

Abstract
Accumulating evidence of G-protein-coupled receptor (GPCR) oligomerization on the one hand and perfect functionality of monomeric receptors on the other creates an impression of controversy. However, the GPCR superfamily is extremely diverse, both structurally and functionally. The life cycle of each receptor includes many stages: synthesis, quality control in the endoplasmic reticulum, maturation in the Golgi, delivery to the plasma membrane (where it can be in the inactive or active state, in complex with cognate G protein, G-protein-coupled receptor kinase or arrestin), endocytosis and subsequent sorting in endosomes. Different GPCR subtypes, and even the same receptor at different stages of its life cycle, most likely exist in different oligomerization states, from monomers to dimers and possibly higher-order oligomers.
Introduction
G-protein-coupled receptors (GPCRs) are the largest family of signaling proteins, encoded in animals by 3%−5% of all genes. Mammals have ∼800−1000 GPCR subtypes, some of which have several forms generated by alternative splicing or mRNA editing. GPCRs have a structurally homologous core of seven transmembrane α helices, but the size and structure of their extracellular and intracellular elements vary wildly, from the most ‘compact’ rhodopsin to receptors with huge extracellular N termini, and others with very large third intracellular loops or C termini [1].
Despite this structural diversity, the first round of signaling initiated by most GPCRs is remarkably uniform: the active receptor catalyzes the exchange of GDP for GTP on heterotrimeric G proteins, whereupon Gα-GTP and released Gβγ activate or inhibit various effectors [1]. Active GPCRs are specifically phosphorylated by G-protein-coupled receptor kinases (GRKs) [2]. Preferential binding of arrestins to active phosphoreceptors precludes further G protein activation [3]. Receptor-bound arrestin recruits two components of internalization machinery, clathrin [4] and AP-2 [5], and a surprising variety of other proteins, initiating the second round of signaling [6,7].
GPCRs respond to various external stimuli (light, odorants) and signals within the body (hormones, neurotransmitters, extracellular Ca2+, protease activity) and are targeted by half of clinically used drugs. The unrivaled biological significance of GPCRs explains the enormous efforts that have been invested into the elucidation of the mechanisms of their function. Although there are many outstanding issues in this area, one question recently came to the fore and became a subject of fierce debate [8,9]:do GPCRs function as monomers, dimers or higher-order oligomers? Here we show that this is, in fact, a series of questions that do not necessarily have the same answer for all GPCRs, or even for a single receptor at different stages of its life cycle.
The birth of the receptor
Like all integral membrane proteins, GPCRs are synthesized in the rough endoplasmic reticulum. Nascent receptors have to pass numerous steps of quality control and posttranslational modifications before they are delivered to the plasma membrane and get their first chance to function. This process has been extensively studied for class C GPCRs. These receptors have two unique properties: they are known to be obligatory dimers [10–12], and their ligand-binding site is localized on the extracellular element formed by the extremely large N terminus [13],in sharp contrast to the majority of GPCRs, where the ligand binds in the pocket formed by transmembrane helices and extracellular loops [14]. Importantly, the composition of the ‘correct’ dimer is always strictly defined. Processing of nascent class A GPCRs is less well understood. Existing evidence allows different interpretations: receptor dimers might be forming soon after synthesis [15], or highly over-expressed receptors might be processed in restricted compartments, existing in close proximity without physical association [16].
The model based on class C receptors predicts that cells could produce only certain strictly defined GPCR combinations. However, this does not seem to be a universal rule: cells make 5−20 endogenous GPCR subtypes, which are mixed and matched in different ways. Virtually any GPCR can be expressed in hugely superphysiological quantities by various cultured cells, where an overexpressed subtype constitutes >95% of the total receptor complement of the cell. Moreover, any combination of GPCRs can be expressed in the same cell. Thus, if class A and B GPCRs are processed and delivered to the cell surface as dimers, then homodimers and/or heterodimers of any composition must be allowable. Interestingly, several observations suggest that some receptors may be delivered to the plasma membrane as oligomers, in which one ‘hitches a ride’ with the other. For example, surface expression of α2C- [17] and α1D-adrenergic receptors [18] is greatly enhanced by coexpression of β2-adrenergic receptor; the correct delivery to the rod outer segment of C-terminally truncated rhodopsin lacking the targeting sequence requires its coexpression with full-length rhodopsin [19].
The life in the plasma membrane
GPCRs might be delivered to their ‘place of employment’ as either monomers or oligomers; we only have an unambiguous answer for class C receptors. Regardless of the form in which they arrive, there remain five questions about their self-association status in the plasma membrane, the answers to which might be different for different receptors and even for a single receptor subtype in different functional states. We need to establish in what form the receptor exists when it is: (i) inactive; (ii) ligand activated; (iii) in complex with G protein; (iv) phosphorylated by GRKs; or (v) bound to arrestin. Another important issue is the stability of the oligomers, which might be permanent (as seen in class C GPCRs), very transient or anything in between. The half-life of the dimer is determined by the KD of GPCR self-association (t1/2 = 6.93 × 10−7/KD). For example, the KD for neurotensin NTS1 receptor self-association was estimated at 2−20 nM [20], which translates into a dimer half-life of 34−340 s, that is, much shorter than the length of most stages in the functional cycle of the receptor.
A large number of studies, mostly using BRET and FRET, indicate that molecules of the same (or two different) receptor are often close enough to generate an energy transfer (ET) signal in nonstimulated cells [15,21,22]. However, we have to keep in mind that ET signal actually means that the two fluorescent moieties are within <60Å of each other. Depending on the receptor subtype and the construct used, this could happen when receptors are in a tight dimer or >500Å apart (Box 1). Thus, ET-based methods faithfully report the proximity of the two receptors, but they cannot distinguish whether the receptors form a structurally defined dimer (Box 1a) or are simply within ‘shouting distance’ of each other (Box 1b). If there is equilibrium in the receptor population where a certain proportion is very close to another at any given moment (e.g. as a result of frequent encounters between overexpressed receptors), the ET signal reflects an ‘average’ distance that has no physical meaning. A recent meticulous FRET study of the dimerization state of neurokinin1 receptor [16] provides an excellent illustration of these points. The authors detected strong FRET at superphysiological, but not at physiological, expression levels and demonstrated that the neurokinin1 receptor exists as a monomer confined to microdomains that constitute ∼1% of the plasma membrane [16].
Box 1. Advantages and limitations of energy transfer-based methods.
Both fluorescence resonance energy transfer (FRET) (between two fluorescent proteins) and bioluminescence resonance energy transfer (BRET) (between luciferase and a fluorescent protein) can be used to detect protein–protein interactions in living cells. Energy transfer-based methods have several limitations, however. The efficiency of energy transfer depends on the relative orientation of the fluorescent moieties and the distance between them, so that absolute distances cannot be measured. Detection of an energy transfer signal indicates that the distance between fluorescent moieties (shown as cylinders at the end of receptor C termini in Figure I) is smaller than 50−60Å [33]. The size of 27 kDa fluorescent proteins and 34 kDa luciferase is comparable to that of the transmembrane core of GPCRs (diameter ∼40Å). These proteins are usually attached to the receptor C terminus, which in different GPCR subtypes varies in length from 25 to 150 amino acids. Polypeptides of this length in extended conformation can cover 80−480Å. Thus, a FRET or BRET signal indicates that two fluorescent tags are within 60Å of each other, which may occur when receptors form a bona fide dimer (Figure Ia) or when they are >500Å apart (Figure Ib). Reliable quantitative FRET and BRET measurements are limited by the quality of the signal and noise level. Fluorescent proteins and luciferase yield background signals arising from incompletely processed proteins inside the cell and high cell autofluorescence in the spectral region used [16]. The use of receptor with an ∼10 kDa acyl carrier protein fused to the extracellular N terminus with subsequent posttranslational enzymatic labeling has several important advantages: the possibility to choose fluorophores with optimal spectral properties, exclusive modification of receptors transported to the plasma membrane, and precise control of donor:acceptor ratio [16]. Proper interpretation of energy transfer data requires quantitative analysis of signal dependence on the donor:acceptor ratio at different levels of receptor expression, estimation of labeling efficiency and possible contribution of unlabeled receptors, including endogenously expressed GPCRs with which labeled receptors may heterodimerize (see Ref. [16] and references therein).
Another method often used to study GPCR oligomerization is disulfide crosslinking. In this paradigm, crosslinking between native or introduced cysteines is interpreted as proof of receptor dimerization [23]. These studies identified multiple positions in several GPCRs as preferred contact sites [23–26]. However, because disulfide formation is irreversible under conditions employed, and crosslinking is usually allowed to proceed for 30−60 min, this method cannot distinguish between a stable bona fide dimer and a transient ‘kiss-and-run’ encounter ‘trapped’ by a covalent bond. This is a common caveat of all crosslinking methods, often used to our advantage to trap transient complexes for coimmunoprecipitation [27].
Rhodopsin constitutes ∼98% of the total protein in rod disc membranes, providing a rare opportunity to look at the state of this receptor in its native milieu. Atomic force microscopy of disc membranes immobilized on mica yielded stunningly beautiful pictures of enormous arrays of hundreds of rhodopsin molecules in evenly spaced military-style formations [28,29]. Because of the regularity of these arrays, the nature of the elementary repeating unit is essentially in the eye of the beholder: one can see it as a monomer, dimer, tetramer, hexamer and so on. Because separated monomers and dimers are few and far between, these data cannot be objectively considered as strong support for one or the other.
Thus, despite persistent efforts of top-notch investigators, we do not know whether inactive GPCRs exist as monomers, dimers, higher-order oligomers or in equilibrium between all of the above.
What is the G-protein-activating unit?
It is important to note that the state of inactive receptor does not provide the answer to this question: upon activation, dimers might dissociate [30,31] or monomers associate [32,33]. Unfortunately, there are no experimental tools to address the most relevant issue: the state of endogenous receptors activating their cognate G proteins in living cells (Figure 1). Therefore, experimentally tractable related questions are asked: first, is a single GPCR molecule sufficient for G protein activation, and second, does the efficiency of G protein activation change when receptors are forced into particular oligomerization states using detergents, and the like [34]? However, the efficiency of receptor–G protein coupling in detergents is significantly lower than that in lipid [20,35], leaving the biological relevance of these comparisons in doubt. A report that the purified leukotriene B4 receptor forms a stable complex consisting of two receptors and one G protein [36] is very intriguing. However, two crucial pieces of information are lacking: first, does this receptor dimer in detergent mimic the putative physiological homodimer in the membrane, and second, does one or both receptors in this complex interact with G protein? It was shown for the leukotriene B4 [37] and class C receptors [13,38] that the activation of one receptor in a dimer is sufficient for G protein coupling.
Figure 1.
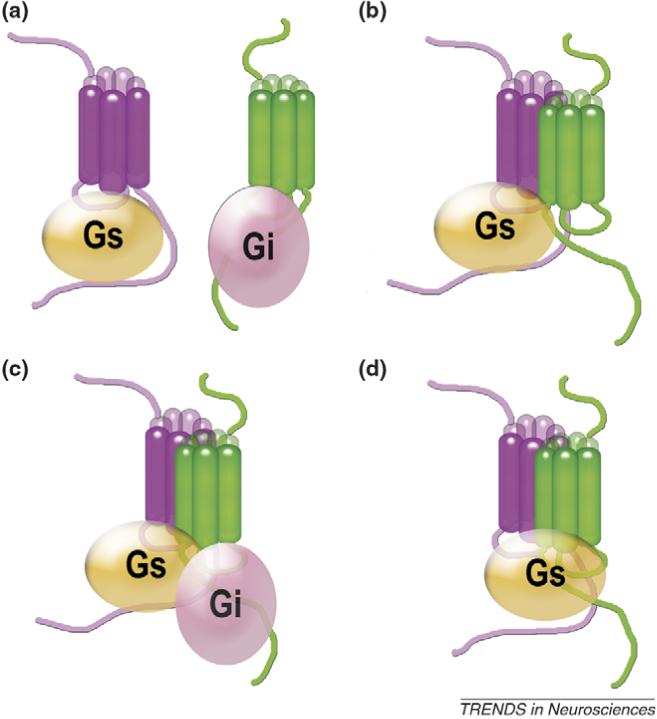
How do GPCRs activate G proteins? Every conceivable model of G protein activation by GPCRs has been proposed: (a) by receptor monomer; (b) by one receptor in a GPCR dimer; (c) by both receptors in a dimer acting via their cognate G proteins; and (d) by both receptors in a dimer docking to a single G protein. These models have important implications for GPCR signaling. Model (a) [9] predicts full functionality of a monomeric receptor, whereas model (d) [29] predicts the opposite, that a receptor dimer is required for G protein activation. Recent conclusive evidence for three different class A GPCRs, rhodopsin [35,39], β2-adrenergic [40] and NTS1 neurotensin receptor [20], demonstrates that a single receptor molecule is sufficient for efficient G protein activation and formation of receptor–G protein complexes with high agonist affinity, strongly supporting model (a) and ruling out model (d). Studies of obligatory heterodimeric class C receptors [38] and the leukotriene receptor [37] suggest that only one receptor within a dimer binds G protein, as in model (b). A two receptor–one G protein complex, as described for the leukotriene receptor in detergent [36], is also compatible with model (b). Reports that the activities of dimeric rhodopsin [35] and the neurotensin receptor [20] are lower than those of the monomeric forms makes model (c) unlikely for these GPCRs. These models equally apply to homo- and heterodimers (for simplicity, only the latter are shown).
Several recent studies compared G protein activation by rhodopsin [35,39], β2-adrenergic [40] and neurotensin NTS1 receptor [20] at very low detergent concentrations or in lipid. Monomeric rhodopsin in low detergent was found to activate transducin as fast as the rate of protein diffusion allows [39]. The NTS1 receptor monomer activated G protein more efficiently than the dimer [20]. Rhodopsin [35] and β2-adrenoreceptor [40] were also reconstituted into high-density lipoprotein particles, tiny patches of lipid bilayer stabilized by a protein ‘belt,’ at one or two receptors per particle. Particles with one or two rhodopsins activated transducin equally [35]. Thus, the second rhodopsin molecule apparently was quite useless, possibly inaccessible for transducin when two rhodopsins are forced close together (Figure 1b). The monomeric β2-adrenoreceptor effectively activated G protein and formed a biologically relevant receptor–G protein complex with high agonist affinity [40].
Collectively, these data clearly demonstrate that a single GPCR molecule is sufficient for G protein binding and activation. However, direct comparison (in a lipid environment) of a receptor that was forced to remain monomeric with one allowed to dimerize (but not necessarily doing so) has been performed only with two GPCRs, rhodopsin and β2-adrenoreceptor [35,40]. These data do not rule out the possibility that two receptors in a dimer interact each with its own G protein (Figure 1c), although the results with rhodopsin [35] and NTS1 receptor [20] make this rather unlikely.
Receptor phosphorylation
The mechanism of GRK action provides a perfect opportunity to test whether receptor kinases preferentially phosphorylate GPCR monomers or oligomers. The enzymatic activity of GRK toward exogenous substrates is greatly increased upon receptor binding, indicating that the active receptor serves as both the GRK activator and as its substrate [41]. In rod disc membranes, where rhodopsin molecules cover ∼50% of the surface, light activation of a few rhodopsins results in phosphorylation of a large number of inactive neighbors [42]. In the disc membrane, rhodopsin molecules are packed so closely that it is impossible to distinguish kinetically stable oligomers from frequent encounters between monomers. However, it is hard to imagine an oligomer in which one active rhodopsin could enable phosphorylation of hundreds of other rhodopsin molecules [42]. In the plasma membrane of cells (even those expressing hugely superphysiological amounts of receptors), the majority of random encounters would involve other proteins, rather than sister receptors. If coexpressed receptors form heterodimers, the activation of one would result in phosphorylation of both (Figure 2). In a stable dimer, the efficiency of crossphosphorylation would be independent of absolute expression level and would reach a maximum at a 1:1 expression ratio of the two receptors (Figure 2a). By contrast, in kiss-and-run dimers, crossphosphorylation would increase with the frequency of random encounters, that is, with the receptor expression level (Figure 2b). Although crossphosphorylation between two visual pigments coexpressed in mouse rods [43] and between chemokine CCR5 and C5a receptors [22] has been reported, conclusive experiments that distinguish between stable dimers and transient encounters have not been performed.
Figure 2.
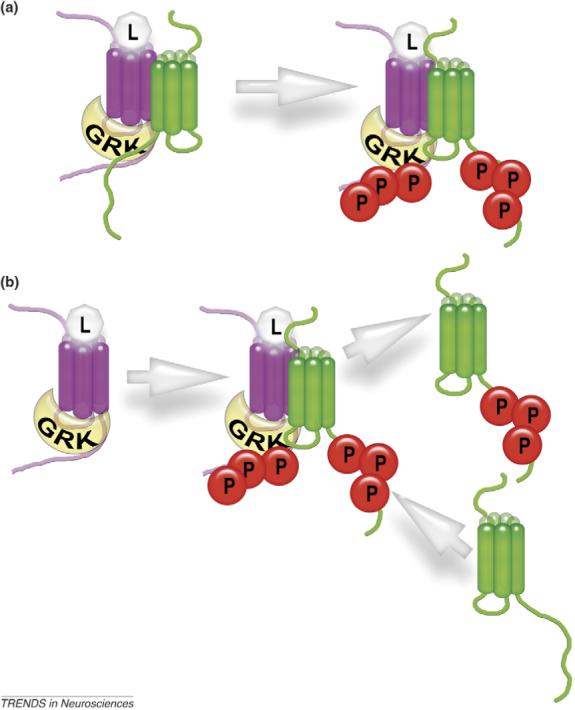
Detecting GPCR oligomerization by crossphosphorylation. The binding to the active receptor dramatically enhances GRK enzymatic activity, even toward exogenous substrates [41]. Thus, when one receptor (magenta) is activated and binds GRK, the other (green) in close proximity is likely to also be phosphorylated. If two receptors form a stable heterodimer (a), one ‘green’ receptor would be phosphorylated for each activated ‘magenta’ receptor, and the efficiency of crossphosphorylation would be independent of the absolute expression level. By contrast, if the half-life of the heterodimer is shorter than the half-life of the receptor–GRK complex, the GRK bound to the magenta receptor would sequentially phosphorylate multiple green receptors (b). In this case, the efficiency of crossphosphorylation would be directly proportional to the expression levels of both receptors. Using receptor mutants that do not have their own phosphorylation sites can simplify the readout of these experiments, ensuring that only crossphosphorylation of the putative dimerization partner is measured. Abbreviations: L in light gray octagon, ligand (agonist); P in red circle, receptor-attached phosphate.
Arrestin binding: stoichiometry matters
One of the arguments advanced in support of GPCR oligomerization was that the fit between known structures of rhodopsin [44,45] and arrestin [46–48] could be best achieved if one arrestin bound two rhodopsins in a dimer [29]. In bright light, arrestin moves to the rhodopsin-containing outer segments of photoreceptor cells and remains there as long as binding-competent rhodopsin is present [49]. Thus, the extent of arrestin translocation provides a quantitative measure of arrestin–rhodopsin interaction under physiologically relevant conditions in a living animal. Rod photoreceptors express enormous amounts of both proteins at an ∼0.8:1 arrestin:rhodopsin ratio [50,51], which can be easily changed in either direction by using hemizygous knockout mice. The stoichiometry of the arrestin–rhodopsin interaction was recently determined in vivo and in vitro [50]. These experiments showed that the molar amount of arrestin in the outer segment at ‘saturation’ exceeds 80% of the molar amount of rhodopsin [50]. in vitro experiments with two purified proteins also yielded saturation at a 0.99:1 arrestin:rhodopsin ratio [50]. This stoichiometry cannot be the result of an arrestin dimer interacting with a rhodopsin dimer because only monomeric arrestin can bind rhodopsin [52]. Thus, each rhodopsin molecule binds its own arrestin. Considering the level of structural [46,47] and functional [53–56] conservation in the arrestin family, all arrestins likely bind individual molecules of their cognate receptors. Indeed, nonvisual arrestins were shown to occupy 90% of m2 muscarinic cholinergic receptor molecules present in the assay at saturation [57]. However, experiments with rhodopsin in native disc membranes [50], where it is free to form any oligomers if it is inclined to do so, cannot distinguish between a rhodopsin monomer binding one arrestin, a dimer binding two, a tetramer binding four and so forth. To determine whether arrestin prefers GPCRs in monomeric or oligomeric form, arrestin binding to lipid entities containing defined numbers of receptor molecules must be compared.
Arrestin-mediated signaling: could receptor oligomers help?
Receptor-bound arrestins serve as scaffolds mobilizing an astonishing variety of signaling proteins [7,58]. The relative size of arrestins and their binding partners suggests that a unitary arrestin–receptor complex can accommodate no more than four to six additional proteins simultaneously [59]. Thus, either there is stiff competition among arrestin-binding proteins for very limited ‘parking space’ (Figure 3a), or the scaffolds include more than one arrestin–receptor complex. Conceivably, receptor oligomerization could serve to dramatically increase signaling capabilities of the complex (Figure 3b). Arrestins also function as adapters linking GPCRs to key components of the internalization machinery: clathrin and AP-2 [4,5]. The C tail of each arrestin molecule contains binding sites for clathrin and AP-2 [60], and the arrestin C tail is released upon receptor binding [61]. Arrestin-mediated anchoring of GPCRs to clathrin/AP-2 may yield large evenly spaced arrays of receptor–arrestin complexes in the clathrin-coated pit (Figure 3c), which could create large scaffolds independently of (or in collaboration with) receptor oligomerization. This might explain why in certain cases receptor internalization appears to be necessary for arrestin-mediated signaling [62], although this is by no means a general rule [63]. The structure and composition of ‘signalosomes’ organized by arrestin–receptor complexes remain to be elucidated. It is possible that unitary arrestin–receptor complexes are sufficient to turn on certain signaling pathways, whereas others may require larger scaffolds organized by internalization machinery and/or receptor oligomerization.
Figure 3.
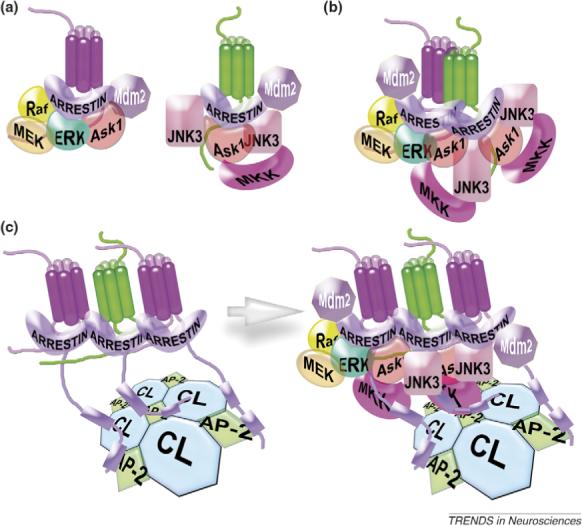
Possible role of GPCR oligomerization in arrestin-mediated signaling. Receptor-bound arrestins were shown to recruit >20 different trafficking and signaling proteins to the complex [7,58]. (a) A comparison of the size of the receptor, arrestin and arrestin interaction partners suggests that a unitary arrestin–receptor complex cannot bind more than four to six proteins at the same time [59]. Arrestin-mediated scaffolding of each MAP kinase pathway (c-Raf-1→MEK1→ERK1/2 or ASK1→MKK4→JNK3) requires simultaneous recruitment of three proteins, where members of the same cascade must be close enough and correctly oriented relative to each other to make these complexes productive. For example, ASK1 in the left complex and one JNK3 in the right complex cannot participate in signaling because their necessary additional partners are absent. (b) It is tempting to speculate that receptor oligomers (a dimer is shown for simplicity), with each receptor bound to its own arrestin [50], increase the size of the scaffolding surface, thereby making the assembly of productive complexes more likely. For example, three complete MAP kinase cascades are assembled here, as opposed to only two on a pair of separate arrestin–receptor complexes shown in (a). Multi-arrestin scaffolds may also ensure ‘economies of scale’: for example, one molecule of E3 ubiquitin ligase Mdm2 might ubiquitinate both arrestins in the complex, saving room for additional partners. (c) Internalization can serve as an alternative mechanism yielding a similar increase in the scaffolding surface. Receptor binding induces the release of the arrestin C tail, which carries clathrin and AP-2-binding sites (shown as blue boxes on the arrestin C tail). Arrestin-mediated GPCR recruitment to the coated pit may create evenly spaced arrays of many arrestin–receptor complexes reproducing spatial organization of the clathrin coat. These multicomplex assemblies might be large enough to recruit the components of both MAP kinase cascades along with other signaling molecules. Finally, mechanisms illustrated in (b) and (c) are not mutually exclusive: arrestin-mediated receptor organization in the coated pit may work together with receptor oligomerization to enable efficient arrestin-mediated signaling. Abbreviations: CL, clathrin; AP-2, clathrin adaptor AP-2 (also termed adaptin2).
In what form are GPCRs internalized?
With the exception of class C receptors, we do not have incontrovertible evidence regarding the oligomerization state of any GPCR at this stage of its life cycle. Several studies suggest that some receptors internalize as oligomers [22,64,65], but these results could also be rationalized without invoking receptor oligomerization. Several types of GPCRs reside in microdomains covering a small fraction of the cell surface [16,66,67]. Crowding of overexpressed receptors in these microdomains is sufficient to yield a FRET signal without oligomerization [16]. If massive phosphorylation of the overexpressed receptor followed by arrestin recruitment and mobilization of clathrin and AP-2 to a particular GPCR subtype induces nucleation of new coated pits in these microdomains, one receptor can drag along another cohabiting in the same microdomain, with resulting co-internalization and subsequent co-sorting of the two receptors.
Do all GPCRs function the same way?
An implicit assumption in the current debate between ‘monomer’ and ‘dimer’ parties [8,9] is that the functional state of all GPCRs remains the same throughout their entire life cycle. However, there is no reason to believe that even a single receptor passes quality control in the endoplasmic reticulum, is delivered to the plasma membrane, exists there before and after activation, binds G protein, GRK, arrestin, is internalized and then undergoes sorting in endosomes in the same oligomerization state. Hundreds of GPCR subtypes share a core structure of the seven-helical bundle, but not much else. Their N termini can be small or huge, do or do not contain ligand-binding elements or tethered ligands; their cytoplasmic loops and C termini wildly vary in size; many receptors do, but some do not require phosphorylation for arrestin binding; different receptors carry GRK phosphorylation sites in the C terminus or any of the cytoplasmic loops [59]. An amazing structural and functional diversity of the GPCR superfamily acquired in more than a billion years of evolution makes it highly unlikely that every receptor performs every function in exactly the same state. Transmembrane helices 1,2,4 [24], 3,4 [26],4 [23], 4,5 [25] and 6 [32] have been implicated in receptor oligomerization in different cases, strongly suggesting that the structure of oligomers formed by different GPCRs might not be the same. Even with the same receptor it might differ depending on its functional state [23]. Therefore, we should avoid overgeneralization. When a particular receptor is found to do something as a monomer or an oligomer, the data mean exactly that and never warrant sweeping conclusions about every function of each member of the enormous GPCR superfamily.
How can we get unambiguous answers?
Another problem that plagues the monomer–dimer debate is the unambiguous interpretation of inherently ambiguous data. Functional crosstalk, energy transfer and receptor crosslinking all allow alternative interpretations. There are very few experimental approaches that can yield clear answers in living cells. The only reason we are fairly sure how class C GPCRs work is that in this case only strictly defined heterodimers can make it to the plasma membrane and function [10–12], which is manifestly not the case with other receptors. One experimental paradigm that can be used in cells and/or purified plasma membranes is forced dimerization via engineered His-based interreceptor metal-binding sites and its controlled release by chelators. Engineered interhelical metal-binding sites within GPCRs were successfully used to study activation-induced movement of α helices [68] and oligomerization of dopamine transporters [69].
Where the receptor can be purified, one can compare individual functional modalities of preparations containing one, two, or more reconstituted receptors per lipid entity. So far, single receptors successfully performed the only function tested, G protein activation [20,35,39,40]. This approach must be extended to GRK phosphorylation, arrestin binding and arrestin-mediated assembly of signalosomes from purified components.
A receptor's propensity to self-associate and the size and stability of the oligomers formed can be determined using site-directed spin labeling and long-range (20−60Å) distance measurements with double electron–electron resonance (DEER) [70]. This approach requires the generation of receptors with a unique cysteine in the position of interest. If the receptor forms stable structurally defined dimers, this method would yield one clear distance. Three to four measured distances between different positions in the receptor would reveal dimer structure. A stable trimer would yield up to three distances, a tetramer up to six, and so forth. DEER measures each distance within range independently [52], in sharp contrast to energy transfer-based methods that, in the case of multiple distances, yield a meaningless average. DEER measurements with inactive as well as active receptor in the absence and presence of purified G proteins, GRKs and arrestins can reveal receptor oligomerization (or lack thereof) in different functional situations. If the receptor in any of these states exists as a monomer (or only transient kiss-and-run dimers form), with two molecules confined to a lipoprotein particle with a diameter of 10 nm [40], DEER would detect no distance or very broad distance distribution. These experiments are labor intensive and require cutting-edge technology, but the clarity of the results well justifies the effort.
To summarize, at this point, the arguments of both sides in the monomer–dimer debate are largely based on very limited, often demonstrably ambiguous evidence and the groundless assumption that all GPCRs at all stages of their functional cycle are in the same oligomerization state. It is very important to devise experiments asking clearly defined questions, use methods that yield conclusive answers and resist the temptation to overinterpret the data.
Figure I.
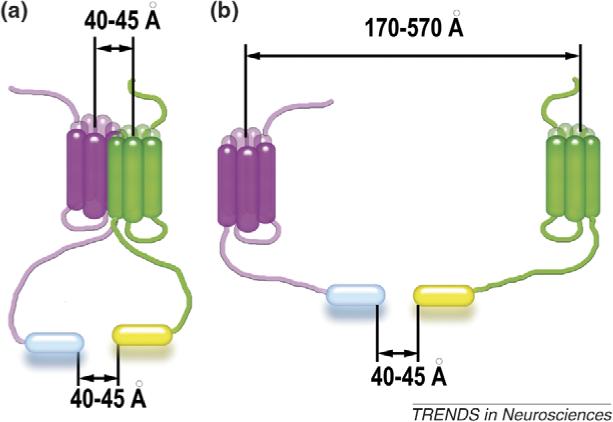
Inter-receptor distance during energy transfer.
Acknowledgements
This work was supported by NIH grants EY011500, GM077561 (V.V.G.) and NS045117 (E.V.G.).
Glossary
- Arrestins
A family of four proteins in mammals that specifically bind active phosphorylated GPCRs, shut down (‘arrest’) G-protein-mediated signaling, promote receptor internalization by linking it to the clathrin coat and redirect the signaling to multiple G-protein-independent pathways.
- G-protein-coupled receptors (GPCRs)
A large family of receptors (encoded by >800 genes in the human genome) that have in common a characteristic bundle of seven membrane-spanning α helices (heptahelical domain) with an extracellular N terminus and intracellular C terminus. GPCRs are sometimes called seven-transmembrane receptors (7TMRs) to acknowledge the fact that some members of this family do not couple to G proteins. According to the most widely used (although not the most comprehensive) classification system, GPCRs are divided into classes (also termed families or groups) A, B and C.
- G-protein-coupled receptors, class A
The largest group of GPCRs, which are often called rhodopsin-like receptors. Small-molecule and peptide agonists of these receptors usually bind within the cavity in the heptahelical domain and/or to the extracellular loops between the helices (with the exception of a few glycoprotein hormone receptors carrying large hormone-binding domains on the extracellular N terminus). The lengths of the C terminus and third intracellular loop in class A are highly variable. Typical examples: rhodopsin, α- and β-adrenergic, muscarinic cholinergic, dopamine and odorant receptors.
- G-protein-coupled receptors, class B
A small group of receptors with relatively large extracellular N-terminal hormone-binding domains. Typical examples: corticotropin-releasing factor receptor, parathyroid hormone receptor.
- G-protein-coupled receptors, class C
A small group of receptors that have been unambiguously shown to function as dimers. The ligand-binding site in these receptors is localized in the extracellular N-terminal Venus flytrap domain. Typical examples: homodimeric metabotrophic glutamate and calcium-sensing receptors, heterodimeric GABAB and sweet and umami taste receptors.
- G proteins
Guanyl nucleotide-binding proteins. Active GPCRs function as guanyl nucleotide exchange factors (GEFs) of heterotrimeric G proteins, promoting the exchange of GDP for GTP on their α subunit.
- GRKs
G-protein-coupled receptor kinases specifically phosphorylate agonist-activated GPCR, initiating the shutoff of G-protein-mediated signaling (desensitization). Humans have seven GRK subtypes (some with a few splice variants).
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author's institution, sharing with colleagues and providing to institution administration. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Kobilka BK. G protein coupled receptor structure and activation. Biochim. Biophys. Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore CA, et al. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 3.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 2004;25:105–111. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Goodman OB,, Jr, et al. β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 5.Laporte SA, et al. The β2-adrenergic receptor/barrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeWire SM, et al. β-Arrestins and cell signaling. Annu. Rev. Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 7.Gurevich EV, Gurevich VV. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fotiadis D, et al. Structure of the rhodopsin dimer: a working model for G-protein-coupled receptors. Curr. Opin. Struct. Biol. 2006;16:252–259. doi: 10.1016/j.sbi.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Chabre M, le Maire M. Monomeric G-protein-coupled receptor as a functional unit. Biochemistry. 2005;44:9395–9403. doi: 10.1021/bi050720o. [DOI] [PubMed] [Google Scholar]
- 10.White JH, et al. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- 11.Kaupmann K, et al. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- 12.Jones KA, et al. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- 13.Pin JP, et al. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 2005;272:2947–2955. doi: 10.1111/j.1742-4658.2005.04728.x. [DOI] [PubMed] [Google Scholar]
- 14.Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Ther. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Bulenger S, et al. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 2005;26:131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Meyer BH, et al. FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2138–2143. doi: 10.1073/pnas.0507686103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prinster SC, et al. α2C-adrenergic receptors exhibit enhanced surface expression and signaling upon association with β2-adrenergic receptors. J. Pharmacol. Exp. Ther. 2006;318:974–981. doi: 10.1124/jpet.106.106526. [DOI] [PubMed] [Google Scholar]
- 18.Uberti MA, et al. Heterodimerization with β2-adrenergic receptors promotes surface expression and functional activity of α1D-adrenergic receptors. J. Pharmacol. Exp. Ther. 2005;313:16–23. doi: 10.1124/jpet.104.079541. [DOI] [PubMed] [Google Scholar]
- 19.Concepcion F, et al. The carboxyl-terminal domain is essential for rhodopsin transport in rod photoreceptors. Vision Res. 2002;42:417–426. doi: 10.1016/s0042-6989(01)00195-x. [DOI] [PubMed] [Google Scholar]
- 20.White JF, et al. Dimerization of the class A G protein-coupled neurotensin receptor NTS1 alters G protein interaction. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12199–12204. doi: 10.1073/pnas.0705312104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huttenrauch F, et al. G protein-coupled receptor kinases promote phosphorylation and β-arrestin-mediated internalization of CCR5 homo- and hetero-oligomers. J. Biol. Chem. 2005;280:37503–37515. doi: 10.1074/jbc.M500535200. [DOI] [PubMed] [Google Scholar]
- 23.Guo W, et al. Crosstalk in G protein-coupled receptors: changes at the transmembrane homodimer interface determine activation. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17495–17500. doi: 10.1073/pnas.0508950102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klco JM, et al. C5a receptor oligomerization. I. Disulfide trapping reveals oligomers and potential contact surfaces in a G protein-coupled receptor. J. Biol. Chem. 2003;278:35345–35353. doi: 10.1074/jbc.M305606200. [DOI] [PubMed] [Google Scholar]
- 25.Kota P, et al. Opsin is present as dimers in COS1 cells: identification of amino acids at the dimeric interface. Proc. Natl. Acad. Sci. U. S. A. 2006;103:3054–3059. doi: 10.1073/pnas.0510982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berthouze M, et al. Two transmembrane Cys residues are involved in 5-HT4 receptor dimerization. Biochem. Biophys. Res. Commun. 2007;356:642–647. doi: 10.1016/j.bbrc.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 27.Luttrell LM, et al. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 28.Fotiadis D, et al. Atomic-force microscopy: rhodopsin dimers in native disc membranes. Nature. 2003;421:127–128. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- 29.Liang Y, et al. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J. Biol. Chem. 2003;278:21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant M, et al. Agonist-dependent dissociation of human somatostatin receptor 2 dimers: a role in receptor trafficking. J. Biol. Chem. 2004;279:36179–36183. doi: 10.1074/jbc.M407310200. [DOI] [PubMed] [Google Scholar]
- 31.Cvejic S, Devi LA. Dimerization of the δ opioid receptor: implication for a role in receptor internalization. J. Biol. Chem. 1997;272:26959–26964. doi: 10.1074/jbc.272.43.26959. [DOI] [PubMed] [Google Scholar]
- 32.Hebert TE, et al. A peptide derived from a β2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 1996;271:16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- 33.Angers S, et al. Detection of β 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. U. S. A. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jastrzebska B, et al. Functional and structural characterization of rhodopsin oligomers. J. Biol. Chem. 2006;281:11917–11922. doi: 10.1074/jbc.M600422200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bayburt TH, et al. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J. Biol. Chem. 2007;282:14875–14881. doi: 10.1074/jbc.M701433200. [DOI] [PubMed] [Google Scholar]
- 36.Baneres JL, Parello J. Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J. Mol. Biol. 2003;329:815–829. doi: 10.1016/s0022-2836(03)00439-x. [DOI] [PubMed] [Google Scholar]
- 37.Damian M, et al. Asymmetric conformational changes in a GPCR dimer controlled by G-proteins. EMBO J. 2006;25:5693–5702. doi: 10.1038/sj.emboj.7601449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hlavackova V, et al. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J. 2005;24:499–509. doi: 10.1038/sj.emboj.7600557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ernst OP, et al. Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. Proc. Natl. Acad. Sci. U. S. A. 2007;104:10859–10864. doi: 10.1073/pnas.0701967104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whorton MR, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palczewski K, et al. Mechanism of rhodopsin kinase activation. J. Biol. Chem. 1991;266:12949–12955. [PubMed] [Google Scholar]
- 42.Binder BM, et al. Light activation of one rhodopsin molecule causes the phosphorylation of hundreds of others. A reaction observed in electropermeabilized frog rod outer segments exposed to dim illumination. J. Biol. Chem. 1990;265:15333–15340. [PubMed] [Google Scholar]
- 43.Shi GW, et al. Light causes phosphorylation of nonactivated visual pigments in intact mouse rod photoreceptor cells. J. Biol. Chem. 2005;280:41184–41191. doi: 10.1074/jbc.M506935200. [DOI] [PubMed] [Google Scholar]
- 44.Li J, et al. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 45.Palczewski K, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 46.Hirsch JA, et al. The 2.8Å crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 47.Han M, et al. Crystal structure of β-arrestin at 1.9Å: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 48.Sutton RB, et al. Crystal structure of cone arrestin at 2.3Å: evolution of receptor specificity. J. Mol. Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 49.Nair KS, et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanson SM, et al. Each rhodopsin molecule binds its own arrestin. Proc. Natl. Acad. Sci. U. S. A. 2007;104:3125–3128. doi: 10.1073/pnas.0610886104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strissel KJ, et al. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J. Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanson SM, et al. Structure and function of the visual arrestin oligomer. EMBO J. 2007;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Celver J, et al. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 54.Kovoor A, et al. Targeted construction of phosphorylation-independent β-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 55.Vishnivetskiy SA, et al. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J. Biol. Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 56.Vishnivetskiy SA, et al. How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- 57.Gurevich VV, et al. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 58.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 59.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol. Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim YM, Benovic JL. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J. Biol. Chem. 2002;277:30760–30768. doi: 10.1074/jbc.M204528200. [DOI] [PubMed] [Google Scholar]
- 61.Hanson SM, et al. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. U. S. A. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daaka Y, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J. Biol. Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 63.DeGraff JL, et al. Role of arrestins in endocytosis and signaling of α2-adrenergic receptor subtypes. J. Biol. Chem. 1999;274:11253–11259. doi: 10.1074/jbc.274.16.11253. [DOI] [PubMed] [Google Scholar]
- 64.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jordan BA, et al. Oligomerization of opioid receptors with β 2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. U. S. A. 2001;98:343–348. doi: 10.1073/pnas.011384898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shcherbakova OG, et al. Organization of β-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J. Cell Biol. 2007;176:521–533. doi: 10.1083/jcb.200604167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brum PC, et al. Differential targeting and function of α2A and α2C adrenergic receptor subtypes in cultured sympathetic neurons. Neuropharmacology. 2006;51:397–413. doi: 10.1016/j.neuropharm.2006.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vilardaga JP, et al. Differential conformational requirements for activation of G proteins and the regulatory proteins arrestin and G protein-coupled receptor kinase in the G protein-coupled receptor for parathyroid hormone (PTH)/PTH-related protein. J. Biol. Chem. 2001;276:33435–33443. doi: 10.1074/jbc.M011495200. [DOI] [PubMed] [Google Scholar]
- 69.Norgaard-Nielsen K, et al. Zn(2+) site engineering at the oligomeric interface of the dopamine transporter. FEBS Lett. 2002;524:87–91. doi: 10.1016/s0014-5793(02)03008-9. [DOI] [PubMed] [Google Scholar]
- 70.Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]