

Abstract
Object
Central nervous system axons regenerate poorly after traumatic brain injury (TBI), partly due to inhibitors such as the protein Nogo-A present in myelin. The authors evaluated the efficacy of anti–Nogo-A monoclonal antibody (mAb) 7B12 administration on the neurobehavioral and cognitive outcome of rats following lateral fluid-percussion brain injury, characterized the penetration of the 7B12 or control antibodies into target brain regions, and evaluated the effects of Nogo-A inhibition on hemispheric tissue loss and sprouting of uninjured motor tracts in the cervical cord. To elucidate a potential molecular response to Nogo-A inhibition, we evaluated the effects of 7B12 on hippocampal GAP-43 expression.
Methods
Beginning 24 hours after lateral fluid-percussion brain injury or sham injury in rats, the mAb 7B12 or control antibody was infused intracrebroventricularly over 14 days, and behavior was assessed over 4 weeks.
Results
Immunoreactivity for 7B12 or immunoglobulin G was detected in widespread brain regions at 1 and 3 weeks postinjury. The brain-injured animals treated with 7B12 showed improvement in cognitive function (p < 0.05) at 4 weeks but no improvement in neurological motor function from 1 to 4 weeks postinjury compared with brain-injured, vehicle-treated controls. The enhanced cognitive function following inhibition of Nogo-A was correlated with an attenuated postinjury downregulation of hippocampal GAP-43 expression (p < 0.05).
Conclusions
Increased GAP-43 expression may be a novel molecular mechanism of the enhanced cognitive recovery mediated by Nogo-A inhibition after TBI in rats.
Keywords: cognition, GAP-43, neurological motor deficits, Nogo-A, regeneration, traumatic brain injury
WIDESPREAD damage to axons and white matter tracts, termed diffuse axonal injury, is an almost universal finding in cases of mild to severe TBI, and even minor degrees of axonal injury may contribute to cognitive and behavioral sequelae of brain-injured patients.2,6 This pathological feature of human TBI has been replicated in clinically relevant animal models of TBI10,25,38 and may contribute to the cognitive and behavioral dysfunction often observed in brain-injured patients.49
Spontaneous regeneration of axons or rearrangement of fiber tracts is limited following traumatic injury to the CNS. This limited regenerative response may be linked to insufficient growth response of surviving neurons to injury, the presence of factors that inhibit axonal outgrowth, and/or the presence of a glial scar.24 Further research has established that myelin contains factors that may contribute to inhibition of axonal outgrowth, including MAG, oligodendrocyte–myelin glycoprotein, and Nogo.17,24 Nogo, MAG, and oligodendrocyte–myelin glycoprotein all bind with high affinity to the neuronal NgR,21,65 which forms a receptor complex with LINGO-1, p75, and/or TAJ/TROY, a member of the tumor necrosis factor receptor superfamily, and activates intracellular guanosine triphosphatases, including RhoA, to mediate axon growth inhibition.58,65 The Nogo-A protein has several components that are important to its function as an inhibitor of axonal outgrowth, including an extracellular 66-amino acid region common to all isoforms of Nogo (Nogo-66), and a Nogo-A specific domain.20,44 In addition, a third active site on the Nogo-A molecule has recently been demonstrated1 that affects mainly fibroblasts, endothelial cells, and smooth muscle cells. Although Nogo-A was originally believed to be pre-dominantly a product of oligodendrocytes, it has also been demonstrated in neuronal subtypes. We have recently reported that Nogo-A expression is increased in both neurons and oligodendrocytes in the ipsilateral cortex, white matter tracts, hippocampus, and thalamus of rats from Days 1 to 7 after a lateral fluid-percussion brain injury.35
Promotion of axonal outgrowth for the treatment of CNS injury has attracted much interest,11,32 and neutralization of myelin inhibitors enhances regenerative sprouting and axonal regeneration following injuries such as SCI and ischemia.8 Administration of the mAb IN-1, shown to bind to and neutralize Nogo-A,16 has consistently improved functional recovery in models of CNS injury including SCI, CST lesion, and cerebral ischemia.8,19,57 Functional recovery after a CNS injury and neutralization of Nogo-A by IN-1 have been attributed to regeneration of severed axonal fiber tracts8,56 or compensatory sprouting from uninjured tracts.60
Growth-associated protein-43 is an important regulator of axonal outgrowth and is increased in vulnerable brain legions, including the hippocampus, following experimental TBI.10,28 The increased expression of GAP-43 after TBI is probably beneficial and may represent an intrinsic attempt by the injured CNS to promote axonal regeneration following TBI.4,15 Overexpression of GAP-43 was observed to overcome myelin inhibition and increase dorsal root ganglion neuron regeneration following SCI.23 The administration of IN-1 was shown to increase the expression of GAP-43 in the spinal cord of adult rats, suggesting a link between Nogo-A inhibition and GAP-43 expression.4
As a step toward clinical applicability, novel affinity-purified mAbs to Nogo-A of the IgG1-subtype (7B12 and 11C7) have been generated44,53 that can be delivered by osmotic minipumps. The mAb 7B12 has recently been shown to improve functional outcome after photothrombotic and focal ischemia in rats64 and in SCI,34 and treatment with the Nogo-inhibitor mAb 11C7 was formerly observed to improve cognitive outcome following lateral fluid-percussion brain injury in rats without influencing hippocampal cell loss.33 Because the mechanisms responsible for the improved cognitive outcome observed with inhibition of Nogo-A following SCI or TBI remain unknown, we evaluated the efficacy of mAb 7B12 administration on the neurobehavioral and cognitive outcome following lateral fluid-percussion brain injury, characterized the penetration of the 7B12 or control antibodies into target brain regions, and evaluated the effects of Nogo-A inhibition on hemispheric tissue loss and sprouting of uninjured motor tracts in the cervical spinal cord. To elucidate a potential molecular response to Nogo-A inhibition, we evaluated the effects of 7B12 on hippocampal GAP-43 expression.
Materials and Methods
Experimental Protocol
Animals were allowed to live for 1, 3, or 6 weeks after sham- or fluid-percussion brain injury. Outcome measures included behavioral analysis (6 weeks survival only) and histological analysis according to the experimental design described in Fig. 1. All procedures described herein were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and were performed in accordance with standards published by the National Research Council.43 All animals were housed in pairs with food and water ad libitum on a 12-hour light/dark cycle. The animals were kept in the colony for 1 week prior to any surgical procedures.
Fig. 1.
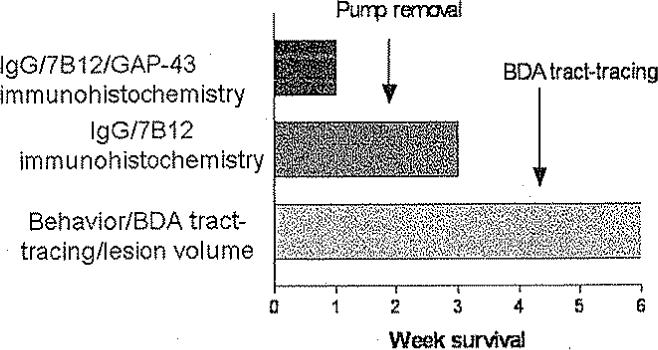
Graph of the experimental design. Animals were allowed to live for 1, 3, or 6 weeks after sham or brain injury. Behavioral analysis was performed in animals with 6–week survival only.
Animals and Surgery
Eighty-nine male Sprague–Dawley rats (mean weight 372 ± 14 g) were anesthetized using sodium pentobarbital (60 mg/kg administered intraperitoneally) and surgically prepared for lateral fluid-percussion brain injury as originally described.39 Briefly, a 5-mm craniotomy was performed over the left parietal cortex, midway between the lambda and the bregma. The dura mater was left intact, and a hollow female Luer-Lok fitting was positioned over the craniectomy and held in place with dental cement. Animals were attached to the fluid percussion device (a saline-filled cylinder) via the Luer-Lok fitting, and brain injury of moderate severity (3.1 ± 0.2 atm) was produced in 55 rats by injecting a bolus of saline at high pressure into the closed cranial cavity. After the injury, the Luer Lok was removed, and the skin was sutured. The 34 sham-injured control animals were anesthetized and surgically prepared as previously described but were not subjected to brain injury. While anesthetized, animal body temperature was maintained with a thermostatically controlled heating pad maintained at 37°C until the animals were able to ambulate. The animals were allowed to live for 1, 3, or 6 weeks (Fig. 1).
Infusion of 7B12 and Implantation of Osmotic Minipumps
At 24 hours after brain injury, immediately following the 24-hour composite neuroscore test, all animals were randomly assigned to receive either highly purified mAb 7B1244 or a control IgG mAb (mouse IgG; Chemicon) in 1X sterile, calcium- and magnesium-free phosphate buffered saline at pH 7.4. The mAb 7B12 was produced against the recombinant prokaryotically produced Nogo-A fragments 1−979, was monospecific to Nogo-A on Western blots, and recognized the C-terminal part of the Nogo-A specific region.34 Osmotic minipumps (ALZET 2ML2), filled with 2 ml mAb solution at 1.75 mg/ml under sterile conditions and delivering 5 μl/hour for 2 weeks, were attached with a 35-mm-long polyethylene catheter to a cannula for intracerebroventricular infusion. The pump assembly was primed overnight at 37°C in 0.9% sodium chloride.
Twenty-four hours after injury, the animals were placed in an induction chamber and anesthetized with 3.5% isoflurane in O2 for 2 minutes and then placed in a stereotactic frame, where 2.5% isoflurane in O2 was delivered through a nose cone. Using a strict aseptic technique, a subcutaneous pocket was created between the scapulae by blunt dissection. A ventricular catheter was inserted through a separate bur hole anterior to the craniectomy and into the left ventricle, using the following coordinates in relation to the bregma: antero-posterior-0.8 mm, lateral-1.3 mm, depth-3.8 mm. The catheter was secured to the bone with tissue adhesive (Vetbond; 3M Corp.), and the wound was sutured after the completion of pump implantation. Following evaluation of the composite neuroscore at 24 hours postinjury, the animals were randomized to receive an intracerebroventricular infusion of either 7B12 (27 brain-injured rats, 16 sham-injured rats) or control IgG (23 brain-injured rats, 18 sham-injured rats) for as long as 2 weeks. At postinjury Day 14, all animals were reanesthetized with isoflurane as described earlier, and the pump assembly was removed. The bur hole was closed with bone wax to prevent cerebrospinal fluid leakage.
Brain Penetration of 7B12 and Control IgG
For immunohistochemical detection of mAb 7B12 and control mouse IgG penetration into brain tissue, three consecutive coronal brain sections (−2.3, −4.3, and −6.0 mm from bregma) from brain- or sham-injured animals with 1 and 3 weeks' survival were selected. The sections were washed in TBS and then treated for 30 minutes with 3% (vol/vol) of 30% hydrogen peroxide in 50% methanol, rinsed, and placed in 5% normal horse serum in TBS for 30 minutes. For detection of 7B12 and IgG, a biotinylated donkey anti–mouse secondary antibody (1:1000; Jackson) dissolved in blocking buffer was applied and incubated overnight at 4°C. The sections were then rinsed with TBS and incubated for 1 hour at room temperature with the ABC reagent (Vector Labs), washed, and developed with diaminobenzidine solution (Vector labs). The sections were rinsed, mounted, cleared using xylenes, coverslipped, and analyzed using a light microscope (Nikon Eclipse 8600). Images were captured using a camera (Magnafire).
Behavioral Evaluation
All motor function and cognitive tests were conducted by trained investigators who were blinded to the treatment regimen and injury status of the animals.
Composite Neuroscore
Animals allowed to live for 6 weeks were subjected to a battery of neurological motor function tests, the composite neuroscore, at 24 hours and weekly at 1, 2, 3, and 4 weeks postinjury. At 24 hours postinjury, the composite neuroscore was used to stratify the animals into two equal treatment groups. The composite neuroscore consisted of forelimb reflex, hindlimb flexion, lateral pulsion, and angle board tests, and a score from 0 to 4 was given for the performance of each task.26 The forelimb reflex test consisted of rapid introduction of a flat surface to the rat when suspended by the tail. The strength and position of the forelimbs were examined and recorded. The hindlimb flexion test examined the strength of a backward extension of the hindlimbs when the animal's tail was lifted and the forelimbs remained on a hard surface. Lateral pulsion evaluated the animal's ability to resist hand pressure to move from left to right. The angle board test evaluated the rats' ability to stand on an inclined plane in a right, left, and vertical position as compared with a baseline performance. Scores for the angle board test were: 4, above or equal to baseline; 3, 2.5° below baseline; 2, 5° below baseline; 1, 7.5° below baseline; and 0, 10° or more below baseline. The angle board tests were averaged across the three positions, and all other tests were added to achieve a maximum composite neuroscore of 28 points.26
Morris Water Maze Test of Cognitive Function
A blinded observer evaluated visuospatial learning ability using the MWM paradigm as previously described.42 The MWM consisted of a circular tank painted black, 1.8 meters in diameter, filled with water maintained at a temperature of 24 to 26°C. A black, circular (10-cm diameter) plexiglas platform, onto which the animals could escape, was submerged 1 cm below the surface of the water and placed in a standard position (southwest quadrant of the tank). The animals' ability to find the platform after being introduced at one of four designated entry points (west, north, east, and south) facing the maze wall was then assessed by subjecting them to 24 training trials over a 3-day interval (8 trials/day, maximal duration 60 sec per trial) at 4 weeks postinjury. The latencies to reach the platform were recorded and averaged for each block of four trials. Seventy-two hours after the last trial, the animals were evaluated in the MWM for their ability to recall the previously learned task (memory probe trial). Each animal was allowed 60 seconds to swim in the maze with the platform removed, while its swim pattern was tracked. The MWM was divided into specified zones, and a memory score was calculated for each animal by multiplying the amount of time spent in these zones by a weighting factor according to its proximity to the platform. These assigned numbers were multiplied by the number of seconds spent in the corresponding zone and totaled according to a paradigm originally described by Smith and colleagues59 and used in numerous previous reports26,36 for the assessment of postinjury memory function. The average of the two trial scores or the first score, if that was higher than the second trial score, was used as the numeric score for the probe trial. Following each MWM trial, the animals were placed under a ceramic lamp to avoid a drop in body temperature.
Evaluation of BDA Tract-Tracing
Following cognitive evaluation at 4 weeks postinjury, a randomly selected subset of animals was flown to the laboratory of Dr. Martin Schwab in Zurich, Switzerland, and assigned to receive injections of BDA (BDA-10,000 Neuronal Tracer Kit; Molecular Probes) into the contralateral (right) sensorimotor cortex for evaluation of the reorganization of corticospinal projections and the number of cervical midline crossings of axon collaterals as previously described in detail.4 The subset of animals consisted of 23 rats; 10 of these rats were from the sham-injured group (five treated with 7B12 and five with mouse IgG) and 13 were from the brain-injured group (seven treated with 7B12, six with mouse IgG). Briefly, animals were anesthetized using a combination of subcutaneous Hypnorm (0.3 mg/kg; Roche) and Dormicum (0.6 mg/kg; Roche) and placed in a stereotactic frame. Pressure injections of 600 nl of a 10% solution of BDA in 0.01 M phosphate buffer (pH 7.4), at a rate of 1 μl/minute were made into the contralateral sensorimotor cortex (using the coordinates of 0.5 and 2 mm posterior to the bregma, 2.5 mm lateral to the midline, and 1.5 mm below the surface of the cortex) through a glass capillary that remained in its position for 2 minutes after the end of the injection.4 Injection variability was controlled for by blinded randomization of the animals and postmortem histological verification of needle placement and injection site.
To kill the animals, all animals received a lethal dose of sodium pentobarbital and transcardially perfused with 0.9% saline containing 1000 U of heparin/L followed by 4% paraformaldehyde. The brains were removed and postfixed overnight at 4°C in paraformaldehyde, transferred into 30% sucrose solution for 3 to 4 days, snap frozen, and stored at −80°C. In animals receiving the contralateral cortical injections of BDA, the spinal cord and brain were removed after perfusion at 6 weeks postinjury and processed as previously described.4 Corticospinal projections were quantified by an observer blinded with respect to the treatment and injury status of the animal. The number of axon fibers crossing the midline in the cervical cord was counted in 40 consecutive transverse sections using a light microscope at a magnification of 200, beginning at the standard C-4 level and continuing for the subsequent 40 sections (cervical and lumbar sections to L-3). To correct for injection variability, quantification of the total number of fibers labeled in the main CST was also performed under light microscopy at a magnification of 400 in three separate sections at C-3 to generate a ratio of sprouting fibers per labeled main CST axon for each animal to account for differences in BDA labeling between individual rats.4
Expression of GAP-43
Brain sections from all animals (one section per animal at bregma −4.5 mm) with 1 week survival were evaluated as previously described.14 Following quenching of endogenous peroxidases and blocking nonspecific binding with 5% normal horse serum, the primary rabbit anti–GAP-43 polyclonal antibody (1:1000; Chemicon) was dissolved in blocking buffer and incubated overnight at 4°C. A secondary biotinylated antibody (1:1000, donkey anti–rabbit; Jackson) was applied for 1 hour at room temperature. Following visualization with ABC reagent and diaminobenzidine (Vector Labs), the sections were mounted, cleared in xylene and coverslipped. All immunohistochemical analyses for GAP-43 in the brains from animals with 1-week survival postinjury were performed at the same time point by the same investigator; the control and injured specimens were prepared under identical conditions in the same batch runs. Negative controls consisted of sections identically treated although with omission of the primary antibody. Because stereological methods could not be used on the present material, we chose to use densitometry to evaluate GAP-43 expression in the stratum oriens, stratum radiatum, stratum lacunosum moleculare, and inner molecular layer of the CA1 region of the hippocampus. The CA1 region was chosen based on our earlier work documenting increased GAP-43 expression with this model of TBI14 and because cells in the CA3 region are significantly more damaged and less morphologically intact in the lateral fluid-percussion model of brain injury. Under low-power light microscopy, an image was captured at a magnification of 40 by a camera (Magnafire), and the exposure time was kept identical for each image. The borders of each region were traced manually and the pixels per area were then automatically calculated using an image analysis software routine (MCID/M4) after defining a threshold for the background that was kept identical for each brain throughout the analysis.4 The percentage of the evaluated area stained for GAP-43 was determined for each animal, and data are presented as a percentage change compared with the mean of the sham-injured, IgG-treated controls as previously described.14
Loss of Hemispheric Tissue
Animals not subjected to BDA tract-tracing were perfused at 6. weeks postinjury, and selected for evaluation of loss of hemispheric tissue as previously described for frozen sections.66 This sample consisted of 17 rats; 10 were in the brain-injured group (five treated with 7B12 and five with mouse IgG), and seven were in the sham-injured group (three treated with 7B12 and four with mouse IgG). Coronal serial frozen sections (40 μm thick) were cut on a sliding microtome (Micron Gmbh) and taken every 1 mm from 0.3 mm to −6.3 mm posterior to the bregma.47 Sections were stained with 0.3% Cresyl violet and imaged using a digital camera (Imaging Research, Inc.) integrated with a light microscope. The periphery of the contralateral and ipsilateral hemispheres was traced on each image by an evaluator blinded to the injury and treatment status of the animals. The area of each hemisphere was calculated using a calibrated image analysis routine (MCID M4; Imaging Research). Based on previous investigations, which showed negligible contralateral tissue loss following lateral fluid-percussion injury, the contralateral hemisphere was used in each section to control for interanimal variation in brain size.66 Hemispheric tissue loss was calculated as a percentage of the contralateral (uninjured) hemisphere volume (Vc) using the following formula: [(Vc – Vi)/(Vc)] × 100, in which Vi represents the volume of the ipsilateral (injured) hemisphere. To calculate hemispheric volume, areas were integrated over the 8 mm rostrocaudal distance. The mean lesion volume per animal, based on calculations of two separate series of sections, is presented (to account for possible tissue shrinkage in frozen tissue).
Statistical Analysis
Data were evaluated for Gaussian (normal) distribution, and parametric data (lesion volume and memory score) are presented in figures as means ± standard deviations (SD), and analyzed using a two-way ANOVA. If the probability value was less than 0.05, the Neumann–Keuls post hoc test was performed.
Nonparametric data (composite neuroscore, tract-tracing ratios, GAP-43 ratios) were analyzed using a Kruskal–Wallis ANOVA and, if significant, were further analyzed using the Mann–Whitney U-test for pairwise comparisons. These data are presented as medians ± the 75th percentile range, or as individual data points.
Morris water maze latencies did not follow a Gaussian distribution and were analyzed using the Kruskal–Wallis ANOVA followed by the Mann–Whitney U-test for each trial block For clarity, MWM latencies are presented as means ± standard error of the means (SEM). A probability value of less than 0.05 was considered significant. Data were analyzed using Statistica (StatSoft) and Prizm (Graph Pad Software) software.
Results
Brain injury consistently produced an apnea immediately postinjury (mean 29.3 ± 10 sec). Five (9%) of 55 animals were excluded due to injury-induced factors (death, apnea more than 60 seconds, or dural breach). In addition, One animal was excluded due to pump infection, eight animals died following pump removal, and two were excluded due to postinjury weight loss.
Penetration of mAb 7B12 and Control Mouse IgG
By 1 week after both sham and brain injury in animals treated with 7B12 or IgG, a diffuse, intense immunoreactive staining for mouse IgG was observed in widespread brain regions with a similar pattern of distribution for 7B12 and control IgG (Fig. 2A). The 7B12 or control antibodies could both be detected close to the ipsilateral ventricle with a gradient into adjacent tissue, extending into the contralateral side with more intense staining in the anterior part of the brain (data not shown). In all animals, IgG staining was observed in the external capsule, thalamus, and hippocampus, whereas no staining or only faint staining was observed in cortical tissue. Compared with sham-injured controls, brain-injured animals showed enhanced IgG accumulation in the external capsule and lesion area, ipsilateral to the injury. By 3 weeks postinjury, immunoreactivity for both IgG and 7B12 was still detectable in approximately 50% of the brains regardless of injury status (Fig. 2B). The IgG immunoreactivity persisted mainly in the periventricular region, external capsule, and hippocampus ipsilateral to the injury, and was more marked in anterior sections close to the infusion site. In a subset of brains, evidence of cellular penetration of 7B12, but not control IgG, was observed within the external capsule (data not shown). These results suggest that the infusion of 7B12 or control IgG reached vulnerable brain regions and that the immunoreactivity persisted up to 3 weeks postinjury (1 week following termination of infusion) in important brain regions and white matter tracts.
Fig. 2.
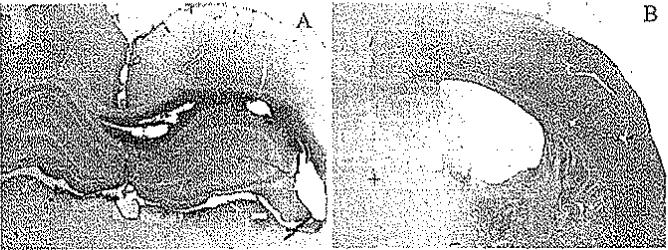
Photomicrographic examples of 7B12 penetration in brain-injured animals after either 1 (A) or 3 (B) weeks of survival. A: At 1 week postinjury, marked bilateral immunoreactivity for IgG was observed, including both hippocampi. B: At 3 weeks postinjury, less marked immunoreactivity for IgG was observed, although still clearly present in the cortex and white matter tract ipsilateral to the injury. An area without immunoreactivity for mouse IgG is also shown (+). Original magnification × 10.
Behavioral Evaluation
Composite Neuroscore
At 24 hours postinjury, a significantly lower composite neuroscore was observed in brain-injured animals when compared with sham-injured animals (p < 0.05; Fig. 3). These scores were used to stratify the animals into treatment groups (median composite neuroscore of 9.5 for 7B12-treated, brain-injured animals compared with 10.0 for brain-injured, IgG-treated animals). At 24 hours and at 1, 2, and 4 weeks postinjury, brain-injured animals, irrespective of treatment status, had a significantly lower composite neuroscore than sham-injured animals (p < 0.05; Fig. 3). No significant differences in composite neuroscore were observed among the 7B12-and IgG-treated groups from 24 hours to 4 weeks postinjury.
Fig. 3.
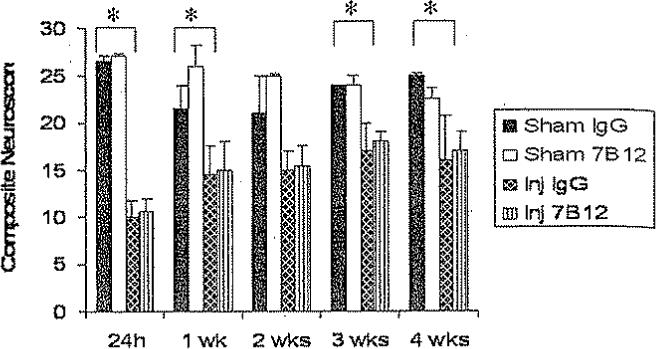
Bar graph comparing the composite neuroscore (medians ± 75th percentile) among the treatment groups. The maximum achievable score was 28. The results at 24 hours were used for stratification into treatment groups. Following initiation of treatment, brain-injured, vehicle-treated animals had consistently lower composite neuroscores than sham-injured controls. *p < 0.05; p = 0.05 at 2 weeks postinjury. There were no significant differences among the 7B12– and IgG-treated groups. Inj = brain-injured.
Cognitive Function
Brain-injured animals, irrespective of treatment status, had longer latencies to reach the platform of the MWM at 4 weeks postinjury (p < 0.05; Fig. 4, upper). The results from the probe trial showed that brain-injured animals had significantly lower memory scores than sham-injured controls (p < 0.05; Fig. 4, lower). The 7B12-treated, brain-injured animals had significantly improved memory scores when compared with brain-injured, IgG-treated controls (p < 0.05; Fig. 4, lower). No differences in swim speed or swim distance were observed among the treatment group (data not shown).
Fig. 4.
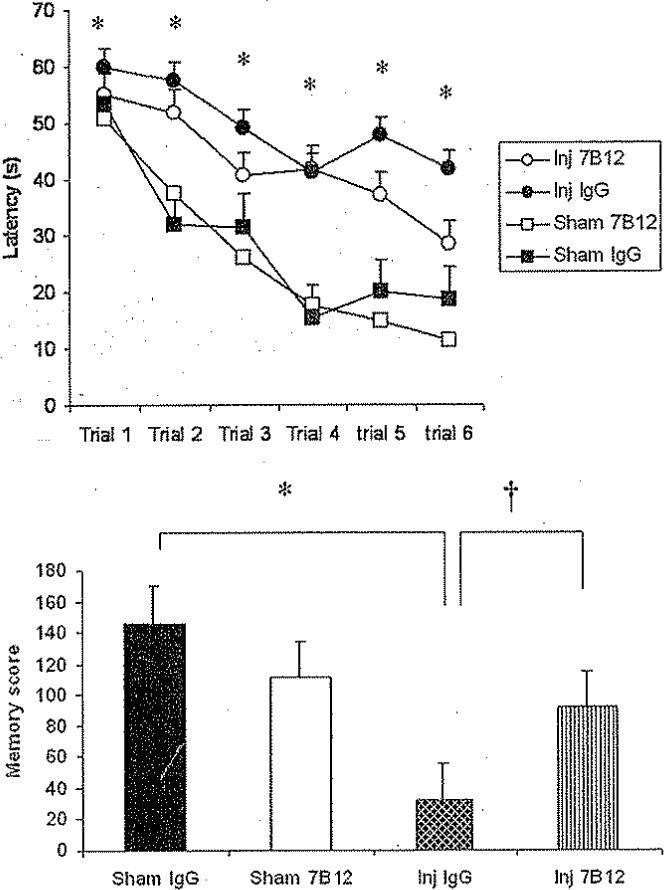
Line graph (upper) and bar graph (lower) showing the MWM trial latencies and memory scores, respectively. Upper: At 4 weeks postinjury, brain-injured, vehicle-treated animals had significantly longer latencies to reach the MWM platform compared with sham-injured, vehicle-treated controls (*p < 0.05). Treatment with 7B12 consistently resulted in shorter latencies when compared with IgG treatment in brain-injured controls. Lower: Three days after the last trial, animals were evaluated in the MWM for their ability to recall the previously learned task (memory probe trial). Sham-injured, vehicle-treated (*p < 0.05) and 7B12-teated animals (†p < 0.05) both had significantly higher memory scores when compared with brain-injured, vehicle-treated animals.
Sprouting of Uninjured CST Axon Collaterals
Lateral fluid-percussion brain injury, regardless of treatment status, induced significant sprouting of unlesioned axon collaterals (at C-4) across the midline, expressed as a ratio of axon collaterals per labeled axon in the main CST (p < 0.05; Fig. 5). Treatment with 7B12 did not significantly alter CST sprouting when compared with IgG-treated, brain-injured controls (Fig. 5A).
Fig. 5.
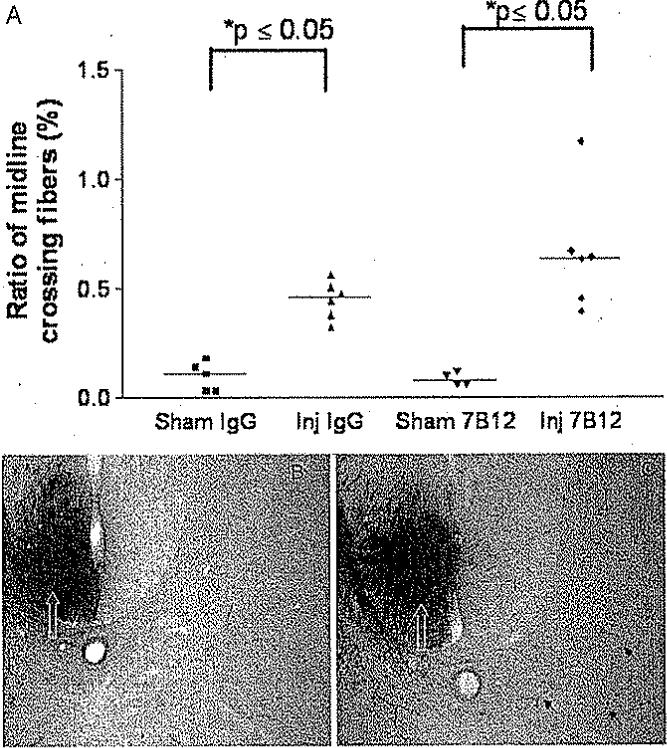
Graph (A) and representative photomicrographs (B and C) of the sprouting of uninjured CST axon collaterals. A: Comparison of the ratio of midline crossing fibers to the total number of labeled axons in the CST, ipsilateral to injury (medians). Fluid-percussion brain injury induced a significant increase in the ratio of midline crossing fibers, regardless of treatment status of the animals. Brain-injured, 7B12-treated animals had a slightly higher ratio when compared with brain-injured, vehicle-treated controls (p = 0.09). B and C: Images showing BDA labeling in the contralateral CST from a sham-injured (B) and brain-injured (C) animal are shown. Midline crossing (sprouting) axonal profiles in B and C are indicated by black arrows, and the decussating CSTs are indicated by white open arrows. Original magnification × 200.
Expression of GAP-43
The immunoreactivity of GAP-43 was observed in the stratum oriens, stratum radiatum, stratum lacunosum moleculare, and inner molecular layer regions of the ipsilateral (injured) hippocampal CA1 subfield of sham- and brain-injured animals (Fig. 6A and B) as previously described;14 there was no visible staining in the negative controls (data not shown). Densitometry analysis showed that 7B12-treated, brain-injured animals had a significantly higher mean GAP-43 expression in the CA1 subregion than brain-injured, vehicle-treated controls (p < 0.05; Fig. 6C).
Fig. 6.
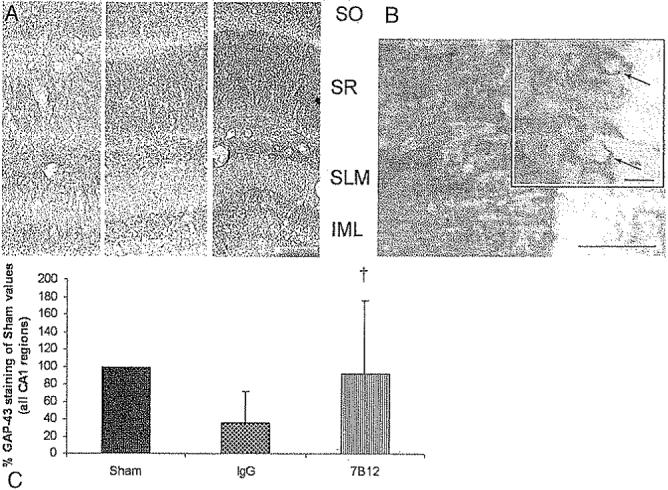
Representative photomicrographs (A and B) and bar graph (C) showing the results of the expression of GAP-43. A: Images showing a highly differentiated laminar pattern of GAP-43 expression at 1 week postinjury in the stratum oriens (SO), stratum radiatum (SR), stratum lacunosum moleculare (SLM), and inner molecular layer (IML) of the CA1 region of the hippocampus, a brain region with high GAP-43 expression. The three images were obtained in a sham-injured, vehicle-treated animal left, a brain-injured, vehicle-treated animal center, and a brain-injured, 7B12-treated animal right. Bar = 500 μm. B: Representative high-power image from the hippocampal CA1 area of a brain-injured, 7B12-treated animal. Staining was more marked in the axonal and dendritic layers than in the neuronal cytoplasm. Inset shows some neurons (arrows) with GAP-43 immunoreactivity in the soma and axon hillock. Bar = 50 μm, 20 μm (inset), C: Graph showing GAP-43 expression relative to sham-injured controls, evaluated by densitometry, in the CA1 region of the hippocampus at 1 week postinjury. The 7B12-treated, brain-injured animals had a significantly higher GAP-43 expression († p < 0.005) than the brain-injured, vehicle-treated animals (IgG).
Loss of Hemispheric Tissue
Fluid-percussion brain injury induced a significant loss of hemispheric tissue in vehicle-treated animals compared with sham-injured controls (p < 0.05; Fig. 7). Administration of 7B12 did not alter the extent of hemispheric tissue loss following TBI compared with IgG-treated, brain-injured controls (Fig. 7).
Fig. 7.
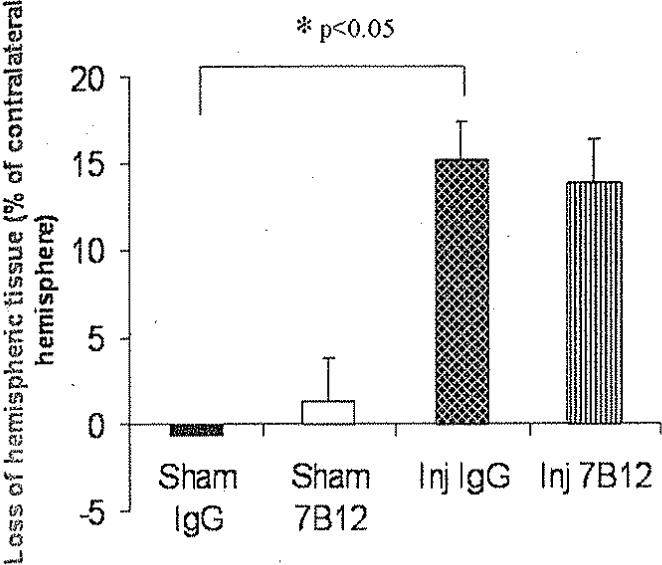
Bar graph showing the loss of hemispheric tissue ipsilateral to the fluid-percussion brain injury at 6 weeks postinjury. Brain injury, regardless of treatment status, caused a marked loss of hemispheric tissue (*p < 0.05) when compared with sham-injured, vehicle-treated controls without significant differences among the brain-injured groups.
Discussion
In the present study, we show that the administration of the novel anti–Nogo-A mAb 7B12, beginning 24 hours and continuing for 2 weeks postinjury, improved cognitive outcome following fluid-percussion brain injury. Unlike previous reports, we present convincing mechanistic data suggesting that significant penetration of 7B12 antibody occurs into target brain areas such as the ipsilateral hippocampus, cortex, and white matter tracts following intracerebroventricular administration into the traumatized brain. We also provide evidence for a novel molecular mechanism for the cognitive recovery mediated by Nogo-A inhibition, by showing that treatment with mAb 7B12 significantly increased the expression of GAP-43 in the hippocampal CA1 region at 1 week postinjury.
We chose the intracerebroventricular route of administration of anti–Nogo-A antibodies based on previous reports of experimental stroke, spinal cord injury, and TBI22,33,64 in which no negative toxic or behavioral effects have been observed. Because 7B12 is rapidly cleared from cerebrospinal fluid, a continuous infusion is needed, and the concentration and volume in combination with a limited diffusion within brain tissue excluded the possibility of intracerebral administration of 7B12. Anti–Nogo-A antibodies will not penetrate into brain tissue in sufficient concentration when administered systemically, and extensive preclinical evaluation, including good laboratory practice safety and toxicity testing in monkeys, has demonstrated a low toxicity following intrathecal infusion (AK Mir, personal communication, 2006). Our observations suggest that the 7B12 antibody is distributed with a gradient from the infusion site and is capable of penetrating into injured brain regions that may be critical for the plasticity observed following cortical lesions and neutralization of Nogo-A.29,46 Also, mAb 7B12 (or control mouse IgG) was found to penetrate into areas such as the cortex, external capsule, hippocampus, and thalamus, areas that have been recently reported to show marked up-regulation of Nogo-A in rats within the first week after fluid-percussion brain injury.35 Although a direct comparison between the degree of mAb penetration and the degree of functional recovery cannot be achieved using the present study design, our data suggest that prolonged neutralization of Nogo-A has beneficial effects on the process causing neurological recovery after TBI. Interestingly, mAb 7B12 has recently been shown to bind to both white and gray matter and to internalize, together with the bound Nogo-A, into neurons and oligodendrocytes from which they are delivered to lysosomes. Following intrathecal infusion of mAb 7B12, the tissue levels of Nogo-A in the spinal cords of intact rats were reduced.34,63
The Nogo-A inhibitory mAb IN-1, an antibody of the IgM/κ subclass, has been evaluated in other models of CNS injury and has frequently been delivered into the CNS via implanted antibody-producing hybridoma cells with concomitant immunosuppression. Although these techniques may cause tumor growth and immunological problems and result in poorly controlled production and secretion of the antibody,16 the present study suggests that the affinity-purified IgG antibody and the use of minipumps is a safe and effective delivery method. Although the stereotactic placement of cannulae into the injured hemisphere may be considered an additional injury, our vehicle-treated controls also had cannulae implanted in an identical fashion to control for these effects.
Although we did not detect an improvement in neurological motor function following TBI and Nogo-A inhibition, other investigators have observed functional motor recovery from 4 to 8 weeks after cerebral ischemia and IN-1 treatment, although not at earlier time points.46 It is therefore possible that longer observation periods may have revealed additional beneficial efficacy on gross motor function. Differential sensitivity for Nogo-A neutralization on the tests for motor function34 or different molecular targets may also contribute to these findings. Improvement of neurological motor function following inhibition of Nogo-A has also been observed following SCI, stroke, pyramidotomy, and CST lesions, even when administered as long as 1 week postinjury, and has been correlated with regeneration of severed fibers or increased sprouting from uninjured CSTs.57 In addition, investigators in previous reports have suggested that not all neuronal subpopulations respond to inhibition of Nogo-A or NgR.31,45 It may be argued that the tests of motor function used in the present report represent a reflexive motor task26 that depends on a functional CST, which has been shown to be insensitive to NgR inhibition.31 Because inhibition of MAG has recently been shown to improve motor outcome following fluid-percussion brain injury in rats,61 there may also be a redundancy among the myelin inhibitors in important motor tracts, thereby reducing the efficacy of Nogo-A inhibition.
In the present study, lateral fluid-percussion brain injury caused increased sprouting of axon collaterals from the uninjured CST that may be linked to the gradual but incomplete functional recovery observed in the lateral fluid-percussion brain injury model.48 Treatment with 7B12 was not associated with significantly increased sprouting of uninjured fiber tracts at 6 weeks postinjury. In a study using a similar tract tracing method after focal cerebral ischemia and administration of 7B12, an increased number of CST fibers crossing the midline and originating in the unlesioned sensorimotor cortex was observed 9 weeks after focal cerebral ischemia.64 Following neutralization of Nogo-A, increased corticostriatal plasticity has also been observed after cerebral ischemia.46 Sprouting of uninjured tracts following a unilateral CST lesion and IN-1 treatment has been shown to be transient (returning to baseline by 6 weeks),4 and marked reorganization of the contralateral uninjured cortex has also been demonstrated following a cortical cut lesion and IN-1 treatment.13 Although we did not observe an increase in CST sprouting following 7B12 administration, the optimal target when evaluating BDA tract tracing and sprouting from injured white matter tracts has not been determined for TBI models. As an example, mice deficient in Nogo-66 receptor showed increased regeneration of raphespinal and rubrospinal tracts following SCI, whereas the CST did not regenerate, suggesting a differential sensitivity to myelin inhibition between motor tracts.34 We cannot exclude the possibility that sprouting, facilitated by Nogo-A inhibition, occurs in regions and neuronal systems other than the upper cervical spinal cord or that sprouting may have been present at other time points postinjury.
In the present report, we observed that inhibition of Nogo-A improved cognitive performance in an MWM paradigm. To our knowledge, these data are the first to demonstrate cognitive efficacy following brain injury and Nogo-A inhibition using 7B12, which belongs to a new generation of purified, monoclonal anti–Nogo-A antibodies. The involvement of the hippocampus in cognitive processes is now well established. The direct monosynaptic pathway from the CA1 and subiculum to the prefrontal cortex can express different forms of plasticity, including long-term potentiation and long-term depression, and is involved in specific aspects of learning and memory consolidation.51 It has been suggested that Nogo-A contributes to hippocampal pathology following kainic acid–induced seizures and perforant pathway lesions,40,41 and regeneration of lesioned acetylcholine-positive septohippocampal axons has been noted after IN-1 treatment following fimbria/fornix lesions.9
Together, available data suggest that Nogo-A may be involved in the development of posttraumatic hippocampal pathology and that inhibition of Nogo-A may increase recovery of hippocampal function after TBI although the underlying molecular mechanisms remain elusive. Hippocampal plasticity has been demonstrated to occur following experimental TBI,30,54 but no previous reports have evaluated the effect of myelin inhibitors on posttraumatic plasticity. To evaluate a possible 7B12-mediated mechanism of cognitive recovery, we next evaluated CA1 hippocampal expression of neuronal GAP-43, an intrinsic determinant of neuronal plasticity5,55 and a molecular indicator of axonal plasticity in the CNS.10,14,28,62 The protein GAP-43, concentrated at growth cones and suggested to play an important role in axonal outgrowth, has been well characterized as an intracellular membrane-associated phosphoprotein, present in neuronal connections during synaptic remodeling and neuronal sprouting.62 To our knowledge, GAP-43 is solely expressed in neurons in the adult rat brain. Increased GAP-43 expression has been postulated to correlate with behavioral recovery following TBI.28 Overexpression of GAP-43 and cytoskeletal-associated protein-23 markedly enhanced spinal cord axon regeneration in vivo,7 and mice that overexpressed GAP-43 showed increased sprouting of adult Purkinje cells.23 Evidence for the role of GAP-43 in hippocampal plasticity was strengthened by a study showing a role in synaptic remodelling in the denervated rat hippocampus.37 Additionally, mice overexpressing GAP-43 show increased neuronal sprouting,3,27 learning, and long-term potentiation.52 In contrast, hippocampal-dependent memory is impaired in heterozygous GAP-43 knockout mice.50
We found a higher GAP-43 expression in the CA1 region of the hippocampus in brain-injured animals at 1 week postinjury compared with mouse IgG-treated controls. Inhibition of Nogo-A and Rho, downstream effectors of Nogo-66/NgR signaling, has previously been shown to increase GAP-43 expression or mRNA following unilateral pyramidotomy and spinal cord injury.4,12 A change in GAP-43 expression has been observed following lateral fluid-percussion brain injury in the CA1 region of the hippocampus.11 Growth-associated protein-43 may be an important promoter of axonal outgrowth following CNS injury.15
Because the expression of GAP-43 was evaluated at 1 week postinjury, in contrast to the cognitive tests at 4 weeks, a direct correlation between immunohistochemistry and behavior cannot be performed. Because increased and/or sustained GAP-43 expression may be related to enhanced intrinsic recovery mechanisms and may be an additional mechanism of overcoming myelin inhibition of axonal outgrowth,18,23 it appears that the early increase in GAP-43 may contribute to the cognitive recovery observed in the present report. Although neuroprotective effects were not observed following TBI and 7B12 administration, we cannot exclude the possibility that earlier initiation of treatment might have generated more beneficial results. Further studies are needed to establish the putative neuroprotective properties of Nogo-A inhibition in TBI.
Conclusions
In the present study, we reported that treatment with the Nogo-A neutralizing antibody 7B12, beginning at a clinically relevant time point postinjury, increased regional expression of GAP-43 protein, believed to be associated with synaptic plasticity, and significantly improved the cognitive outcome following experimental brain trauma. These novel observations suggest that Nogo-A may be an important contributor to the pathophysiology of TBI, and treatment strategies targeting Nogo-A may be a suitable approach for further development into the clinical stage.
Acknowledgments
We express our gratitude to Mrs. Jeanne Marks for editing the manuscript and to Novartis Inc., Basel, Switzerland, for kindly supplying the antibody. We also wish to thank Rishi Puri, Rachel Hoover, Marie Millard, Carrie Keck, David LeBold, Zacharias Spangler, Kristie Soltesz, and Asenia McMillan for their excellent technical support.
This work was supported by NIH Grant No. NS R01–40978, a merit review grant from the Veterans Administration, and by NIH Grant No. NS P50–08803. Niklas Marklund is partly supported by a grant from the Swedish Brain Injury Foundation.
Abbreviations used in this paper
- ANOVA
analysis of variance
- BDA
biotinylated dextran amine
- CNS
central nervous system
- CST
corticospinal tract
- GAP
growth-associated protein
- Ig
immunoglobulin
- mAb
monoclonal antibody
- MAG
myelin-associated glycoprotein
- MWM
Morris water maze
- NgR
Nogo-66 receptor
- SCI
spinal cord injury
- TBI
traumatic brain injury
- TBS
Tris-buffered saline.
References
- 1.Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, et al. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–388. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- 2.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 3.Aigner L, Arber S, Kapfhammer JP, Laux T, Schneider C, Botteri F, et al. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 4.Bareyre FM, Haudenschild B, Schwab ME. Long-lasting sprouting and gene expression changes induced by the monoclonal antibody IN-1 in the adult spinal cord. J Neurosci. 2002;22:7097–7110. doi: 10.1523/JNEUROSCI.22-16-07097.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- 6.Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet. 1994;344:1055–1056. doi: 10.1016/s0140-6736(94)91712-4. [DOI] [PubMed] [Google Scholar]
- 7.Bomze HM, Bulsara KR, Iskandar BJ, Caroni P, Skene JH. Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci. 2001;4:38–43. doi: 10.1038/82881. [DOI] [PubMed] [Google Scholar]
- 8.Buchli AD, Schwab ME. Inhibition of Nogo: a key strategy to increase regeneration, plasticity and functional recovery of the lesioned central nervous system. Ann Med. 2005;37:556–567. doi: 10.1080/07853890500407520. [DOI] [PubMed] [Google Scholar]
- 9.Cadelli D, Schwab ME. Regeneration of lesioned septohippocampal acetylcholinesterase-positive axons is improved by antibodies against the myelin-associated neurite growth inhibitors NI-35/250. Eur J Neurosci. 1991;3:825–832. doi: 10.1111/j.1460-9568.1991.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 10.Christman CW, Salvant JB, Walker SA, Povlishock JT. Characterization of a prolonged regenerative attempt by diffusely injured axons following traumatic brain injury in the adult cat: a light and electron microscopic immunocytochemical study. Acta Neuropathol. 1997;94:329–337. doi: 10.1007/s004010050715. [DOI] [PubMed] [Google Scholar]
- 11.David S, Lacroix S. Molecular approaches to spinal cord repair. Annu Rev Neurosci. 2003;26:411–440. doi: 10.1146/annurev.neuro.26.043002.094946. [DOI] [PubMed] [Google Scholar]
- 12.Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emerick AJ, Neafsey EJ, Schwab ME, Kartje GL. Functional reorganization of the motor cortex in adult rats after cortical lesion and treatment with monoclonal antibody IN-1. J Neurosci. 2003;23:4826–4830. doi: 10.1523/JNEUROSCI.23-12-04826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emery DL, Raghupathi R, Saatman KE, Fischer I, Grady MS, McIntosh TK. Bilateral growth-related protein expression suggests a transient increase in regenerative potential following brain trauma. J Comp Neurol. 2000;424:521–531. [PubMed] [Google Scholar]
- 15.Emery DL, Royo NC, Fischer I, Saatman KE, McIntosh TK. Plasticity following injury to the adult central nervous system: is recapitulation of a developmental state worth promoting? J Neurotrauma. 2003;20:1271–1292. doi: 10.1089/089771503322686085. [DOI] [PubMed] [Google Scholar]
- 16.Fiedler M, Horn C, Bandtlow C, Schwab ME, Skerra A. An engineered IN-1 F(ab) fragment with improved affinity for the Nogo-A axonal growth inhibitor permits immunochemical detection and shows enhanced neutralizing activity. Protein Eng. 2002;15:931–941. doi: 10.1093/protein/15.11.931. [DOI] [PubMed] [Google Scholar]
- 17.Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- 18.Fischer D, He Z, Benowitz LI. Counteracting the Nogo receptor enhances optic nerve regeneration if retinal ganglion cells are in an active growth state. J Neurosci. 2004;24:1646–1651. doi: 10.1523/JNEUROSCI.5119-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fouad K, Klusman I, Schwab ME. Regenerating corticospinal fibers in the Marmoset (Callitrix jacchus) after spinal cord lesion and treatment with the anti-Nogo-A antibody IN-1. Eur J Neurosci. 2004;20:2479–2482. doi: 10.1111/j.1460-9568.2004.03716.x. [DOI] [PubMed] [Google Scholar]
- 20.Fournier AE, Grandpré T, Gould G, Wang X, Strittmatter SM. Nogo and the Nogo-66 receptor. Prog Brain Res. 2002;137:361–369. doi: 10.1016/s0079-6123(02)37027-4. [DOI] [PubMed] [Google Scholar]
- 21.Fournier AE, Grandpré T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- 22.Freund P, Schmidlin E, Wannier T, Bloch J, Mir A, Schwab ME, et al. Nogo-A-specific antibody treatment enhances sprouting and functional recovery after cervical lesion in adult primates. Nat Med. 2006;12:790–792. doi: 10.1038/nm1436. [DOI] [PubMed] [Google Scholar]
- 23.Gianola S, Rossi F. GAP-43 overexpression in adult mouse Purkinje cells overrides myelin-derived inhibition of neurite growth. Eur J Neurosci. 2004;19:819–830. doi: 10.1111/j.0953-816x.2004.03190.x. [DOI] [PubMed] [Google Scholar]
- 24.Grados-Munro EM, Fournier AE. Myelin-associated inhibitors of axon regeneration. J Neurosci Res. 2003;74:479–485. doi: 10.1002/jnr.10803. [DOI] [PubMed] [Google Scholar]
- 25.Graham DI, Raghupathi R, Saatman KE, Meaney D, McIntosh TK. Tissue tears in the white matter after lateral fluid percussion brain injury in the rat: relevance to human brain injury. Acta Neuropathol. 2000;99:117–124. doi: 10.1007/pl00007414. [DOI] [PubMed] [Google Scholar]
- 26.Hoover RC, Motta M, Davis J, Saatman KE, Fujimoto ST, Thompson HJ, et al. Differential effects of the anticonvulsant topiramate on neurobehavioral and histological outcomes following traumatic brain injury in rats. J Neurotrauma. 2004;21:501–512. doi: 10.1089/089771504774129847. [DOI] [PubMed] [Google Scholar]
- 27.Hulo S, Alberi S, Laux T, Muller D, Caroni P. A point mutant of GAP-43 induces enhanced short-term and long-term hippocampal plasticity. Eur J Neurosci. 2002;15:1976–1982. doi: 10.1046/j.1460-9568.2002.02026.x. [DOI] [PubMed] [Google Scholar]
- 28.Hulsebosch CE, DeWitt DS, Jenkins LW, Prough DS. Traumatic brain injury in rats results in increased expression of GAP-43 that corelates with behavioral recovery. Neurosci Lett. 1998;255:83–86. doi: 10.1016/s0304-3940(98)00712-5. [DOI] [PubMed] [Google Scholar]
- 29.Kartje GL, Schulz MK, Lopez-Yunez A, Schnell L, Schwab ME. Corticostriatal plasticity is restricted by myelin-associated neurite growth inhibitors in the adult rat. Ann Neurol. 1999;45:778–786. doi: 10.1002/1531-8249(199906)45:6<778::aid-ana12>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 30.Kharatishvili I, Nissinen JP, McIntosh TK, Pitkänen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140:685–697. doi: 10.1016/j.neuroscience.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Lee DH, Strittmatter SM, Sah DW. Targeting the Nogo receptor to treat central nervous system injuries. Nat Rev Drug Discov. 2003;2:872–878. doi: 10.1038/nrd1228. [DOI] [PubMed] [Google Scholar]
- 33.Lenzlinger PM, Shimizu S, Marklund N, Thompson HJ, Schwab ME, Saatman KE, et al. Delayed inhibition of Nogo-A does not alter injury-induced axonal sprouting but enhances recovery of cognitive function following experimental traumatic brain injury in rats. Neuroscience. 2005;134:1047–1056. doi: 10.1016/j.neuroscience.2005.04.048. [DOI] [PubMed] [Google Scholar]
- 34.Liebscher T, Schnell L, Schnell D, Scholl J, Schneider R, Gullo M, et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann Neurol. 2005;58:706–719. doi: 10.1002/ana.20627. [DOI] [PubMed] [Google Scholar]
- 35.Marklund N, Fulp CT, Shimizu S, Puri R, McMillan A, Strittmatter SM, et al. Selective temporal and regional alterations of Nogo-A and small proline-rich repeat protein 1A (SPRR1A) but not Nogo-66 receptor (NgR) occur following traumatic brain injury in the rat. Exp Neurol. 2006;197:70–83. doi: 10.1016/j.expneurol.2005.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marklund N, Keck C, Hoover R, Soltesz K, Millard M, LeBold D, et al. Administration of monoclonal antibodies neutralizing the inflammatory mediators tumor necrosis factor alpha and interleukin-6 does not attenuate acute behavioral deficits following experimental traumatic brain injury in the rat. Restor Neurol Neurosci. 2005;23:31–42. [PubMed] [Google Scholar]
- 37.Masliah E, Fagan AM, Terry RD, DeTeresa R, Mallory M, Gage FH. Reactive synaptogenesis assessed by synaptophysin immunoreactivity is associated with GAP-43 in the dentate gyrus of the adult rat. Exp Neurol. 1991;113:131–142. doi: 10.1016/0014-4886(91)90169-d. [DOI] [PubMed] [Google Scholar]
- 38.Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- 39.McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 40.Meier S, Bräuer AU, Heimrich B, Schwab ME, Nitsch R, Savaskan NE. Molecular analysis of Nogo expression in the hippocampus during development and following lesion and seizure. FASEB J. 2003;17:1153–1155. doi: 10.1096/fj.02-0453fje. [DOI] [PubMed] [Google Scholar]
- 41.Mingorance A, Fontana X, Solé M, Burgaya F, Urena JM, Teng FY, et al. Regulation of Nogo and Nogo receptor during the development of the entorhino-hippocampal pathway and after adult hippocampal lesions. Mol Cell Neurosci. 2004;26:34–49. doi: 10.1016/j.mcn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Morris R. Development of a water maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 43.National Research Council . Guide for the Care and Use of Laboratory Animals. National Academy Press; Washington, DC: 1996. [Google Scholar]
- 44.Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, et al. Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci. 2003;23:5393–5406. doi: 10.1523/JNEUROSCI.23-13-05393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oudega M, Rosano C, Sadi D, Wood PM, Schwab ME, Hagg T. Neutralizing antibodies against neurite growth inhibitor NI-35/250 do not promote regeneration of sensory axons in the adult rat spinal cord. Neuroscience. 2000;100:873–883. doi: 10.1016/s0306-4522(00)00350-x. [DOI] [PubMed] [Google Scholar]
- 46.Papadopoulos CM, Tsai SY, Alsbiei T, O'Brien TE, Schwab ME, Kartje GL. Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN-1 antibody treatment in the adult rat. Ann Neurol. 2002;51:433–441. doi: 10.1002/ana.10144. [DOI] [PubMed] [Google Scholar]
- 47.Paxinos G, Watson S. The Rat Brain in Stereotaxic Coordinates. Academic Press; Sydney: 1986. [Google Scholar]
- 48.Pierce JES, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral, and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- 49.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 50.Rekart JL, Meiri K, Routtenberg A. Hippocampal-dependent memory is impaired in heterozygous GAP-43 knockout mice. Hippocampus. 2005;15:1–7. doi: 10.1002/hipo.20045. [DOI] [PubMed] [Google Scholar]
- 51.Riedel G, Micheau J. Function of the hippocampus in memory formation: desperately seeking resolution. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:835–853. doi: 10.1016/s0278-5846(01)00153-1. [DOI] [PubMed] [Google Scholar]
- 52.Routtenberg A, Cantallops I, Zaffuto S, Serrano P, Namgung U. Enhanced learning after genetic overexpression of a brain growth protein. Proc Natl Acad Sci U S A. 2000;97:7657–7562. doi: 10.1073/pnas.97.13.7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, et al. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp Neurol. 1997;147:418–427. doi: 10.1006/exnr.1997.6629. [DOI] [PubMed] [Google Scholar]
- 54.Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt-Kastner R, Bedard A, Hakim A. Transient expression of GAP-43 within the hippocampus after global brain ischemia in rat. Cell Tissue Res. 1997;288:225–238. doi: 10.1007/s004410050808. [DOI] [PubMed] [Google Scholar]
- 56.Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- 57.Seymour AB, Andrews EM, Tsai SY, Markus TM, Bollnow MR, Brenneman MM, et al. Delayed treatment with monoclonal antibody IN-1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. J Cereb Blood Flow Metab. 2005;25:1366–1375. doi: 10.1038/sj.jcbfm.9600134. [DOI] [PubMed] [Google Scholar]
- 58.Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, et al. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45:353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 59.Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- 60.Thallmair M, Metz GA, Z'Graggen WJ, Raineteau O, Kartje GL, Schwab ME. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nature Neurosci. 1998;1:124–131. doi: 10.1038/373. [DOI] [PubMed] [Google Scholar]
- 61.Thompson HJ, Marklund N, LeBold DG, Morales DM, Keck CA, Vinson M, et al. Tissue sparing and functional recovery following experimental traumatic brain injury is provided by treatment with an anti-myelin-associated glycoprotein antibody. Eur J Neurosci. 2006;24:3063–3072. doi: 10.1111/j.1460-9568.2006.05197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006;201:253–265. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 63.Weinmann O, Schnell L, Ghosh A, Montani L, Wiessner C, Wannier T, et al. Intrathecally infused antibodies against Nogo-A penetrate the CNS and downregulate the endogenous neurite growth inhibitor Nogo-A. Mol Cell Neurosci. 2006;32:161–173. doi: 10.1016/j.mcn.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 64.Wiessner C, Bareyre FM, Allegrini PR, Mir AK, Frentzel S, Zurini M, et al. Anti-Nogo-A antibody infusion 24 hours after experimental stroke improved behavioral outcome and corticospinal plasticity in normotensive and spontaneously hypertensive rats. J Cereb Blood Flow Metab. 2003;23:154–165. doi: 10.1097/01.WCB.0000040400.30600.AF. [DOI] [PubMed] [Google Scholar]
- 65.Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM. A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci. 2002;5:1302–1308. doi: 10.1038/nn975. [DOI] [PubMed] [Google Scholar]
- 66.Zhang C, Raghupathi R, Saatman KE, Smith DH, Stutzmann JM, Wahl F, et al. Riluzole attenuates cortical lesion size, but not hippocampal neuronal loss, following traumatic brain injury in the rat. J Neurosci Res. 1998;52:342–349. doi: 10.1002/(SICI)1097-4547(19980501)52:3<342::AID-JNR10>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]