

Abstract
Background & Aims
We studied the role of protease-activated receptor 2 (PAR2) and its activating enzymes, trypsins and tryptase, in Clostridium difficile toxin A (TxA)-induced enteritis.
Methods
We injected TxA into ileal loops in PAR2 or dipeptidyl peptidase I (DPPI) knockout mice or in wild-type mice pretreated with tryptase inhibitors (FUT-175 or MPI-0442352) or soybean trypsin inhibitor. We examined the effect of TxA on expression and activity of PAR2 and trypsin IV messenger RNA in the ileum and cultured colonocytes. We injected activating peptide (AP), trypsins, tryptase, and p23 in wild-type mice, some pretreated with the neurokinin 1 receptor antagonist SR140333.
Results
TxA increased fluid secretion, myeloperoxidase activity in fluid and tissue, and histologic damage. PAR2 deletion decreased TxA-induced ileitis, reduced luminal fluid secretion by 20%, decreased tissue and fluid myeloperoxidase by 50%, and diminished epithelial damage, edema, and neutrophil infiltration. DPPI deletion reduced secretion by 20% and fluid myeloperoxidase by 55%. In wild-type mice, FUT-175 or MPI-0442352 inhibited secretion by 24%−28% and tissue and fluid myeloperoxidase by 31%−71%. Soybean trypsin inhibitor reduced secretion to background levels and tissue myeloperoxidase by up to 50%. TxA increased expression of PAR2 and trypsin IV in enterocytes and colonocytes and caused a 2-fold increase in Ca2+ responses to PAR2 AP. AP, tryptase, and trypsin isozymes (trypsin I/II, trypsin IV, p23) caused ileitis. SR140333 prevented AP-induced ileitis.
Conclusions
PAR2 and its activators are proinflammatory in TxA-induced enteritis. TxA stimulates existing PAR2 and up-regulates PAR2 and activating proteases, and PAR2 causes inflammation by neurogenic mechanisms.
Protease-activated receptor 2 (PAR2) belongs to a family of G protein–coupled receptors that are activated by proteolytic cleavage within their extracellular N-terminal domain.1,2 PAR2 is highly expressed in the gastrointestinal tract, where it is present on enterocytes, myocytes, myenteric and submucosal neurons, and extrinsic sensory nerves.2–4 Proteases that activate PAR2 include enzymes from inflammatory cells (eg, mast cell tryptase, neutrophil proteinase 3) and epithelial tissues (pancreatic and extrapancreatic trypsins, kallikreins), coagulation factors (VIIa, Xa), and proteases from dust mites and bacteria.2,3,5 Trypsins and tryptase are prominent agonists of PAR2 in the gastrointestinal tract. Pancreatic trypsin in the intestinal lumen may activate PAR2 at the apical membrane of enterocytes.5 However, intestinal epithelial cells express trypsinogen IV, which is activated by enteropeptidase to form trypsin IV, an isozyme that also activates PAR2.6 Tryptase released from mast cells can activate PAR2 on myocytes, neurons, and epithelial cells.3,7 Mast cells require the papain-like cysteine protease dipeptidyl peptidase I (DPPI) for full expression of active tryptase,8 and high levels of DPPI are constitutively expressed in the small and large intestine of the mouse, emphasizing its potential role in intestinal inflammation.9 Mice lacking DPPI exhibit a 75% reduction in the normal tryptase levels in mast cells and show increased survival from sepsis, which is directly due to the lack of DPPI in mast cells.8,10 Proteases that activate PAR2 are released and activated during inflammation and injury, and PAR2 controls responses to these insults, with effects on gastrointestinal secretion, motility, inflammation, and pain.1,3,5,11–13
We investigated the role of PAR2 and proteases that activate PAR2 in enteritis induced by toxin A (TxA) from Clostridium difficile. TxA-induced enteritis in rodents parallels the human infection, a common iatrogenic illness in patients receiving antibiotics. Despite its prevalence, nothing is known about the role of PAR2 and proteases in TxA-induced enteritis. However, TxA and PAR2 agonists induce intestinal inflammation by similar mechanisms involving many of the same cell types, suggesting a major role for PAR2 in TxA-induced enteritis. Enterocytes, intrinsic and extrinsic neurons, and inflammatory cells of the lamina propria participate in the host response to TxA in rodents.14 TxA induces disruption of tight junctions of enterocytes and compromises the integrity of the epithelial layer.15,16 Degranulation of mast cells causes release of calcitonin gene-related peptide and substance P, which activate enteric neurons to increase intestinal secretion, leading to diarrhea.14 Cytokines, such as interleukin 1 and tumor necrosis factor α, recruit neutrophils, which cause acute inflammation.14,16 Similarly to TxA, PAR2 agonists cause redistribution of tight junctional proteins and increased paracellular permeability of intestinal epithelial cells,7 with secretion of prostaglandin E2.5 PAR2 agonists also cause fluid and electrolyte secretion in the intestine and promote inflammation of the intestine and other tissues by neurogenic mechanisms involving calcitonin gene-related peptide and substance P.1,12,17 In view of the similar proinflammatory effects of TxA and PAR2 agonists in the intestine, and because animals deficient in mast cells or receiving mast cell stabilizers have diminished inflammation in response to TxA,18,19 we hypothesized that PAR2 and PAR2-activating proteases would have a proinflammatory role in this model.
To test this hypothesis, we examined the role of PAR2 and the enzymes that activate it, tryptase and trypsin, in TxA-induced enteritis, using both a genetic and a pharmacologic approach. We assessed inflammation by measuring intestinal secretion, measuring myeloperoxidase (MPO) activity in tissue and luminal fluid, and histologic assessment of tissues. Our aims were to (1) investigate the effect of deletion of PAR2 or the mast cell–processing enzyme DPPI on TxA-induced ileitis, (2) determine the effects of the tryptase inhibitors nafamostat mesilate (FUT-175) and MPI-044235220 and of soybean trypsin inhibitor (SBTI) on TxA-induced inflammation, (3) evaluate the effects of TxA on expression of PAR2 and of PAR2-activating proteases, and (4) determine whether proteases known to activate PAR2 induce intestinal inflammation.
Materials and Methods
PAR2 Agonists and Protease Inhibitors
Pancreatic trypsin I/II was from Sigma Chemical Co (T1426, 10,000 U/mg; St Louis, MO), and recombinant human mast cell tryptase was from Promega (34.5 U/mg; Madison, WI). Trypsinogen IV and prop23 were expressed in Escherichia coli, purified by affinity chromatography, and activated by enterokinase.21 Purified trypsin IV (145 U/mg) and p23 (165 U/mg) were used for all experiments. Trypsin IV and p23 activity was determined using the substrate tosyl-GPR-pNA (150 μmol/L, 100 mmol/L Tris-HCl, 1 mmol/L CaCl2, pH 8, 25°C, with 1 U defined as the enzymatic activity converting 1 μmol of substrate per minute). PAR2-activating peptide (AP; SLIGRL-NH2, corresponding to the tethered ligand that is exposed by proteolytic activation) and the inactive reverse peptide (LRGILS-NH2, control) were from CPC Scientific (San Jose, CA). SBTI was from Fluka (93619) and Sigma Chemical Co (T9128) (Sigma, St. Louis, MO). FUT-175 was from Calbiochem (San Diego, CA). MPI-0442352 was a gift from C. Pleiman (Myriad Genetics, Salt Lake City, UT). A β strand template library22–24 was used to develop the tryptase inhibitor MPI-0442352 (mol wt, 784.94 with C40H48N8O7S). Tryptase inhibition was quantified using the release of pNA from the chromogenic substrate S-2366, L-pyroGlu-Pro-Arg-pNA (diaPharma, West Chester, OH), monitored at 405 nm and has been described.25 In brief, MPI-0442352 (0−600 nmol/L) and human lung tryptase (0.2 nmol/L, in Tris, pH 8.0) (Elastin Products, Owensville, MO) were incubated at room temperature for 30 minutes, and S-2366 (50−800 μmol/L) was added to initiate the reaction. MPI 0443252, purified to >95%, had a Ki of 90 nmol/L.
Animals
The Institutional Animal Care and Use Committee of the University of California at San Francisco approved all animal protocols. Mice in which the Par2 gene, also known as F2rl1, was deleted26 were maintained in a 50/50 mixed 129SVJae/C57BL6 strain background. Littermate Par2+/+, Par2+/−, and Par2−/− mice from heterozygote crosses were used for studies. DPPI+/+ and DPPI−/− mice from Dr Timothy Ley27 were backcrossed 10 generations into a C57BL/6 background and bred as homozygous knockouts. C57/Bl6 mice were from Jackson Laboratory (Davis, CA). Mice were maintained under climate-controlled (22 ± 4°C) and light-controlled (12-hour light/12-hour dark cycle) conditions in a barrier facility. Male and female mice were studied at 6−12 weeks of age.
Induction of Intestinal Inflammation With TxA
C difficile TxA was purified as described.28 Genetically altered mice were coded before experimentation and studied in a blinded manner. Mice were fasted overnight but allowed access to water. They were anesthetized with isoflurane, and TxA (0.5 or 1 μg, whichever was a submaximal dose in the strain of mice selected for study) or vehicle (0.05 mol/L Tris, pH 7.4, control) in 100 μL was injected into ileal loops as described.29 The animals received buprenorphine (0.05 mg/kg subcutaneously), were allowed to regain consciousness, and were killed after 3 hours (pentobarbital sodium 200 mg/kg intraperitoneally). Intestinal loops were removed, and the loop length and weight were recorded. Data are reported as milligrams per centimeters and as a percentage of the wild-type or positive control group. Loop fluid and a portion of the ileum were frozen separately for measurement of MPO activity. The remaining tissue was fixed in 10% formalin and paraffin embedded, after which sections were stained with H&E for histologic grading. Some C57/Bl6 mice received FUT-175 (10 mg/kg, in 0.25 mmol/L Tris, pH 7.4, injected into the loop 15 minutes before addition of TxA or vehicle [control]), MPI-0442352 (15 mg/kg, in dimethylacetamide/PEG300/EtOH/water at 3%/40%/12%/45% intraperitoneally 30 minutes before TxA or vehicle), SBTI (10 or 20 mg in 0.025 mol/L Tris, pH 7.4, injected into the loop 15 minutes before TxA or vehicle), or corresponding vehicle. The number of animals used was 4−13 in control groups and 6−26 in experimental groups.
Induction of Intestinal Inflammation With Activators of PAR2
Ileal loops were surgically prepared in anesthetized wild-type mice as described. AP (6.6 μg/mouse), trypsin II (12 μg/mouse), tryptase (4 μg/mouse), trypsin IV (25 μg/mouse), p23 (2.3 μg/mouse), reverse peptide, or vehicle (0.05 mol/L Tris, pH 7.4, controls) (all in 100 μL) was injected into the loop. Inflammation was assessed after 4 hours. In a separate group of experiments, mice were pretreated with the neurokinin 1 receptor antagonist SR140333 (Dr Xavier Edmonds-Alt, Sanofi Recherche, Montpellier, France) (2 mg/kg) or its carrier (1% dimethyl sulfoxide) intraperitoneally (0.2 mL) 60−90 minutes before formation of ileal loops and injection with AP. The number of mice used was 6 in each group.
MPO Assay
Both tissue and loop fluid were assayed. Samples were homogenized and sonicated, and the homogenate was centrifuged (12,000g, 15 minutes). MPO activity in the supernatant was quantified in triplicate at 2 dilutions with a microtiter plate assay using 5-O-dianiside (Aldrich, Milwaukee, WI) as the substrate and human neutrophil MPO (Calbiochem, San Diego, CA) as a standard.29 Supernatants were assayed for protein in triplicate with a bicinchoninic acid microtiter plate assay (Pierce, Rockford, IL). Results obtained with tissue are expressed as units of MPO per milligram of protein. Results obtained with fluid are expressed as units of MPO per milliliter of fluid. Results between groups are compared as percentages.
Microscopy and Histologic Scoring
Sections were observed by using a Zeiss Axioplan microscope (Zeiss, Thornwood, NJ) and a Zeiss Fluar ×20 (numerical aperture, 1.0) or Plan Apochromat ×40 (numerical aperture, 1.4). Images were imported via a SPOT Insight camera (Diagnostic Instruments, Sterling Heights, MI). Contrast and brightness were adjusted using Adobe Photoshop CS (Adobe Systems, Mountain View, CA). The severity of inflammation was scored in coded slides by a histologist blinded to experimental groups on a scale of 1 (mild) to 3 (severe) for epithelial damage, edema, and neutrophil infiltration as described.28
Localization of PAR2
In a separate series of experiments, TxA (1.0 μg) or vehicle (control) was injected into ileal loops formed in C57/Bl6 wild-type mice as described. After 4 hours, the loop tissue was fixed in 4% paraformaldehyde in 100 mmol/L phosphate-buffered saline, pH 7.4, overnight at 4°C, washed, placed in 30% sucrose for 16 hours at 4°C, and embedded in OCT. Frozen sections (10 μm) were incubated in phosphate-buffered saline containing 0.1% Triton X-100 and 5% normal goat serum. Sections were incubated with rabbit anti-mouse PAR2 antiserum (9717,30 generated by CURE, University of California at Los Angeles; 1:400, 16 hours at 4°C) and washed. For immunofluorescence, sections were incubated with a goat anti-rabbit anti–immunoglobulin G conjugated to fluorescein isothiocyanate (FITC; 1:200, 2 hours; Jackson ImmunoResearch, West Grove, PA).31 Tissue sections in which the primary antibody was omitted served as controls. Single confocal sections were acquired using a Zeiss LSM510 Meta equipped with an argon laser and an Axiovert 200M microscope using a Plan Apochromat ×20 (numerical aperture, 0.80), 488λ excitation, and a long-pass 505 filter. Acquisition parameters were adjusted to be less sensitive in the PAR2 antiserum-stained tissue to ensure that the brighter-staining TxA-treated tissue was not at saturation and more sensitive in the secondary antibody only tissue to enable visualization of the tissue. Images used were unmodified. Pixel intensity was quantified using LSM510 software on unmodified images acquired using identical parameters.
Cell Culture and Treatment of NCM460 Cells With TxA
NCM460 cells, an epithelial cell line from normal human colon,6 were maintained in Ham's F-12 medium with 20% heat-inactivated fetal bovine serum, 0.5 mg/mL insulin-transferrin-selenium (Invitrogen, Carlsbad, CA), 0.4 mg/mL hydrocortisone (Sigma Chemical Co), and 0.58 μg/ml l-glutamine in 95% air/5% CO2 at 37°C. Cells were plated at 5 × 105/mL onto plastic for polymerase chain reaction (PCR) and onto glass coverslips for measurement of intracellular calcium concentration ([Ca2+]i). After 48 hours, they were washed with phosphate-buffered saline containing Ca2+ and Mg2+ and placed in Ham's F-12 containing 0.1% bovine serum albumin. Cells were treated at 37°C with vehicle or TxA (5 nmol/L) for 0, 6, or 24 hours or with vehicle or TxA (5 μmol/L) for 30−360 minutes. Vehicle consisted of the equivalent dilution of Tris 0.05 mol/L in NCM culture medium (1/1300 or 1/1,300,000 for 5 μmol/L and 5 nmol/L of TxA, respectively). Cells were then used for PCR and Ca2+ signaling assays.
Measurement of [Ca2+]i
NCM460 cells were washed and incubated in Hanks' balanced salt solution with 0.1% bovine serum albumin, 20 mmol/L HEPES (pH 7.4), and 5 μmol/L of fura-2/AM (Molecular Probes, Eugene, OR) for 45−60 minutes at 37°C. Washed coverslips were mounted in an open chamber at 37°C. Fluorescence of individual cells was measured at 340 nm and 380 nm excitation and 510 nm emission using a Zeiss Axiovert microscope, an ICCD video camera (Stanford Photonics, Stanford, CA), and a video microscopy acquisition program (Axon Instruments, Inc, Union City, CA). Test substances (50 μL injection) were added to the chamber. Results are expressed as the 340/380-nm emission ratio, which is proportional to the [Ca2+]i. The magnitude of the response to PAR2-AP was determined by subtraction of the baseline response; thus, each cell served as an internal control.
Real-Time Quantitative PCR Analysis
Total RNA was extracted from NCM460 cells using the RNeasy kit (Qiagen, Valencia, CA), treated with deoxyribonuclease (Ambion, Austin, TX) for 60 minutes and repurified. The reverse-transcriptase reaction (100 μL final using 1 μg of RNA) was conducted as follows: 25°C for 10 minutes, 48°C for 30 minutes, and 99°C for 5 minutes using random hexamers. The PCR reaction mixture contained 25 μL of 2× Universal PCR master mix, 0.5 μL of template complementary DNA, and 2.5 μL of primers/probe (Applied Biosystems, Foster City, CA) in 50 μL in a 96-well hard-shell reaction plate (MJ Research, Waltham, MA). Reactions were amplified using a continuous fluorescence detector (DNA Engine Opticon 2; MJ Research) and performed in triplicate. For PAR2 and β-glucuronidase, primers and probes were from Applied Biosystems (cat. no. Hs001173741 and Hs99999908, respectively). Primers and probes for mesotrypsin/trypsin IV were from Applied Biosystems (primer/probes sequences were as follows: forward, 5′-ccaccctaaatacaacagggacac-3′; reverse, 5′-tggacacgcgggcatt-3′; probe, 5′atcatgctgatcaaactctcctcacctg-3′). The relative quantity messenger RNA was obtained of by the comparative threshold cycle (Ct) method with β-glucuronidase as an endogenous control. The Ct indicates the cycle number at which the amount of amplified target reaches a fixed threshold above background. Opticon Monitor Analysis software (version 2.02; MJ Research) was used to calculate a Ct value for each reaction, which was normalized for amplification by subtracting the Ct value of β-glucuronidase. The fold increase was calculated relative to the Ct value of unstimulated cells.
Statistics
Results are reported as means ± SE. Data were compared using analysis of variance and multiple comparison testing with the Student–Newman–Keuls t test or Student t test, with P < .05 considered significant.
Results
Deletion of PAR2 Ameliorates TxA-Induced Fluid Secretion and Granulocyte Infiltration
We compared enteritis with a half-maximal dose of TxA (0.5 μg for the Par2+/+ mice) in Par2+/+, Par2+/−, and Par2−/− mice using accumulation of fluid in the intestinal lumen and MPO activity in both the intestinal wall and fluid as end points. Basal fluid secretion in vehicle-treated control loops was similar in Par2+/+, Par2+/−, and Par2−/− mice (55 ± 4, 48 ± 4, and 53 ± 9 mg/cm, respectively) (Figure 1A). In Par2+/+ mice, TxA significantly increased secretion from basal levels by 2.5-fold (135 ± 8 mg/cm; P < .05). In comparison, TxA-induced secretion in Par2+/− mice was decreased by ∼15% (116 ± 10 mg/cm), and TxA-induced secretion in Par2−/− mice was significantly decreased by ∼20% (109 ± 8 mg/cm; P < .05) (Figure 1A). Granulocyte infiltration (assessed by MPO assays of tissue and loop fluid) was similar in controls among Par2+/+, Par2+/−, and Par2−/− mice (tissue: 0.07 ± 0.04, 0.07 ± 0.03, and 0.05 ± 0.03 U MPO/mg protein; loop fluid: 0.08 ± 0.02, 0.07 ± 0.03, and 0.07 ±0.04 U MPO/mL, respectively) (Figure 1B and C). TxA also caused a significant 30-fold increase in tissue MPO in Par2+/+ mice (tissue: 2.15 ± 0.22 U MPO/mg protein; loop fluid: 0.8 ± 0.02 U MPO/mL; P < .05). Compared with wild-type mice, Par2+/− and Par2−/− mice had decreased responses to TxA, with a 15% and 49% reduction in tissue MPO and a 20% and 56% decrease in fluid MPO, respectively (tissue: 1.85 ± 0.22 and 1.10 ± 0.14 U MPO/mg protein; loop fluid: 0.64 ± 0.1 and 0.35 ± 0.06 U MPO/mL, respectively) (Figure 1B and C). Once again, these decreases were statistically significant in the Par2−/− mice (P < .05). Thus, PAR2 deletion decreases TxA-induced intestinal secretion and granulocyte infiltration in a gene/dose-dependent manner, with heterozygote mice having a smaller decrease from wild-type mice than knockout mice. Deletion of PAR2 did not eliminate inflammation, which in this case must be due to activation of other receptors.
Figure 1.

Deletion of PAR2 inhibits TxA-induced fluid secretion and granulocyte infiltration. Ileal loops were injected with vehicle (vehicle control, open bars) or 0.5 μg TxA (closed bars). (A) Intestinal secretion expressed as milligrams per centimeter of loop. (B) Tissue MPO expressed as units MPO per milligram protein. (C) MPO in loop fluid expressed as units per milliliter fluid. *P < .05 between vehicle and TxA loops in Par2+/+ mice. †P < .05 between results obtained with TxA in Par2+/+ and Par2+/− or Par2−/− mice.
Deletion of PAR2 Ameliorates TxA-Induced Pathologic Changes
Histologic assessment confirmed that epithelial damage, edema, and neutrophil infiltration induced by TxA were diminished in Par2+/− mice and significantly decreased in Par2−/− mice relative to Par2+/+ animals (Table 1 and Figure 2; P < .05). In Par2−/− mice treated with TxA, there was significantly diminished epithelial damage, edema, and infiltration of neutrophils (P < .05) and a striking retention of mucus in goblet cells. Thus, deletion of PAR2 diminishes all characteristics of TxA-induced enteritis examined.
Table 1.
Histologic Score for TxA-Induced Enteritis in Par2+/+, Par2+/−, and Par2>−/− Mice
| Mice genotype and treatment | Epithelial damage | Edema congestion | Polymorphonuclear leukocyte infiltration | Total score |
|---|---|---|---|---|
| Par2+/+ + vehicle control | 0.16 ± 0.1 | 0.50 ± 0.1 | 0.68 ± 0.3 | 1.34 ± 0.4 |
| Par2+/+ + TxA | 0.91 ± 0.1a | 1.35 ± 0.1a | 2.21 ± 0.1a | 4.47 ± 0.3a |
| Par2+/− + vehicle control | 0.06 ± 0.1 | 0.25 ± 0.1 | 0.81 ± 0.1 | 1.13 ± 0.2 |
| Par2+/− + TxA | 0.89 ± 0.2 | 1.00 ± 0.1 | 1.73 ± 0.2b | 3.61 ± 0.4 |
| Par2−/− + vehicle control | 0.35 ± 0.2 | 0.45 ± 0.2 | 0.75 ± 0.1 | 1.55 ± 0.5 |
| Par2−/− + TxA | 0.25 ± 0.1b | 0.75 ± 0.2b | 1.70 ± 0.2b | 2.70 ± 0.3b |
P < .05 compared with Par2+/+ vehicle control group.
P < .05 compared with Par2+/+ TxA group.
Figure 2.

Deletion of PAR2 inhibits TxA-induced enteritis. Images show the ileum. (A) Par2+/+ vehicle control, (B) Par2+/+ TxA, (C) Par2+/− vehicle control, (D) Par2+/− TxA, (E) Par2−/− vehicle control, (F) Par2−/− TxA. Note the normal villous structure in control groups. In Par2+/+ mice, TxA caused pathologic changes including loss of villi, infiltration of granulocytes, and severe necrosis (arrows). Scale bar = 100 μm.
Deletion of DPPI Ameliorates TxA-Induced Fluid Secretion and Granulocyte Infiltration
Responses of DPPI+/+ and DPPI−/− mice to a half-maximal dose of TxA (1 μg in this strain, DPPI+/+ mice were less sensitive to TxA) showed that basal fluid secretion did not differ between DPPI+/+ (46 ± 2 mg/cm) and DPPI−/− mice (54 ± 4 mg/cm) (Figure 3A). TxA caused a robust increase in intestinal secretion over that seen with vehicle control (139 ± 7 mg/cm), similar to that seen in Par2+/+ mice (Figure 1A). In DPPI−/− animals, intestinal secretion induced by TxA was significantly reduced to 78% of that seen in wild-type animals (109 ± 6 mg/cm; P < .05). Basal granulocyte infiltration, as determined by MPO assays, was similar in DPPI+/+ and DPPI−/− animals (tissue: 0.06 ± 0.01 and 0.05 ± 0.01 U MPO/mg protein; loop fluid: 0.03 ± 0.01 and 0.03 ± 0.01 mU MPO/mL, respectively). Granulocyte infiltration in response to TxA, as determined by tissue MPO, was not different between the strains (1.9 ± 0.2 and 1.8 ± 0.2 mU MPO/mg protein in DPPI+/+ and DPPI−/− animals, respectively) (Figure 3B), but fluid MPO was significantly decreased; levels seen in DPPI−/− mice were 45% of those in DPPI+/+ mice (1.7 ± 0.2 and 0.79 ± 0.1 mU MPO/mL in DPPI+/+ and DPPI−/− animals, respectively; P < .05) (Figure 3C). Thus, deletion of DPPI decreases TxA-induced intestinal secretion and fluid MPO but not tissue MPO. Again, residual inflammation induced by TxA must be due to other receptors and agonists that participate in host responses.
Figure 3.

Deletion of DPPI diminishes TxA-induced fluid secretion and MPO activity in loop fluid. Ileal loops were injected with vehicle (vehicle control, open bars) or 1.0 μg TxA (closed bars). (A) Intestinal secretion expressed as milligram per centimeter of loop. (B) Tissue MPO, expressed as units MPO per milligram protein. (C) MPO in loop fluid expressed as units per milliliter of fluid. *P < .05 between buffer and TxA loops in DPPI+/+ mice. †P < .05 between results obtained with TxA in DPPI+/+ and DPPI−/− mice.
Deletion of DPPI Ameliorates TxA-Induced Pathologic Changes
Histologic assessment showed that DPPI+/+ and DPPI−/− mice appeared similar under basal conditions (Figure 4 and Table 2). In DPPI+/+ mice, TxA caused pathologic changes, including a loss of villi, edema, neutrophil infiltration, and necrosis, that were diminished in DPPI−/− mice, with a significantly reduced edema (P < .05) and a reduced total score. Thus, deletion of DPPI diminishes the characteristics of TxA-induced enteritis.
Figure 4.
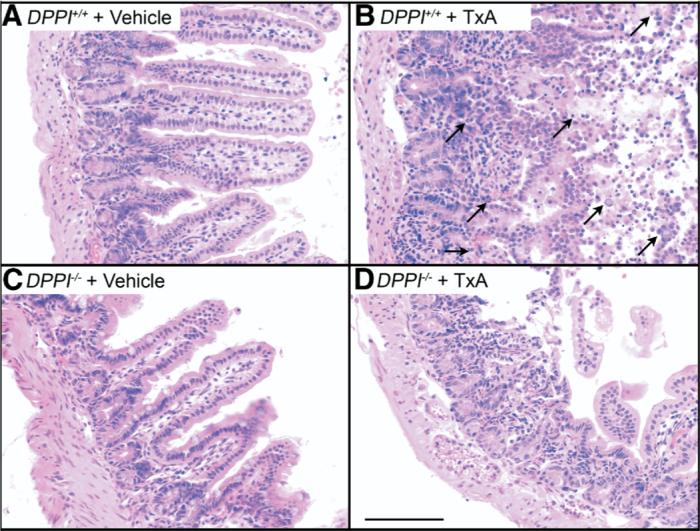
Deletion of DPPI inhibits TxA-induced enteritis. Images show the ileum. (A) DPPI+/+ vehicle control, (B) DPPI+/+ TxA, (C) DPPI−/− vehicle control, (D) DPPI−/− TxA. Note the normal villous structure in control groups. In DPPI+/+ mice, note loss of villi, infiltration of granulocytes, and severe necrosis (arrows), pathologic effects that were diminished in DPPI−/− mice. Scale bar = 100 μm.
Table 2.
Histologic Score for TxA-Induced Enteritis in DPPI+/+ and DPPI−/− Mice
| Mice genotype and treatment | Epithelial damage | Edema congestion | Polymorphonuclear leukocyte infiltration | Total score |
|---|---|---|---|---|
| DPPI+/+ + vehicle control | 0.18 ± 0.1 | 1.28 ± 0.2 | 0.90 ± 0.2 | 2.35 ± 0.4 |
| DPPI+/+ + TxA | 1.64 ± 0.2a | 2.13 ± 0.2a | 2.38 ± 0.2a | 6.08 ± 0.5a |
| DPPI−/− + vehicle control | 0.16 ± 0.1 | 0.64 ± 0.1 | 0.73 ± 0.1 | 1.52 ± 0.2 |
| DPPI−/− + TxA | 1.32 ± 0.1 | 1.65 ± 0.1b | 2.32 ± 0.1 | 5.38 ± 0.3 |
P < .05 compared with DPPI+/+ vehicle control group.
P < .05 compared with DPPI+/+ TxA group.
Tryptase and Trypsin Inhibitors Ameliorate TxA-Induced Intestinal Secretion and Granulocyte Infiltration
We determined if pharmacologic blockade of tryptase or trypsin would diminish TxA responses in wild-type C57/Bl6 mice. In control experiments, mice treated with vehicle before a submaximal dose of TxA (1.0 μg in C57/Bl6) had a strong secretory response (122 ± 7 mg/cm) (Figure 5A). Pretreatment with the tryptase inhibitors FUT-175 or MPI-0442352 significantly decreased secretion by 24% and 28%, respectively (93 ± 9 and 89 ± 13 mg/cm, respectively; P < .05). Pretreatment with 10 or 20 mg SBTI also significantly decreased secretory responses to TxA by 62% and 38%, respectively, with a return to basal or almost basal levels of secretion (47 ± 3 and 76 ± 5 mg/cm, respectively; P < .05). Tissue MPO activity was prominent in mice treated with vehicle before TxA (1.72 ± 0.1 U MPO/g protein) (Figure 5B) but was significantly decreased by 47% and 31%, respectively, in groups pretreated with FUT-175 or MPI-0442352 (0.92 ± 0.1 and 1.19 ± 0.12 U MPO/mg protein, respectively; P < .05). Pretreatment with 10 and 20 mg SBTI also significantly decreased tissue MPO by 56% and 30%, respectively (0.76 ± 0.16 and 1.21 ± 0.2 U MPO/mg protein, respectively; P < .05), but increased basal tissue MPO. Fluid MPO activity was increased in mice treated with vehicle before TxA (0.45 ± 0.09 U MPO/mL with TxA) (Figure 5C). Pretreatment with FUT-175 and MPI-0442352 significantly diminished responses by 71% and 38%, respectively (0.13 ± 0.04 and 0.28 ± 0.06 U MPO/mL, respectively; P < .05). We were unable to quantify fluid MPO in SBTI-pretreated mice, because SBTI interacted with the substrate to cause spurious results in the MPO assay. Thus, inhibitors of tryptase and trypsin suppress TxA-induced ileitis, but again some residual inflammation remained in many experimental groups.
Figure 5.
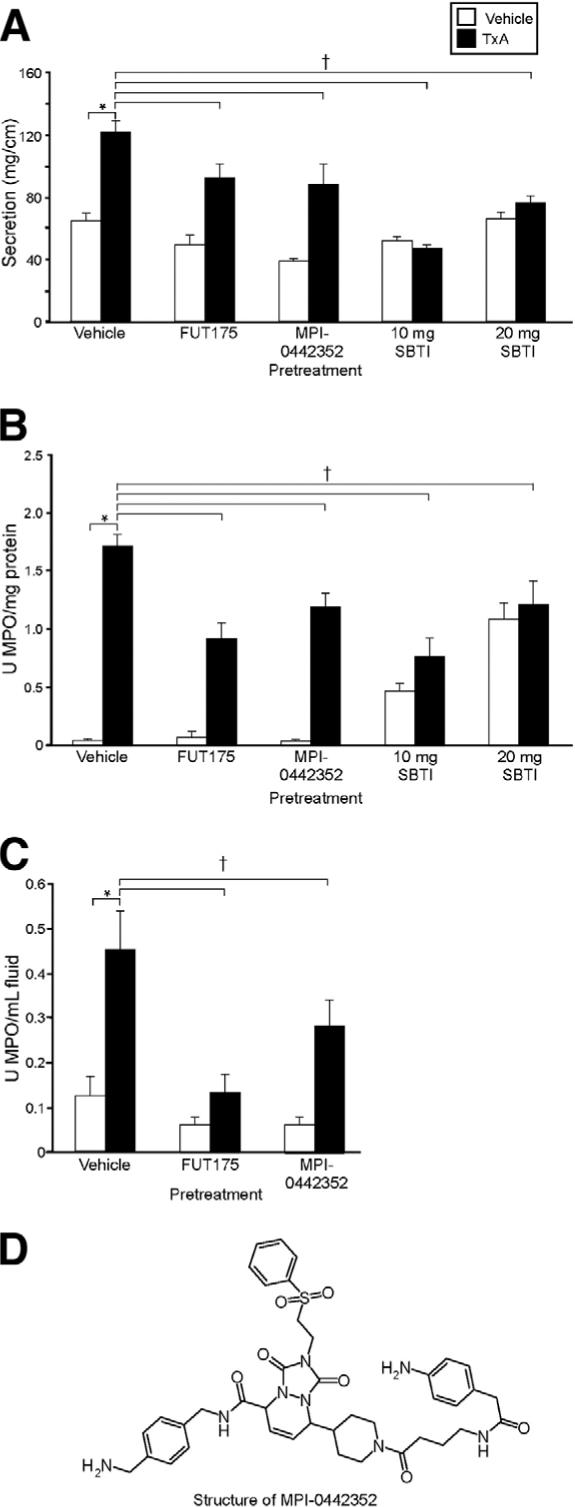
Effect of pretreatment with tryptase and trypsin inhibitors on TxA-induced enteritis. Mice were preinjected with FUT-175, MPI-0442352, or SBTI before injection with vehicle (vehicle control, open bars) or 1.0 μg TxA (closed bars). (A) Intestinal secretion expressed as milligram per centimeter of loop. (B) Tissue MPO expressed as units per milligram protein. (C) MPO in loop fluid expressed as units per milliliter of fluid. (D) Structure of MPI-0442352. *P < .05 between vehicle control and TxA loops in vehicle-pretreated mice. †P < .05 between results obtained with TxA in mice pretreated with vehicle compared with protease inhibitors.
Tryptase and Trypsin Inhibitors Ameliorate TxA-Induced Pathologic Changes
In vehicle-treated control mice, TxA caused marked epithelial damage, edema, and neutrophil infiltration (Figure 6 and Table 3). Pretreatment with FUT-175 significantly decreased both epithelial damage and total histologic score (P < .05), but MPI-0442352 did not alter the pathologic changes induced by TxA. SBTI significantly decreased epithelial damage, edema, and total score. Granulocyte infiltration occurred in limited areas (Figure 6), which was not reflected in the total score (Table 3).
Figure 6.

Effect of pretreatment with protease inhibitors on TxA-induced pathologic changes. Images show the ileum. (A) Vehicle control, (B) TxA, (C) FUT-175 plus TxA, (D) MPI-0442352 plus TxA, (E) SBTI (10 mg) plus TxA, (F) SBTI (20 mg) plus TxA. TxA caused a marked loss of villi, edema, neutrophil infiltration, and necrosis (arrows). SBTI pretreatment decreased epithelial damage and edema, but neutrophil infiltration was sometimes prominent (arrow). Scale bar = 100 μm.
Table 3.
Histologic Score for TxA-Induced Enteritis in Mice Pretreated With Protease Inhibitors
| Pretreatment + treatment | Epithelial damage | Edema congestion | Polymorphonuclear leukocyte infiltration | Total score |
|---|---|---|---|---|
| Vehicle + vehicle control | 0.44 ± 0.1 | 0.97 ± 0.1 | 1.29 ± 0.2 | 2.73 ± 0.4 |
| Vehicle + TxA | 1.61 ± 0.1a | 1.77 ± 0.1a | 1.90 ± 0.1 | 5.21 ± 0.2a |
| FUT-175 + vehicle control | 1.02 ± 0.1 | 0.02 ± 0.1 | 0.88 ± 0.1 | 2.65 ± 0.2 |
| FUT-175 + TxA | 1.03 ± 0.2a | 1.51 ± 0.2 | 1.77 ± 0.2 | 4.35 ± 0.3a |
| MPI-0442352 + vehicle control | 0.56 ± 0.1 | 0.48 ± 0.02 | 0.56 ± 0.1 | 1.60 ± 0.2 |
| MPI-0442352 + TxA | 1.67 ± 0.2 | 1.93 ± 0.2 | 1.98 ± 0.2 | 5.55 ± 0.5 |
| 10 mg SBTI + vehicle control | 0.30 ± 0.1 | 1.30 ± 0.1 | 1.46 ± 0.1 | 3.07 ± 0.2 |
| 10 mg SBTI + TxA | 0.53 ± 0.1b | 1.17 ± 0.3b | 1.63 ± 0.1 | 3.32 ± 0.3b |
| 20 mg SBTI + vehicle control | 0.43 ± 0.1 | 0.56 ± 0.1 | 0.79 ± 0.1 | 1.87 ± 0.2 |
| 20 mg SBTI + TxA | 0.86 ± 0.1b | 1.06 ± 0.1b | 1.24 ± 0.1 | 3.13 ± 0.3b |
NOTE. Vehicle refers to vehicle for protease inhibitors, and vehicle control refers to vehicle for TxA.
P < .05 compared with inhibitor vehicle + vehicle control group.
P < .05 compared with inhibitor vehicle + TxA group.
TxA Up-Regulates Immunoreactive PAR2 in the Mouse Ileum
We determined the effect of TxA on levels of immunoreactive PAR2 in the intestine. Under basal conditions, PAR2 was detected on colonocytes in the ileum (Figure 7A). In mice treated with 1 μg TxA for 4 hours, the intensity of PAR2 immunoreactivity in enterocytes was significantly increased (Figure 7B). PAR2 staining in tissue obtained from mice treated with buffer had 100,785 ± 48,537 pixels above baseline, whereas PAR2 staining in tissue obtained from mice treated with TxA had 346,401 ± 58,338 pixels above baseline (P < .05). Tissue sections incubated only with anti-rabbit immunoglobulin G conjugated to FITC had no staining (Figure 7C and D). To enable visualization of the tissue, we acquired these images with a higher gain than that used for sections stained for PAR2 (compare black levels in Figure 7A and B vs Figure 7C and D). Thus, exposure to TxA significantly increased PAR2 expression.
Figure 7.
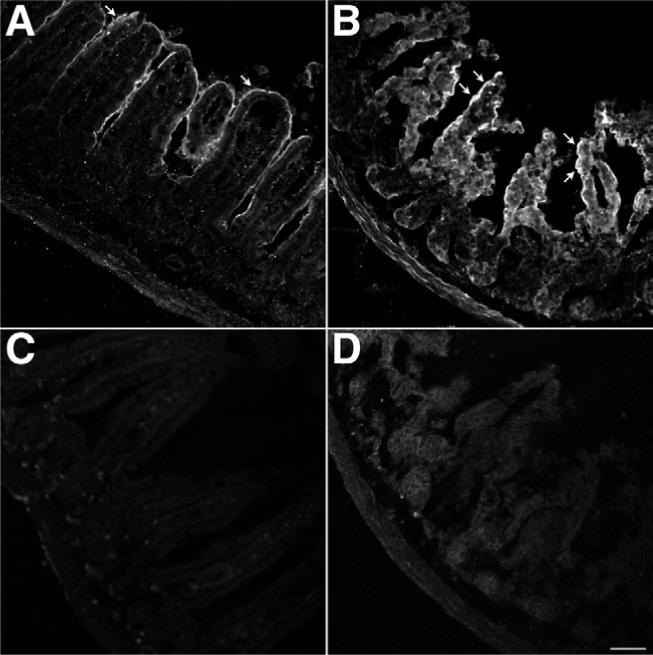
Effects of TxA on PAR2 expression. Ileal loops formed in wild-type mice were injected with (A and C) vehicle or (B and D) 1.0 μg TxA. Tissue was stained for PAR2 using a mouse anti-PAR2 antibody 9717 and (A and B) a goat anti-rabbit secondary antibody conjugated to FITC or (C and D) the goat anti-rabbit secondary antibody conjugated to FITC alone. Images were acquired by confocal microscopy and are unmodified. The images shown in A and B were acquired under identical conditions to optimize the quantification of pixel intensity above baseline. The images shown in C and D were acquired under identical conditions to optimize tissue visualization. (A) Basal PAR2 expression with positive enterocytes (arrow). (B) TxA markedly up-regulated PAR2 expression in enterocytes (arrows). Omission of the mouse anti-PAR2 antibody 9717 eliminated specific staining in both (C) vehicle-treated ileum and (D) TxA-treated ileum. Scale bar = 50 μm.
TxA Enhances PAR2 and Trypsin IV Expression and Activity of PAR2 in Human Colonocytes
To evaluate in more detail the effects of TxA on expression and activity of PAR2 and PAR2-activating proteases, we exposed NCM460 cells to TxA or vehicle (control). We assessed expression of PAR2 and trypsin IV messenger RNA using real-time quantitative PCR with a Ct method and β-glucuronidase as a control. Trypsin IV is an extrapancreatic trypsin that is expressed in epithelial cell lines and normal human intestine and can activate PAR2.6 Results above threshold were obtained after 28−30 cycles. Exposure to TxA (5 μmol/L) caused a statistically significant increase in expression of PAR2 messenger RNA to 174% of control levels at 2 hours and 199% at 4 hours (Figure 8A) but after 6 hours declined to 136% of control (P < .05). TxA also caused a striking increase in trypsin IV expression to 149% after 2 hours and 211% after 6 hours (Figure 8B). Thus, TxA causes a doubling in RNA levels of PAR2 and trypsin IV, which remain elevated for a prolonged period.
Figure 8.

Effects of TxA on NCM460 cells. (A) Real-time quantitative PCR of PAR2 levels. Results of 3 experiments are shown. After 120 minutes of exposure to TxA, PAR2 levels were increased and remained increased even after 360 minutes. (B) Real-time quantitative PCR of trypsin IV levels. Results of 3 experiments are shown. After 120 minutes of exposure to TxA, trypsin IV levels were increased and remained increased even after 360 minutes. (C) Ca2+ mobilization in response to PAR2 AP or vehicle (1/1300 dilution of 0.05 mol/L Tris in NCM culture medium) in cells pretreated with 5 nmol/L or 5 μmol/L TxA. Ca2+ responses to PAR2-AP (10−5 mol/L) are expressed as a 340/380 ratio (peak baseline; n = 38−45 cells). TxA significantly enhanced responses to PAR2-AP. *P < .05 between control or 0 minutes and TxA-exposed cells. †P = .055 between control or 0 minutes and TxA-exposed cells.
To assess whether TxA up-regulated functional PAR2, we analyzed PAR2-dependent signaling in NCM460 cells by measuring changes in [Ca2+]i in response to AP, a PAR2-selective agonist. Because TxA may cause transient changes in [Ca2+]i, we determined that levels had returned to basal before stimulation with AP. Prolonged exposure to TxA did not significantly increase the baseline [Ca2+]i (0.76 ± 0.4, 0.57 ± 0.1, 0.69 ± 0.3, and 0.81 ± 0.4 in cells exposed to vehicle, 5 nmol/L TxA for 6 hours, 5 nmol/L TxA for 24 hours, and 5 μmol/L TxA for 1 hour with 24-hour recovery, respectively). In comparison with vehicle (peak baseline, 0.087 ± 0.006), exposure to TxA (5 nmol/L) for 6 hours significantly increased responses to AP (10−5 mol/L) by 190% (0.166 ± 0.018; Figure 8C; P < .05). This increase was stable and did not rise further after 24 hours with TxA, when it was 184% of control (0.160 ± 0.013). To determine if a prolonged effect would occur, we incubated cells with a higher dose of TxA (5 μmol/L) for 1 hour and tested PAR2 Ca2+ responses after a 24-hour recovery. Responses were 156% of control (0.136 ± 0.013; P < .05). Thus, exposure of colonocytes to TxA causes a marked increase in PAR2 signaling that remains for 24 hours after TxA is withdrawn.
Activation of PAR2 Induces Inflammation in the Intestine
Because TxA-induced inflammation was diminished in mice with deletion or inhibition of PAR2 or enzymes that activate PAR2 and because TxA increased PAR2 expression and function in cell lines and animals, we determined whether PAR2 activation alone would induce ileitis. Mice were treated with AP, tryptase, pancreatic trypsin II, extrapancreatic human trypsin IV, and p23, a rat trypsin isozyme that resembles trypsin IV in its resistance to endogenous inhibitors.21 Intestinal secretion and MPO activity were measured after 4 hours. AP significantly stimulated intestinal secretion (111 ± 20 mg/cm; Figure 9A; P < .05 vs vehicle control). All enzymes tested, tryptase, trypsin II, trypsin IV, and p23, significantly increased intestinal secretion, albeit to a lower level than AP (76 ± 4, 78 ± 4, 80 ± 11, and 89 ± 7 mg/cm, respectively; Figure 9A). AP also significantly stimulated granulocyte infiltration (tissue MPO, 0.20 ± 0.03 U MPO/mg protein; fluid MPO, 0.26 ± 0.02 U MPO/mL fluid; P < .05 vs vehicle control). All enzymes tested, tryptase, trypsin II, trypsin IV, and p23, significantly increased tissue MPO levels (0.18 ± 0.05, 0.23 ± 0.04, 0.17 ± 0.01, and 0.19 ± 0.03 U MPO/mg protein, respectively; Figure 9B; P < .05). Fluid MPO levels were maximally increased with tryptase and trypsin II (0.22 ± 0.04 and 0.26 ± 0.07 U MPO/mL fluid, respectively; Figure 9C; P < .05 vs vehicle control), while trypsin IV and p23 induced a weaker response (0.14 ± 0.29 and 0.15 ± 0.04 U MPO/mL fluid, respectively; Figure 9C; P < .05 vs vehicle control). Reverse peptide did not induce a significant inflammatory response (results not shown; fluid secretion, 46.4 ± 6.6 mg/cm; tissue MPO, 0.07 ± 0.02 U/mg protein; fluid MPO, 0.03 ± 0.01 U/mL). Thus, AP and the enzymes that activate PAR2 can induce ileitis.
Figure 9.
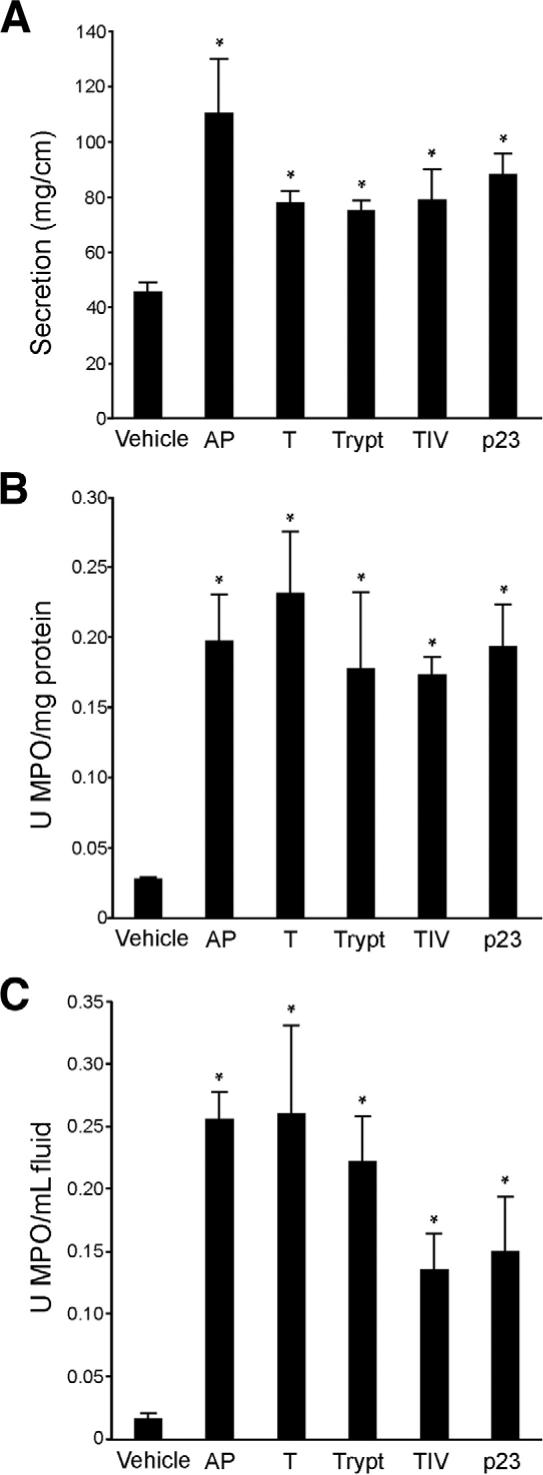
Effect of activation of PAR2 on inflammation in the ileum. Mouse ileum was injected with AP (SLIGRL), pancreatic trypsin (T), tryptase (Trypt), trypsin IV (TIV), p23, or vehicle. Inflammation was assessed as before (Figures 1, 3, and 5). (A) All agents increased intestinal secretion, expressed as milligrams per centimeter of loop, with AP having the largest effect. (B) All agents increased tissue MPO, expressed as units MPO per milligram protein, to similar levels. (C) Fluid MPO, expressed as units MPO per milliliter fluid, was most strikingly increased by AP, trypsin, and tryptase. *P < .05 between control and results obtained by activating agents.
PAR2-Induced Ileitis Occurs via Neurogenic Mechanisms
Because substance P and the neurokinin 1 receptor in large part mediate TxA-induced ileitis,28,32 we also examined their role in PAR2-induced inflammation. We treated mice with the neurokinin 1 receptor antagonist SR140333 or vehicle (control) before induction of ileitis with AP. Mice treated with vehicle had robust levels of intestinal secretion and tissue and fluid MPO activity (intestinal secretion, 74 ± 0.03 mg/cm; tissue MPO, 0.32 ± 0.05 U/mg protein; fluid MPO, 0.37 ± 0.09 U/mL fluid; Figure 10A–C). SR140333 decreased intestinal secretion to basal levels and significantly diminished tissue and fluid MPO activity by 59% and 71%, respectively (intestinal secretion, 47 ± 0.02 mg/cm; tissue MPO, 0.13 ± 0.01 U/mg protein; fluid MPO, 0.11 ± 0.04 U/mL fluid; P < .05; Figure 10A–C; P < .05). Thus, PAR2-induced ileitis occurs via neurogenic mechanisms involving activation of the neurokinin 1 receptor.
Figure 10.

Effect of NK1R inhibition on PAR2-induced inflammation in the ileum. Mouse ileum was injected with carrier (1% dimethyl sulfoxide) or SR140333 60 minutes before AP. (A) Intestinal secretion, expressed as milligram per centimeter of loop, (B) tissue MPO, expressed as units MPO per milligram protein, and (C) fluid MPO, expressed as units MPO per milliliter fluid, were all significantly decreased by pretreatment with SR140333. *P < .05 between AP and results obtained by pretreatment with SR140333 followed by AP.
Discussion
In our experiments, deletion of PAR2 resulted in diminished inflammatory responses to TxA, as reflected by decreases in intestinal secretion, granulocyte infiltration into tissues and luminal fluid, and improved histologic assessment. Deletion and inhibition of PAR2-activating proteases also reduced TxA-induced ileitis. DPPI-deficient mice, which have reduced levels of active tryptase, had decreased intestinal secretion, fluid MPO activity, epithelial damage, and edema. The serine protease inhibitors FUT-175 and MPI-0442352, as well as SBTI, all diminished inflammation in response to TxA. TxA increased immunoreactive PAR2 on enterocytes of the ileum, while exposure of human colonocytes to TxA sensitized responses to PAR2 agonists and up-regulated messenger RNA for PAR2 and trypsin IV. PAR2 activators, such as AP, tryptase, and trypsins, also induced ileitis by a mechanism that depends on activation of the neurokinin 1 receptor. Together, these results indicate that PAR2 and enzymes that can activate PAR2 play a proinflammatory role in enteritis induced by TxA from C difficile. The mechanism may involve both activation of existing PAR2 and up-regulation of PAR2 and trypsin IV expression in enterocytes. Moreover, PAR2-induced inflammation occurs via a neurogenic mechanism involving activation of the neurokinin 1 receptor.
Contribution of PAR2 to C difficile TxA-Induced Enteritis
Although the inflammatory response to TxA was strong in Par2+/+ mice, it diminished significantly in Par2−/− mice and was intermediate in Par2+/− mice, suggesting a gene/dosage effect. These results suggest that PAR2 plays a proinflammatory role in C difficile enteritis. In support of this role for PAR2, deletion of PAR2 also protects against colitis induced by Citrobacter rodentium, allergic inflammation of the airways, and arthritis.33–36 In contrast, pretreatment with PAR2-AP protects against chemically induced colitis due to the release of calcitonin gene-related peptide.11 Although deletion of PAR2 significantly diminished MPO, TxA-induced pathologic changes were more completely blocked. This result may be due to the difference in recruitment versus activation of neutrophils. Activation of neutrophils is responsible for TxA-induced tissue damage, and an MPO assay would not discriminate between recruitment and activation of neutrophils.
We found that PAR2 activation by AP, tryptase, and several isozymes of trypsin (bovine pancreatic trypsin I/II, human trypsin IV, and rat p23) induced ileitis, with statistically significant increases in intestinal secretion and tissue and fluid MPO activity. Neurogenic mechanisms participate in PAR2-induced inflammation, because our results show that blockade of neurokinin 1 receptors prevents AP-induced ileitis. Thus, the participation of PAR2 in inflammatory responses to TxA is likely due, in part, to release of substance P and activation of the neurokinin 1 receptor. These results confirm observations in the intestine and skin that PAR2 agonists cause inflammation by a neurogenic mechanism.12,37 Similar neurogenic mechanisms also mediate TxA-induced enteritis, because antagonism or deletion of substance P and calcitonin gene-related peptide prevents TxA-induced enteritis.28,29,32,38,39 During TxA-induced enteritis, substance P and calcitonin gene-related peptide are up-regulated both locally and in dorsal root ganglia, emphasizing their role in the inflammatory response.39,40 Neurokinin 1 receptors are also up-regulated and activated during TxA-induced entritis.38 PAR2 activation of enteric neurons increases short-circuit current, indicative of chloride ion secretion,17 which would lead to intestinal secretion, which we found diminished with deletion of PAR2. Thus, both extrinsic and intrinsic neurons can mediate modulation of the inflammatory response to TxA by PAR2.
Further support for involvement of PAR2 in C difficile enteritis is provided by our finding that TxA up-regulates PAR2 immunoreactivity in enterocytes in the ileum, PAR2 responsiveness, and PAR2 and trypsin IV messenger RNA in colonocytes. These findings also suggest that the enterocyte is one of the key cells that participate in modulation of TxA responses by PAR2. Our results are consistent with other reports that PAR2 is up-regulated in cell lines by specific inflammatory mediators as well as in inflamed tissues.33,41–43 PAR2 up-regulation during C difficile infection could magnify the proinflammatory actions of proteases on epithelial cells, because activation of PAR2 on enterocytes disrupts tight junctions to increase paracellular permeability, which can result in bacterial translocation and colonic inflammation.1,7 TxA similarly disrupts tight junctions of enterocytes.15
PAR2 has other roles in inflammation that may participate in TxA-induced enteritis, but these have not been examined. For example, PAR2 is present on endothelial cells and neutrophils, and PAR2 agonists promote interleukin-6 secretion, l-selectin shedding by granulocytes, and leukocyte rolling and adhesion.26,44,45 Recruited neutrophils secrete proteases, including neutrophil elastase, that activate PAR2 on epithelial and nonepithelial cells to induce release of interleukin-8 and other cytokines important in TxA-induced inflammation.46 These potential mechanisms require further investigation.
Contributions of Proteases to C difficile TxA-Induced Enteritis
Our use of knockout mice and protease inhibitors to study the involvement of proteases in TxA-induced colitis showed that although deletion of DPPI reduced TxA-induced intestinal secretion, fluid MPO activity, and tissue edema, tissue MPO activity, neutrophil infiltration, and epithelial damage were unaltered. Thus, DPPI may influence the secretion of fluid and neutrophils into the lumen, without a marked effect on migration and activation of granulocytes in the tissues. Therefore, deletion of DPPI protects against TxA-induced enteritis, but to a smaller extent than PAR2 deletion. This is not surprising, because PAR2 is activated by several proteases,2,3,5 and DPPI−/− mice retain ∼25% of normal tryptase levels.8
Understanding the mechanism by which deletion of DPPI protects against TxA-induced enteritis is complicated by the fact that deletion of DPPI affects the activity of several proteases, only some of which can activate PAR2. DPPI knockout mice show diminished activity of tryptase, neutrophil elastase, and proteinase 3 but are completely deficient in activity of cathepsin G, mast cell chymase, and the granzymes A and B of cytotoxic lymphocytes.8,27,47
The serine protease inhibitors FUT-175 and MPI-0442352 inhibited TxA-induced enteritis, as evidenced by decreased intestinal fluid secretion and granulocyte infiltration into tissues and the intestinal lumen. FUT-175, originally described as a synthetic inhibitor of complement and serine proteases such as thrombin and trypsin, is a potent and selective inhibitor of human tryptase, with a Ki of 95 pmol/L.20 At 10 mg/kg, the dose used in this study, FUT-175 inhibits scratching induced by tryptase but not by histamine or serotonin in mice.48 Our results obtained with FUT-175 may be largely due to tryptase inhibition, because results obtained with an alternative human tryptase inhibitor, MPI-0442352, were similar, although less striking. The broad-spectrum inhibitor of both tryptic and nontryptic serine peptidases, SBTI, caused the most dramatic decrease in intestinal secretion, granulocyte infiltration, and histologic changes in response to TxA. In support of our results, SBTI also reduces infectious colitis in mice.36 The source and isozyme of tryptic peptidase that mediates TxA-induced enteritis remains to be determined. However, one possibility is trypsin IV, which we found to be up-regulated by TxA. Trypsin IV is a prominent protease that is widely expressed by extrapancreatic epithelial cells and tissues, including normal human colon, and can activate PAR2.6 Because trypsin IV is resistant to endogenous polypeptide trypsin inhibitors and degrades these inhibitors, if released, it could signal for prolonged periods by cleaving PARs. Because there are marked differences in the human and mouse trypsin genes, we did not attempt to characterize the forms of trypsin that are up-regulated by TxA in mice. However, we did observe that both trypsin IV and p23, a rat trypsin isozyme that is also resistant to endogenous inhibitors,21 induced ileitis in mice.
Although deletion of PAR2 and DPPI diminished responses to TxA, a substantial inflammatory response remained, which must be due to other mechanisms. These include other receptors known to participate in TxA-induced enteritis, such as the neurotensin receptor 1 and corticotrophin-releasing hormone receptors 1 and 2, both of which are up-regulated in response to TxA, and the CC chemokine receptor (CCR1).49–51
In summary, our use of overlapping genetic and pharmacologic approaches showed that PAR2 and its activators are proinflammatory in TxA-induced enteritis. Although each of these approaches by themselves has limitations, considered together our results show that deletion of PAR2 and the proteases that activate this receptor protect against inflammatory responses to TxA. Intestinal inflammation mediated by PAR2 involves a neurogenic mechanism. Moreover, TxA increases PAR2 immunoreactivity, messenger RNA, and signaling in intestinal epithelial cells. Thus, our results show, for the first time, that PAR2, DPPI, tryptase, and trypsin mediate the proinflammatory effects of TxA from C difficile. Because PAR2 and tryptase are up-regulated in human intestinal disease, including irritable bowel syndrome and ulcerative colitis,52–54 PAR2 and the enzymes that activate it may be a general mechanism in the pathophysiology of intestinal inflammation, including inflammatory bowel disease.
Acknowledgments
Sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases; National Heart, Lung, and Blood Institute; and Crohn's and Colitis Foundation of America (grants DK52388 (to EFG), DK57840, DK43207, DK39957 [to NWB], DK47343 [to CHP], HL024136 [to GHC], and CURE Peptidomics, Proteomics & RIA core, DK41301, and CCFA #1730 [to EFG]).
Abbreviations used in this paper
- AP
activating peptide
- Ct
comparative threshold cycle
- DPPI
dipeptidyl peptidase I
- FITC
fluorescein isothiocyanate
- MPO
myeloperoxidase
- PAR2
protease-activated receptor 2
- PCR
polymerase chain reaction
- SBTI
soybean trypsin inhibitor
- TxA
toxin A.
References
- 1.Vergnolle N. Modulation of visceral pain and inflammation by protease-activated receptors. Br J Pharmacol. 2004;141:1264–1274. doi: 10.1038/sj.bjp.0705750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunnett NW, Cottrell GS. Protease-activated receptors in gastrointestinal function and disease. In: Lendeckel U, Hooper NM, editors. Proteases in gastrointestinal tissues. Vol. 5. Dordrecht: Springer: 2006. pp. 1–32. [Google Scholar]
- 3.Corvera CU, Dery O, McConalogue K, Bohm SK, Khitin LM, Caughey GH, Payan DG, Bunnett NW. Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J Clin Invest. 1997;100:1383–1393. doi: 10.1172/JCI119658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reed DE, Barajas-Lopez C, Cottrell G, Velazquez-Rocha S, Dery O, Grady EF, Bunnett NW, Vanner SJ. Mast cell tryptase and protein-ase-activated receptor 2 induce hyperexcitability of guinea-pig submucosal neurons. J Physiol. 2003;547:531–542. doi: 10.1113/jphysiol.2002.032011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong W, McConalogue K, Khitin LM, Hollenberg MD, Payan DG, Bohm SK, Bunnett NW. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc Natl Acad SciUSA. 1997;94:8884–8889. doi: 10.1073/pnas.94.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. 2004;279:13532–13539. doi: 10.1074/jbc.M312090200. [DOI] [PubMed] [Google Scholar]
- 7.Jacob C, Yang PC, Darmoul D, Amadesi S, Saito T, Cottrell GS, Coelho AM, Singh P, Grady EF, Perdue M, Bunnett NW. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and beta-arrestins. J Biol Chem. 2005;280:31936–31948. doi: 10.1074/jbc.M506338200. [DOI] [PubMed] [Google Scholar]
- 8.Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH. Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem. 2001;276:18551–18556. doi: 10.1074/jbc.M100223200. [DOI] [PubMed] [Google Scholar]
- 9.Pham CT, Armstrong RJ, Zimonjic DB, Popescu NC, Payan DG, Ley TJ. Molecular cloning, chromosomal localization, and expression of murine dipeptidyl peptidase I. J Biol Chem. 1997;272:10695–10703. doi: 10.1074/jbc.272.16.10695. [DOI] [PubMed] [Google Scholar]
- 10.Mallen-St Clair J, Pham CT, Villalta SA, Caughey GH, Wolters PJ. Mast cell dipeptidyl peptidase I mediates survival from sepsis. J Clin Invest. 2004;113:628–634. doi: 10.1172/JCI19062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiorucci S, Mencarelli A, Palazzetti B, Distrutti E, Vergnolle N, Hollenberg MD, Wallace JL, Morelli A, Cirino G. Proteinase-activated receptor 2 is an anti-inflammatory signal for colonic lamina propria lymphocytes in a mouse model of colitis. Proc Natl Acad SciUSA. 2001;98:13936–13941. doi: 10.1073/pnas.241377298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen C, Coelho AM, Grady E, Compton SJ, Wallace JL, Hollenberg MD, Cenac N, Garcia-Villar R, Bueno L, Steinhoff M, Bunnett NW, Vergnolle N. Colitis induced by proteinase-activated receptor-2 agonists is mediated by a neurogenic mechanism. Can J Physiol Pharmacol. 2003;81:920–927. doi: 10.1139/y03-080. [DOI] [PubMed] [Google Scholar]
- 13.Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, Cirino G, Gerard N, Basbaum AI, Andrade-Gordon P, Hollenberg MD, Wallace JL. Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nat Med. 2001;7:821–826. doi: 10.1038/89945. [DOI] [PubMed] [Google Scholar]
- 14.Pothoulakis C, Castagliuolo I, LaMont JT. Nerves and intestinal mast cells modulate responses to enterotoxins. News Physiol Sci. 1998;13:58–63. doi: 10.1152/physiologyonline.1998.13.2.58. [DOI] [PubMed] [Google Scholar]
- 15.Chen ML, Pothoulakis C, LaMont JT. Protein kinase C signaling regulates ZO-1 translocation and increased paracellular flux of T84 colonocytes exposed to Clostridium difficile toxin A. J Biol Chem. 2002;277:4247–4254. doi: 10.1074/jbc.M109254200. [DOI] [PubMed] [Google Scholar]
- 16.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vergnolle N, Macnaughton WK, Al-Ani B, Saifeddine M, Wallace JL, Hollenberg MD. Proteinase-activated receptor 2 (PAR2)-activating peptides: identification of a receptor distinct from PAR2 that regulates intestinal transport. Proc Natl Acad SciUSA. 1998;95:7766–7771. doi: 10.1073/pnas.95.13.7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wershil BK, Castagliuolo I, Pothoulakis C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology. 1998;114:956–964. doi: 10.1016/s0016-5085(98)70315-4. [DOI] [PubMed] [Google Scholar]
- 19.Pothoulakis C, Karmeli F, Kelly CP, Eliakim R, Joshi MA, O'Keane CJ, Castagliuolo I, LaMont JT, Rachmilewitz D. Ketotifen inhibits Clostridium difficile toxin A-induced enteritis in rat ileum. Gastroenterology. 1993;105:701–707. doi: 10.1016/0016-5085(93)90886-h. [DOI] [PubMed] [Google Scholar]
- 20.Mori S, Itoh Y, Shinohata R, Sendo T, Oishi R, Nishibori M. Nafamostat mesilate is an extremely potent inhibitor of human tryptase. J Pharmacol Sci. 2003;92:420–423. doi: 10.1254/jphs.92.420. [DOI] [PubMed] [Google Scholar]
- 21.Fukuoka S, Nyaruhucha CM. Expression and functional analysis of rat P23, a gut hormone-inducible isoform of trypsin, reveals its resistance to proteinaceous trypsin inhibitors. Biochim Biophys Acta. 2002;1588:106–112. doi: 10.1016/s0925-4439(02)00153-9. [DOI] [PubMed] [Google Scholar]
- 22.Ogbu CO, Qabar MN, Boatman PD, Urban J, Meara JP, Ferguson MD, Tulinsky J, Lum C, Babu S, Blaskovich MA, Nakanishi H, Ruan F, Cao B, Minarik R, Little T, Nelson S, Nguyen M, Gall A, Kahn M. Highly efficient and versatile synthesis of libraries of constrained beta-strand mimetics. Bioorg Med Chem Lett. 1998;8:2321–2326. doi: 10.1016/s0960-894x(98)00420-x. [DOI] [PubMed] [Google Scholar]
- 23.Kahn M. B-Sheet mimetics and use thereof as protease inhibitors. 2000 U.S. Patent 6,020,331.
- 24.Kahn M, Ogbu CO, Eguchi M, Kim HO, Boatmen PD., Jr B-Sheet mimetics and use thereof as inhibitors of biologically active peptides or proteins. 2001 U.S. Patent 6,245,764 B1.
- 25.Oh SW, Pae CI, Lee DK, Jones F, Chiang GK, Kim HO, Moon SH, Cao B, Ogbu C, Jeong KW, Kozu G, Nakanishi H, Kahn M, Chi EY, Henderson WR., Jr Tryptase inhibition blocks airway inflammation in a mouse asthma model. J Immunol. 2002;168:1992–2000. doi: 10.4049/jimmunol.168.4.1992. [DOI] [PubMed] [Google Scholar]
- 26.Lindner JR, Kahn ML, Coughlin SR, Sambrano GR, Schauble E, Bernstein D, Foy D, Hafezi-Moghadam A, Ley K. Delayed onset of inflammation in protease-activated receptor-2-deficient mice. J Immunol. 2000;165:6504–6510. doi: 10.4049/jimmunol.165.11.6504. [DOI] [PubMed] [Google Scholar]
- 27.Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad SciUSA. 1999;96:8627–8632. doi: 10.1073/pnas.96.15.8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pothoulakis C, Castagliuolo I, LaMont JT, Jaffer A, O'Keane JC, Snider RM, Leeman SE. CP-96,345, a substance P antagonist, inhibits rat intestinal responses to Clostridium difficile toxin A but not cholera toxin. Proc Natl Acad SciUSA. 1994;91:947–951. doi: 10.1073/pnas.91.3.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirkwood KS, Bunnett NW, Maa J, Castagliolo I, Liu B, Gerard N, Zacks J, Pothoulakis C, Grady EF. Deletion of neutral endopeptidase exacerbates intestinal inflammation induced by Clostridium difficile toxin A. Am J Physiol Gastrointest Liver Physiol. 2001;281:G544–G551. doi: 10.1152/ajpgi.2001.281.2.G544. [DOI] [PubMed] [Google Scholar]
- 30.Cuffe JE, Bertog M, Velazquez-Rocha S, Dery O, Bunnett N, Korbmacher C. Basolateral PAR-2 receptors mediate KCl secretion and inhibition of Na+ absorption in the mouse distal colon. J Physiol. 2002;539:209–222. doi: 10.1113/jphysiol.2001.013159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cottrell GS, Roosterman D, Marvizon JC, Song B, Wick E, Pikios S, Wong H, Berthelier C, Tang Y, Sternini C, Bunnett NW, Grady EF. Localization of calcitonin receptor-like receptor and receptor activity modifying protein 1 in enteric neurons, dorsal root ganglia, and the spinal cord of the rat. J Comp Neurol. 2005;490:239–255. doi: 10.1002/cne.20669. [DOI] [PubMed] [Google Scholar]
- 32.Castagliuolo I, Riegler M, Pasha A, Nikulasson S, Lu B, Gerard C, Gerard NP, Pothoulakis C. Neurokinin-1 (NK-1) receptor is required in Clostridium difficile-induced enteritis. J Clin Invest. 1998;101:1547–1550. doi: 10.1172/JCI2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, Smith AJ, Hunter GD, McLean JS, McGarry F, Ramage R, Jiang L, Kanke T, Kawagoe J. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidlin F, Amadesi S, Dabbagh K, Lewis DE, Knott P, Bunnett NW, Gater PR, Geppetti P, Bertrand C, Stevens ME. Protease-activated receptor 2 mediates eosinophil infiltration and hyper-reactivity in allergic inflammation of the airway. J Immunol. 2002;169:5315–5321. doi: 10.4049/jimmunol.169.9.5315. [DOI] [PubMed] [Google Scholar]
- 35.Su X, Camerer E, Hamilton JR, Coughlin SR, Matthay MA. Protease-activated receptor-2 activation induces acute lung inflammation by neuropeptide-dependent mechanisms. J Immunol. 2005;175:2598–2605. doi: 10.4049/jimmunol.175.4.2598. [DOI] [PubMed] [Google Scholar]
- 36.Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, Wallace JL, Hollenberg MD, Vergnolle N. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci U S A. 2005;102:8363–8368. doi: 10.1073/pnas.0409535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 38.Pothoulakis C, Castagliuolo I, Leeman SE, Wang CC, Li H, Hoffman BJ, Mezey E. Substance P receptor expression in intestinal epithelium in Clostridium difficile toxin A enteritis in rats. Am J Physiol. 1998;275:G68–G75. doi: 10.1152/ajpgi.1998.275.1.G68. [DOI] [PubMed] [Google Scholar]
- 39.Keates AC, Castagliuolo I, Qiu B, Nikulasson S, Sengupta A, Pothoulakis C. CGRP upregulation in dorsal root ganglia and ileal mucosa during Clostridium difficile toxin A-induced enteritis. Am J Physiol. 1998;274:G196–G202. doi: 10.1152/ajpgi.1998.274.1.G196. [DOI] [PubMed] [Google Scholar]
- 40.Castagliuolo I, Keates AC, Qiu B, Kelly CP, Nikulasson S, Leeman SE, Pothoulakis C. Increased substance P responses in dorsal root ganglia and intestinal macrophages during Clostridium difficile toxin A enteritis in rats. Proc Natl Acad SciUSA. 1997;94:4788–4793. doi: 10.1073/pnas.94.9.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamilton JR, Frauman AG, Cocks TM. Increased expression of protease-activated receptor-2 (PAR2) and PAR4 in human coronary artery by inflammatory stimuli unveils endothelium-dependent relaxations to PAR2 and PAR4 agonists. Circ Res. 2001;89:92–98. doi: 10.1161/hh1301.092661. [DOI] [PubMed] [Google Scholar]
- 42.Buddenkotte J, Stroh C, Engels IH, Moormann C, Shpacovitch VM, Seeliger S, Vergnolle N, Vestweber D, Luger TA, Schulze-Osthoff K, Steinhoff M. Agonists of proteinase-activated receptor-2 stimulate upregulation of intercellular cell adhesion molecule-1 in primary human keratinocytes via activation of NF-kappa B. J Invest Dermatol. 2005;124:38–45. doi: 10.1111/j.0022-202X.2004.23539.x. [DOI] [PubMed] [Google Scholar]
- 43.Dattilio A, Vizzard MA. Up-regulation of protease activated receptors in bladder after cyclophosphamide induced cystitis and co-localization with capsaicin receptor (VR1) in bladder nerve fibers. J Urol. 2005;173:635–639. doi: 10.1097/01.ju.0000143191.55468.1d. [DOI] [PubMed] [Google Scholar]
- 44.Vergnolle N. Proteinase-activated receptor-2-activating peptides induce leukocyte rolling, adhesion, and extravasation in vivo. J Immunol. 1999;163:5064–5069. [PubMed] [Google Scholar]
- 45.Shpacovitch VM, Varga G, Strey A, Gunzer M, Mooren F, Buddenkotte J, Vergnolle N, Sommerhoff CP, Grabbe S, Gerke V, Homey B, Hollenberg M, Luger TA, Steinhoff M. Agonists of proteinase-activated receptor-2 modulate human neutrophil cytokine secretion, expression of cell adhesion molecules, and migration within 3-D collagen lattices. J Leukoc Biol. 2004;76:388–398. doi: 10.1189/jlb.0503221. [DOI] [PubMed] [Google Scholar]
- 46.Uehara A, Muramoto K, Takada H, Sugawara S. Neutrophil serine proteinases activate human nonepithelial cells to produce inflammatory cytokines through protease-activated receptor 2. J Immunol. 2003;170:5690–5696. doi: 10.4049/jimmunol.170.11.5690. [DOI] [PubMed] [Google Scholar]
- 47.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ui H, Andoh T, Lee JB, Nojima H, Kuraishi Y. Potent pruritogenic action of tryptase mediated by PAR-2 receptor and its involvement in anti-pruritic effect of nafamostat mesilate in mice. Eur J Pharmacol. 2006;530:172–178. doi: 10.1016/j.ejphar.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 49.Castagliuolo I, Wang CC, Valenick L, Pasha A, Nikulasson S, Carraway RE, Pothoulakis C. Neurotensin is a proinflammatory neuropeptide in colonic inflammation. J Clin Invest. 1999;103:843–849. doi: 10.1172/JCI4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wlk M, Wang CC, Venihaki M, Liu J, Zhao D, Anton PM, Mykoniatis A, Pan A, Zacks J, Karalis K, Pothoulakis C. Corticotropin-releasing hormone antagonists possess anti-inflammatory effects in the mouse ileum. Gastroenterology. 2002;123:505–515. doi: 10.1053/gast.2002.34783. [DOI] [PubMed] [Google Scholar]
- 51.Morteau O, Castagliuolo I, Mykoniatis A, Zacks J, Wlk M, Lu B, Pothoulakis C, Gerard NP, Gerard C. Genetic deficiency in the chemokine receptor CCR1 protects against acute Clostridium difficile toxin A enteritis in mice. Gastroenterology. 2002;122:725–733. doi: 10.1053/gast.2002.31873. [DOI] [PubMed] [Google Scholar]
- 52.Kim JA, Choi SC, Yun KJ, Kim DK, Han MK, Seo GS, Yeom JJ, Kim TH, Nah YH, Lee YM. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm Bowel Dis. 2003;9:224–229. doi: 10.1097/00054725-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Raithel M, Winterkamp S, Pacurar A, Ulrich P, Hochberger J, Hahn EG. Release of mast cell tryptase from human colorectal mucosa in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:174–179. doi: 10.1080/003655201750065933. [DOI] [PubMed] [Google Scholar]
- 54.Barbara G, Stanghellini V, De Giorgio R, Cremon C, Cottrell GS, Santini D, Pasquinelli G, Morselli-Labate AM, Grady EF, Bunnett NW, Collins SM, Corinaldesi R. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693–702. doi: 10.1053/j.gastro.2003.11.055. [DOI] [PubMed] [Google Scholar]