:33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- 25.Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. Biochemistry. 1998;37:3575–3580. doi: 10.1021/bi972999h. [DOI] [PubMed] [Google Scholar]
- 26.Horton JK, Prasad R, Hou E, Wilson SH. J. Biol. Chem. 2000;275:2211–2218. doi: 10.1074/jbc.275.3.2211. [DOI] [PubMed] [Google Scholar]
- 27.Pascucci B, Stucki M, Jonsson ZO, Dogliotti E, Hubscher U. J. Biol. Chem. 1999;274:33696–33702. doi: 10.1074/jbc.274.47.33696. [DOI] [PubMed] [Google Scholar]
- 28.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 29.Wilson SH, Kunkel TA. Nat. Struct. Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 30.Caldecott KW, Aoufouchi S, Johnson P, Shall S. Nucleic Acids Res. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dianova II, Sleeth KM, Allinson SL, Parsons JL, Breslin C, Caldecott KW, Dianov GL. Nucleic Acids Res. 2004;32:2550–2555. doi: 10.1093/nar/gkh567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 33.Vidal AE, Boiteux S, Hickson ID, Radicella JP. EMBO J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Ahn J, Wilson SH, Prives C. EMBO J. 2001;20:914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kedar P, Kim S, Robertson A, Hou E, Prasad R, Horton J, Wilson S. J. Biol. Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- 36.Wilson SH. Mutat. Res. 1998;407:203–215. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- 37.Beard WA, Wilson SH. Chem. Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 38.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. DNA Repair (Amst) 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 39.Bennett RA, Wilson DM, III, Wong D, Demple B. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong D, Demple B. J. Biol. Chem. 2004;279:25268–25275. doi: 10.1074/jbc.M400804200. [DOI] [PubMed] [Google Scholar]
- 41.Sokhansanj BA, Rodrigue GR, Fitch JP, Wilson DM., III Nucleic Acids Res. 2002;30:1817–1825. doi: 10.1093/nar/30.8.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strauss PR, Beard WA, Patterson TA, Wilson SH. J. Biol. Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 43.Beard WA, Wilson SH. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 44.Gill SC, von Hippel PH. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 45.Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkinson AE, Wilson SH. J. Biol. Chem. 1996;271:16000–16007. doi: 10.1074/jbc.271.27.16000. [DOI] [PubMed] [Google Scholar]
- 46.Sukhanova MV, Khodyreva SN, Lebedeva NA, Prasad R, Wilson SH, Lavrik OI. Nucleic Acids Res. 2005;33:1222–1229. doi: 10.1093/nar/gki266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vande Berg BJ, Beard WA, Wilson SH. J. Biol. Chem. 2001;276:3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
- 49.Jackson EB, Theriot CA, Chattopadhyay R, Mitra S, Izumi T. Nucleic Acids Res. 2005;33:3303–3312. doi: 10.1093/nar/gki641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramana CV, Boldogh I, Izumi T, Mitra S. Proc. Natl. Acad. Sci. U. S. A. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beard WA, Shock DD, Wilson SH. J. Biol. Chem. 2004;279:31921–31929. doi: 10.1074/jbc.M404016200. [DOI] [PubMed] [Google Scholar]
- 52.Prasad R, Beard WA, Strauss PR, Wilson SH. J. Biol. Chem. 1998;273:15263–15270. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 53.Cistulli C, Lavrik OI, Prasad R, Hou E, Wilson SH. DNA Repair (Amst) 2004;3:581–591. doi: 10.1016/j.dnarep.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 54.Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. J. Biol. Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 55.Horton JK, Stefanick DF, Wilson SH. DNA Repair (Amst) 2005;4:1111–1120. doi: 10.1016/j.dnarep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 56.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. J. Biol. Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 57.Dimitriadis EK, Prasad R, Vaske MK, Chen L, Tomkinson AE, Lewis MS, Wilson SH. J. Biol. Chem. 1998;273:20540–20550. doi: 10.1074/jbc.273.32.20540. [DOI] [PubMed] [Google Scholar]