

Abstract
Calpastatin, the endogenous inhibitor of calpain, is an intrinsically unstructured protein proposed to undergo folding transitions upon binding to the enzyme. As this feature has never been experimentally tested, we have set out to characterize the conformation of two peptides corresponding to its conserved subdomains, A and C, known to interact with calpain in a Ca2+-dependent manner. The peptides are disordered in water but show a high propensity for α-helical conformation in the presence of trifluoroethanol. The conformational transition is sensitive to Ca2+, and is clearly seen upon binding of the peptides to the enzyme. Secondary-structure prediction of all calpastatin sequences shows that the helix-forming potential within these regions is a conserved feature of the inhibitor. Furthermore, quantitative data on the binding strength of calpastatin fragments reveal that binding of the inhibitor is accompanied by a large decrease in its configurational entropy. Taken together, these observations point to significant binding-induced local folding transitions in calpastatin, in a way that ensures highly specific, yet reversible, action of the inhibitor.
Keywords: Calpastatin, calpain inhibitor, local folding, induced folding, disorder-to-order transition, intrinsically unstructured protein, protein disorder, natively unfolded protein
Calpastatin is a specific, endogenous inhibitor of calpain, the Ca2+-dependent intracellular cysteine protease (Takano and Maki 1999). A typical calpastatin molecule contains five functional domains: The first domain (domain L) regulates L-type Ca2+ channels (Hao et al. 2000); the other four, homologous domains (domains I–IV) are inhibitory domains, each with the potency of inhibiting one calpain molecule (Maki et al. 1987; Emori et al. 1988). The inhibitory domains contain three short, conserved segments (subdomains A, B, and C), which are primarily responsible for calpain inhibition. Although the structure of the enzyme-inhibitor complex has not been described at atomic resolution, biochemical studies have shown that subdomain B binds to the active site of the enzyme (Ma et al. 1993; Takano et al. 1995), whereas subdomains A and C potentiate this binding by anchoring the inhibitor to the enzyme in a Ca2+-dependent manner. These two only bind to the active conformation of the enzyme (Tompa et al. 2002), probably through the calmodulin-like domains of its large and small subunits, respectively (Ma et al. 1993; 1994; Yang et al. 1994; Takano et al. 1995). Based on prediction analysis of their sequence, it has been suggested that subdomains A and C assume an α-helical conformation upon binding to their target region on calpain (Ma et al. 1993). Although this assumption has never been experimentally verified, it is still considered generally correct (Takano and Maki 1999).
The issue of the exact binding mode of calpastatin is gaining momentum again with the recent recognition that many proteins and protein domains lack a well-defined three-dimensional (3D) structure in their native, functional state (Wright and Dyson 1999; Dunker et al. 2002). Although these intrinsically unstructured proteins (IUPs) resemble the highly denatured, random coil-like states of globular proteins, their function often stems from molecular recognition and permanent binding to their target molecules (Tompa 2002). Upon binding, they undergo local or extended folding transitions, in which part of their polypeptide chain assumes an ordered conformation. This mode of binding makes highly specific interactions reversible, which is fundamental for regulation. This simple yet powerful molecular scheme is widely encountered in protein–DNA, protein–RNA, and protein–protein interactions (Demchenko 2001; Dyson and Wright 2002).
Calpastatin has long been known to lack a well-defined 3D structure. Its 1H-NMR spectrum is characterized by little chemical shift dispersion, which is indicative of a highly flexible chain with no structural order (Uemori et al. 1990; Konno et al. 1997). Circular dichroism (CD) spectroscopy studies have led to similar conclusions, i.e., that the conformation of calpastatin is best approximated by a random coil (Uemori et al. 1990; Konno et al. 1997), with possibly traces of structured elements (Hackel et al. 2000). Other observations, such as its extended conformation by small-angle X-ray scattering (Konno et al. 1997) and size exclusion chromatography (Takano and Maki 1999), extreme proteolytic sensitivity, unusual SDS-PAGE mobility (Takano et al. 1988; Ma et al. 1993) and resistance to denaturing conditions such as heat or strong acids (Takano and Maki 1999), all attest to its open and highly flexible conformational state. By all such structural criteria (see Wright and Dyson 1999; Dunker et al. 2002; Uversky 2002a,b), calpastatin has been classified to belong to IUPs (Tompa 2002; Uversky 2002a). Thus, identification of secondary structural elements within its structure may significantly advance our understanding of the functional fine-tuning of this protein class.
To this end, the conformation and possible structural transitions upon changes in the environment of the peptides representing subdomains A and C have been studied by secondary structure predictions, CD spectroscopy, and analysis of quantitative binding data. Chou-Fasman predictions show that the α-helix is the preferred conformation within these segments of calpastatin. According to CD spectra, both peptides are disordered in water but have a preference for the α-helical conformation in the presence of trifluoroethanol (TFE) and also upon binding to calpain. Finally, the binding of calpastatin to calpain is accompanied by its significant disorder-to-order transition, as witnessed by quantitative binding data from the literature. These various pieces of evidence together imply the delicate balance of calpastatin structure between structural order and disorder, which is probably fundamental to the effective control of the calpain–calpastatin system.
Results
CD spectra of peptides A and C in water–TFE mixtures
The secondary structure of peptides A and C was studied by CD spectroscopy. The spectra were first recorded in water–TFE mixtures containing 0%, 8%, 16%, 35%, 50%, and 100% TFE (Fig. 1 ▶). In pure water, both spectra indicate the lack of well-defined and characteristic secondary structures; the curves correspond to typical U-type CD spectra, characterized by an intensive negative peak at ~200 nm and a shoulder at 220 nm (Perczel and Hollósi 1996). In TFE solution, on the other hand, both peptides assume an ordered secondary structure containing relatively high proportions of α-helix and expanded β-sheet conformers, as indicated by a strong maximum at about λ = 195 nm and by the location and ratio of pi–pi* (λ = 208 nm) and n–pi* (λ = 220 nm) maxima (θ220/θ208 = 0.82 for peptide A and θ220/θ208 = 0.71 for peptide C; Fig. 1 ▶).
Figure 1.
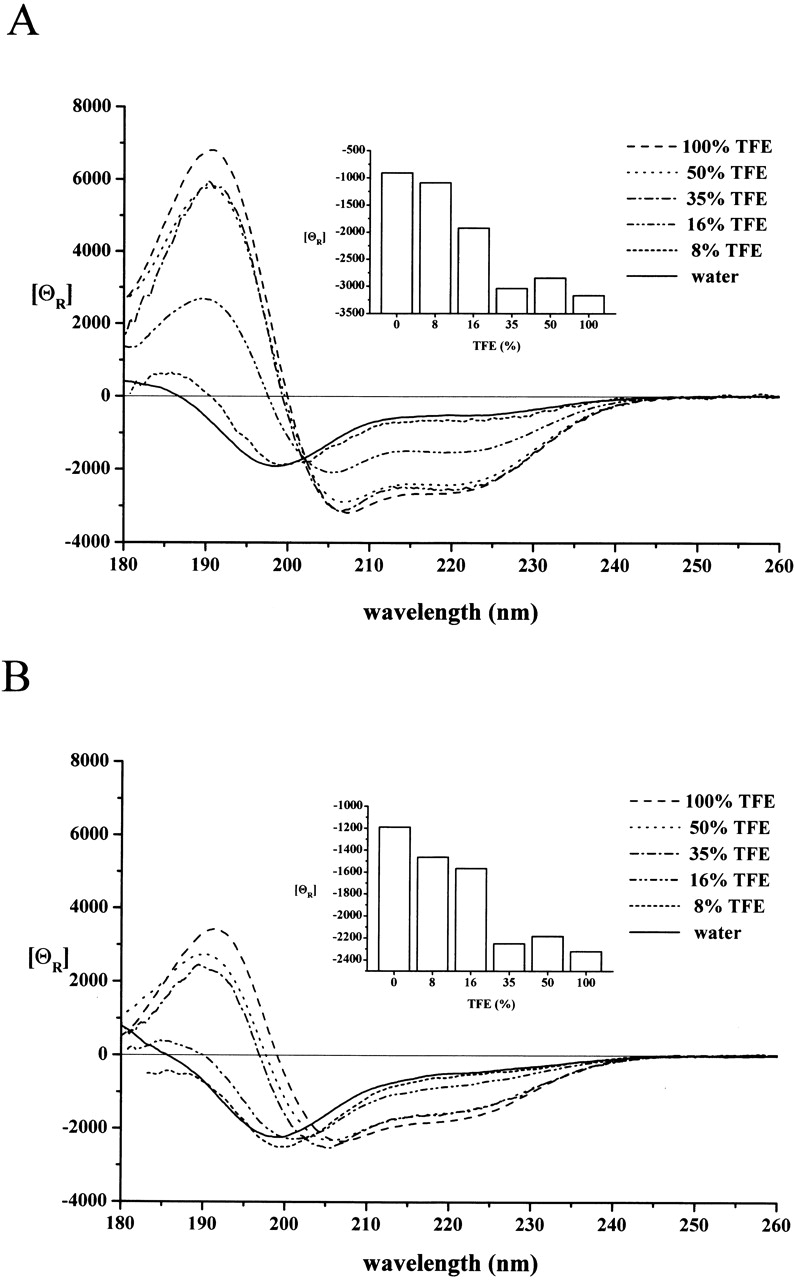
CD spectra of peptides A and C in water–TFE solutions. The CD spectrum of peptide A (A) and peptide C (B) at 0.7 mM concentration was recorded in water–TFE mixtures containing TFE at the percentages indicated. The conformational change in both cases corresponds to a transition to an α-helix conformation. The molar ellipticity values recorded at 208 nm, mainly contributed by the α-helix conformation of the peptides, reach half-maximal values at TFE concentrations of 16% for peptide A and ~25% for peptide C (insets).
The transition to the ordered, mainly α-helical, structure occurs at relatively low TFE concentrations for both peptides, at half-maximal TFE concentrations of 16% and ~25% for peptide A and C, respectively (cf. Fig. 1 ▶ insets). This points to a significant propensity for ordering, which has been found to be rather insensitive to experimental conditions, as a similar tendency could be observed at pH 3 and pH 7.4 and ionic strengths 0.15 and 1 M NaCl (data not shown).
The effect of Ca2+ on the conformation of peptides
Direct binding experiments have shown that the binding of calpastatin to calpain requires Ca2+ ions (Takano and Maki 1999), that is, the inhibitor only recognizes the active conformation of the enzyme that forms with Ca2+ binding. This ability to distinguish between inactive and active conformations is maintained within subdomains A and C (Tompa et al. 2002) and, thus, it is of interest whether Ca2+ has a direct effect on their conformation. Experiments performed in TFE in the presence of increasing amount of Ca2+ indicate that the α-helix type spectra of both peptides obtained in TFE without Ca2+ changed and U-type spectra resembling those obtained in water were detected (Fig. 2 ▶). Ca2+ was very effective in eliciting this conformational change; in the case of peptide A, the half-maximal effect occurs at 1 equivalent of the ion. With peptide C, the effect is somewhat weaker: the half-maximal effect takes 2 equivalents of Ca2+ ions. It should be noted that Ca2+ ions have no influence on the shape of the CD spectra in water (not shown).
Figure 2.
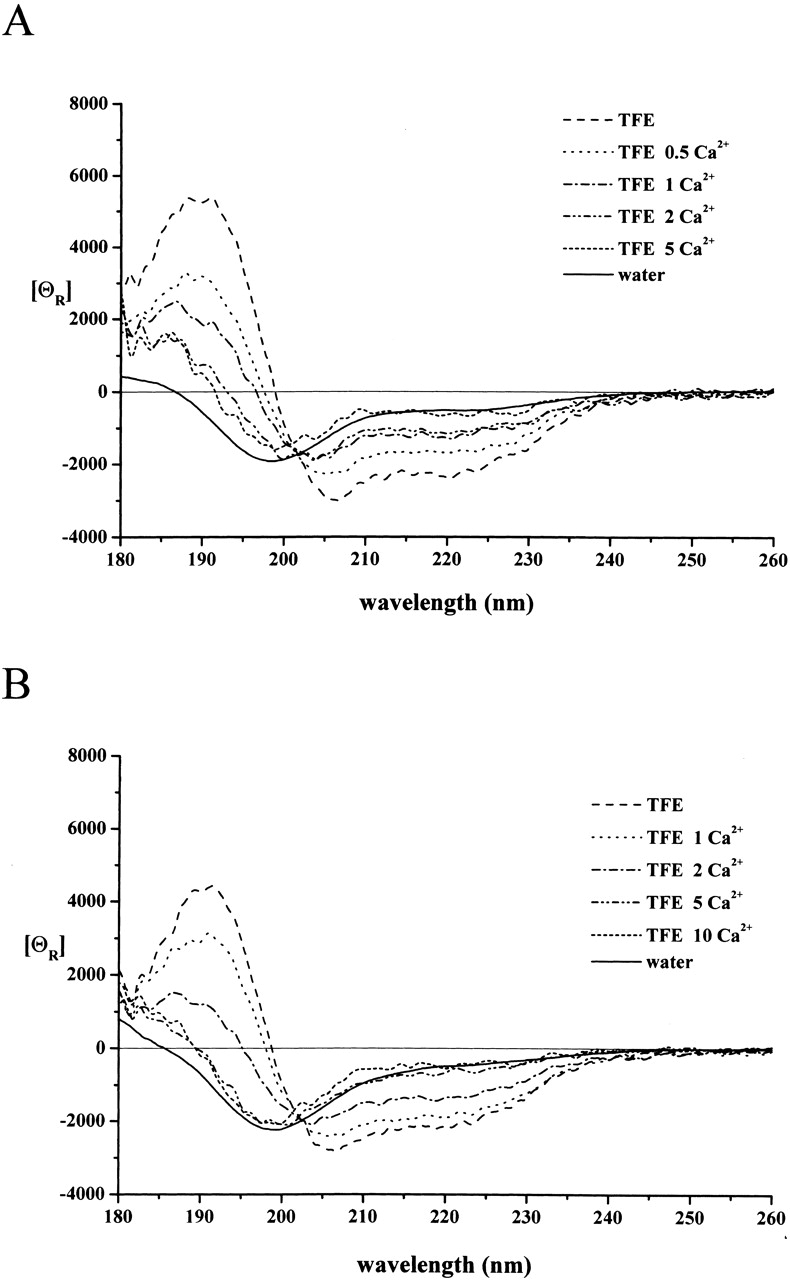
The effect of Ca2+ ions on the conformation of peptides A and C. The CD spectrum of peptide A (A) and peptide C (B) was recorded at 10 μM concentrations in TFE in the absence and presence of Ca2+ ions (given in molar equivalence). With both peptides, Ca2+ ions cause a conformational change toward the unstructured state observed in water. In the case of peptide A, the conformational transition occurs with a half-effective concentration that corresponds to 1 equivalent. With peptide C, the binding of Ca2+ is weaker; the half-effective stoichiometry is ~2 equivalents. For a comparison, the spectra in water, taken from Figure 1 ▶, are also shown.
The effect of Ca2+ ions on peptide conformation can be ascribed to specific binding, as demonstrated by recording the spectra in the presence of Na+ ions (no effect at up to 10 equivalents of Na+ on either peptide, whether in the absence or presence of TFE) or Mg2+ ions (a marginal effect on peptide A, no effect on peptide C at concentrations up to 10 equivalents; data not shown).
Structural change of subdomains A and C with calpain binding
The CD spectrum of the truncated small subunit of m-calpain (21K) with and without 3 mM Ca2+ ions was recorded. In accord with structural data (Hosfield et al. 1999; Strobl et al. 2000), the spectrum points to a high proportion of α-helix conformation within the protein. The shape of the curve does not change significantly after the addition of Ca2+ ions (not shown; cf. Blanchard et al. 1997 and Lin et al. 1997). In the presence of Ca2+ ions the spectra were also recorded by adding an equimolar amount of peptide A (Fig. 3A ▶) or peptide C (Fig. 4A ▶) to the small subunit.
Figure 3.

Peptide A assumes α-helical structure upon binding to the small subunit of m-calpain. (A) The CD spectrum of the small subunit of human m-calpain (21K) at 0.4 mg/mL was recorded in the absence (I) and presence (II) of an equimolar concentration of peptide A at 3 mM Ca2+ in water. (B) The difference spectrum obtained by subtracting the spectrum of the small subunit from the joint spectrum (III). For a comparison, the spectra of peptide A in TFE (IV) and water (V), taken from Figure 1A ▶ and normalized to the given peptide concentration, are also shown.
Figure 4.

Peptide C assumes α-helical structure upon binding to the small subunit of m-calpain. (A) The CD spectrum of the small subunit of m-calpain (21K) at 0.4 mg/mL was recorded in the absence (I) and presence (II) of an equimolar concentration of peptide C at 3 mM Ca2+ in water. (B) The difference spectrum obtained by subtracting the spectrum of the small subunit from that of peptide C + small subunit mixture (III). For a comparison, the spectra of peptide C in TFE (IV) and water (V), taken from Figure 1B ▶ and normalized to the given peptide concentration, are also shown.
In both cases a difference spectrum was obtained by subtracting the CD spectrum of the small subunit from that of the peptide + small subunit mixture (Figs. 3B ▶ and 4B ▶, respectively). This subtraction was performed by considering that the binding constant between 21K and subdomain C is in the nanomolar range (Takano et al. 1995) and therefore we assumed that the concentration of free calpain and of peptide is negligible in solution. Although the interaction between subdomain A of calpastatin and 21K is somewhat weaker (Takano et al. 1995), the subtraction is still allowed in this case. The difference spectra indicate the presence of ordered peptides, generated with binding to the small subunit of m-calpain. In the case of subdomain C (Fig. 4B ▶), ordering does occur toward a helical structure, in accord with recent X-ray data showing that the carboxy-terminal half of this subdomain assumes an α-helix, whereas the amino-terminal half remains disordered, in complex with the small subunit of calpain (Todd et al. 2003). Although a similar structure has been suggested for subdomain A by homology modeling, the difference spectrum (Fig. 3B ▶) is also compatible with the β-structure or a mixture of the two, suggesting that ordering of the two subdomains may follow somewhat different routes.
Helix-forming potential within subdomains A and C is conserved in evolution
As seen in the previous section, subdomains A and C assume an ordered, predominantly α-helical conformation with binding to the small subunit of the enzyme. Apparently, if this structural transition is essential for the recognition, it also has to manifest itself in the evolutionary conservation of the predicted secondary structure within regions A and C. In accord, the values averaged for all calpastatin inhibitory domain sequences show a significant bias for the α-helix structure in the carboxy-terminal region of both subdomains, that is, where these regions probably contact the enzyme (Todd et al. 2003; Fig. 5 ▶). For subdomain A, α-helix propensity dominates in the region 16–26 (positions 6–16 within the peptide). In the case of subdomain C, a weaker but still significant preference is seen, especially in the region 96–106 (positions 11–21 within the peptide). A further argument for the significance of this evolutionary tendency might come from the standard deviation values associated with these averages, as they are significantly lower for the α-helix within these regions than for the turn conformation. This difference implies that mutations decreasing the helix-forming potential have been poorly tolerated in evolution, whereas turn values have undergone accidental changes, being functionally less important. Intriguingly, helix propensity also dominates within subdomain B, but gives way to the β-turn conformation toward its carboxyl terminus. This region is the most conserved part of the inhibitory subdomain, which has been shown to be primarily responsible for inhibition by a mutagenesis screen (Betts et al. 2003) and to be prone to β-turn conformation by NMR (Ishima et al. 1991). By all these observations, structural preferences of conserved regions of calpastatin appear to presage their functional conformations.
Figure 5.

α-Helicity is preferred within subdomains A and C of calpastatin. All known calpastatin sequences have been downloaded from the Swiss-Prot database; their individual inhibitory domains have been separated and subjected to secondary structure prediction as given in the Materials and Methods section. Inhibitory domains were then aligned by ClustalW to allow the calculation of averages of secondary structure propensity values obtained; where gaps in one or more sequences were generated by the alignment, averaging was done for the remaining sequences. The average for α-helix (solid line) and β-turn (dotted line) is shown for the region encompassing the conserved subdomains A, B, and C marked by thick horizontal lines. For clarity, the significantly lower β-sheet values (~0.8) are not shown. Standard deviation values are seen at the bottom of the figure. Pairs of thin vertical lines mark the regions within each subdomain that are probably in direct contact with calpain in the calpastatin–calpain complex (see text for details).
Binding data suggest significant folding induced by binding
Because the 3D structure of the calpain–calpastatin complex is not available yet, the extent of disorder-to-order transition of the inhibitor with binding cannot be directly studied and verified. From data on the binding properties of various calpastatin fragments, however, one can assess the overall magnitude of conformational changes, namely ordering, with binding. To this end, relevant data for the inhibitory domain and subdomains A, B, and C have been collected. Intriguingly, all the data fall into the low to medium nanomolar range; the reported values are Kd = 3 nM (Yang et al. 1994) and 7 nM (Ma et al. 1994) for subdomain A, IC50 = 30 nM (Maki et al. 1989) and 80 nM (Ma et al. 1993) for subdomain B, Kd = 30 nM (Takano et al. 1995) for subdomain C, and KI = 3 nM (Maki et al. 1988) and IC50 = 6 nM (Ma et al. 1993) and 18 nM (Maki et al. 1988) for the whole inhibitory domain encompassing all three subdomains. Thus, the inhibitory domain that binds through all three of its subdomains does not bind any stronger than one subdomain alone. This observation points to the essentially disordered structure of the inhibitor before binding and a large decrease in its configurational entropy with the association with the enzyme.
Discussion
A noted functional feature of IUPs is that their induced folding with target binding uncouples specificity from binding strength, making highly specific binding interactions reversible (Dunker et al. 2002; Dyson and Wright 2002; Tompa 2002). Evidently, this effect requires the structure of an IUP to be balanced between order and disorder before binding, as too much order would make the interaction too strong for the effective disruption of the complex. Too little order, on the other hand, would reduce the speed of interaction, due to having to select the right form out of a great number of conformers. Because both reversibility and speed of the interaction are essential for the function of a regulatory protein, such as calpastatin, we have sought signs of this balance with this inhibitor. Our data show that we have a good case for such a behavior.
The CD spectra presented show that subdomains A and C of calpastatin are unstructured in water, but tend to assume an α-helical conformation in the presence of increasing amounts of TFE or with binding to calpain. Ten to 30% of TFE is not unusual to induce a transition to α-helical conformation in the case of proteins, but it is rather low for peptides. Because peptides A and C comprise only 19 amino acid residues, they are too short to adopt longer stretches of α-helix, and even less for the necessarily structureless state of chain termini. In our experience, the mid-size (~30-residue) peptides usually undergo transition to the α-helical conformation at TFE concentrations in the range of 40%–50%, but not at less than that (Perczel and Hollósi 1996). Thus, as the transition to the α-helix in the case of peptides A and C occurs at rather low TFE concentrations, it is conceivable that these regions already have a tendency toward this conformation in the unbound state of the inhibitor. This is in accord with earlier observations that calpastatin is not fully disordered (Hackel et al. 2000). For subdomain B, a similar tendency for ordering has been observed; this region may assume a β-turn with binding to the active site of the enzyme (Ishima et al. 1991), exactly where β-turn conformational preference is maximal within this subdomain. These data indicate that the functional subdomains of calpastatin have a preference for the same ordered structure they assume with binding to the enzyme, and such conformers may already be present at a low level in the free inhibitor. This conclusion is underscored by the second observation (made while our manuscript was under review), that the caroxy-terminal half of subdomain C does attain an α-helical conformation when bound to the small subunit of calpain (Todd et al. 2003). It is tempting to conclude that calpastatin, in accord with the above principles, is not fully unstructured, but is structurally primed for efficient interaction with calpain. Such a behavior has already been suggested for several other IUPs, such as FlgM (Daughdrill et al. 1998) and p27Kip1 (Bienkiewicz et al. 2002), or the transactivator domain of CREB (Hua et al. 1998) and p53 (Lee et al. 2000), and may turn out to be a common phenomenon among IUPs which function in molecular recognition, as considered in Dunker et al. 2002; Dyson and Wright 2002; and Uversky 2002a. The tendency of the α-helical propensity to be evolutionarily conserved within subdomains A and C underlines that this is a functional feature of the protein.
It also occurred to us that these conclusions could be corroborated by correlating the potency of individual inhibitory domains of calpastatin (D1–D4) with the average helical propensity values of the binding segments of their subdomains A and C. This could be done for two cases, where a semiquantitative comparison of the individual inhibitory domains has been reported. In these, all four inhibitory domains possess similar, but quantitatively different, potency against calpain. With pig calpastatin, the potency (with helix-forming propensity averaged for the binding segments of subdomains A and C in parentheses) decreases in the order D1 (1.080) > D3 (1.112) > D2 (1.152) > D4 (1.102; Maki et al. 1987); with rabbit calpastatin, the order differs significantly: D2 (1.141) > D4 (1.064) > D1 (1.068) > D3 (1.132; Emori et al. 1988). As seen, the helical propensity shows hardly any correlation with inhibitory potency, which potentially undermines our point that α-helicity is important for the binding of the inhibitor. Two considerations pertain here, however. First, the helix-forming propensity within these binding segments is very conserved with low SD values (cf. Fig. 5 ▶): For the 33 domain sequences, the lowest value is 1.046 (bovine D1) and the highest is 1.169 (sheep D2). Thus, there are no naturally occurring outlying examples to clearly demonstrate correlation. We do know, however, that α-helicity does matter in terms of inhibitory activity, as drastic helix-breaking mutations within subdomains A and C do abolish inhibition by calpastatin (Ma et al. 1994). Another intriguing explanation for the apparent lack of correlation between inhibitory activity and helicity within the naturally occurring range recalls a similar observation with another intrinsically unstructured inhibitor, p27Kip1: increasing or reducing the stability of its preformed α-helix with point mutations did not affect the strength of the inhibitory complex but stabilization of the helix actually hindered kinetically the interaction of the inhibitor with its partners (Bienkiewicz et al. 2002). Formation of the helix, thus, is not rate limiting to the interaction and the inhibitor derives a kinetic advantage from intrinsic structural disorder; the same may apply to calpastatin.
The observed effect of Ca2+ may also contribute to setting the fine conformational balance inferred earlier. With the free peptides, Ca2+ appears to bind to the disordered conformers and, thus, to oppose the conformational transition required for the interaction of the inhibitor with the enzyme. As previous observations have clearly established that calpastatin only binds to calpain in the presence of Ca2+ ions (Yang et al. 1994; Takano et al. 1995), in a way that sensitizes the enzyme to Ca2+ (Tompa et al. 2002), this interaction may be rationalized by the bipartite coordination of Ca2+ by the enzyme and inhibitor in their ternary complex. Subdomains A and C contain numerous acidic residues and thus binding of Ca2+, but not Mg2+ or Na+, by the free peptides is reasonable. In light of previous work, in which no Ca2+ binding by whole calpastatin has been observed (see Takano and Maki 1999), this interaction must be transient and must occur by rather disordered conformations of the peptides. The calcium binding-induced unfolding is likely due to the repeating appearance of residues (serines, aspartates, threonine) capable of Ca2+ binding. Side-chain and backbone (amide C = O) ligands jointly may have a strong conformational effect that gives rise to unfolding of the helical conformation. This is consistent with general structural data on Ca2+ binding (Pidcock and Moore 2001), in which the actual segment donating coordinating groups often assumes a loop or turn conformation wrapped around the ion. This loose coordination may only become permanent in the presence of calpain, when subdomains A and C undergo a transition to the preferred α-helical conformation and coordination of Ca2+ is partially taken over by residues of the enzyme.
The comparison of quantitative binding data reveals that binding of the subdomains is about as strong as binding of an entire inhibitory domain that binds through all three of these subdomains. This is contradictory to the principle of multidentate binding, which in many instances brings about a fundamental increase in binding strength as demonstrated by high avidity in antibody–antigen interactions or the chelate effect in the binding of ions. Thus, calpastatin is largely disordered before binding and its binding cannot be regarded as a classic interaction of two proteins: local—and possibly global—folding effects feature in the binding process. The association reaction, therefore, is accompanied by a large unfavorable decrease in conformational entropy that allows calpastatin to contact the enzyme over a large surface area without excessive binding strength.
All these observations and considerations, together with other structural data taken from the literature, imply the overall disorder and limited local order of calpastatin in its unbound state. In accord with the principles outlined in the introductory section, this special structural feature may guarantee facile, specific yet readily reversible, regulation of calpain by calpastatin. Whether this phenomenon is general within the class of IUPs, remains to be seen.
Materials and methods
Materials
Trifluoroethanol, calcium chloride (CaCl2), magnesium chloride (MgCl2), calcium perchlorate [Ca(ClO4)2], and magnesium perchlorate [Mg(ClO4)2] of analytical purity were purchased from Merck. The expression vectors of rat m-calpain encoding an intact large (80 kD) and a truncated, but fully functional, small (21 kD) subunit were kindly provided by Professor John Elce (Department of Biochemistry, Queen’s University, Kingston, Ontario, Canada). The truncated small subunit (denoted as 21K) was subcloned from vector pACpET-21K into a pET-22b (Novagene) by PCR. 21K with a carboxy-terminal His6 tag was purified as described earlier for whole calpain (Schád et al. 2002). The 19-mer peptides corresponding to subdomains A (SGKSGMDAALDDLIDTLGG) and C (SKPIGPDDAIDALSSDFTS) of human calpastatin domain I have been synthesized as described previously (Tompa et al. 2002).
CD spectroscopy
The CD measurements were performed over the wavelength range of 180–300 nm on a Jasco-810 spectropolarimeter at room temperature in nitrogen atmosphere. The samples were dissolved in TFE (in the absence or presence of Ca2+, Mg2+, or Na+ salt at appropriate concentration) or in distilled water and mixed to achieve the desired TFE/water ratio. Peptide concentration was typically 0.7 mM or 10 μM for Ca2+ titration experiments; the optical path length was set to 0.02 cm. In the spectra reported [θ]R represents the ellipticity value per mole of peptide residue (deg.cm2/dmole) averaged for 3 (0.7 mM) or 10 (10 μM) individual scans; the difference spectra were expressed in Δɛ (dm3mol−1 cm) units. To determine the ratio of conformations in various water–TFE mixtures, the spectra were smoothed by the Savitzky-Golay method and decomposed by the Jasco software, by using the method of Yang (Yang et al. 1986).
Secondary structure prediction
All calpastatin sequences known (mouse, rat, rabbit, bovine, sheep, pig, grivet, human, and Xenopus) have been downloaded from the Swiss-Prot database. Ttheir individual inhibitory domains (33 overall) have been separated and subjected to secondary structure prediction by the Chou-Fasman method (Prevelige and Fasman 1990). Inhibitory domains were then aligned by ClustalW to allow the calculation of averages of secondary structure propensity values over all homologous sites. At positions where gaps in one or more sequences were generated by the alignment, averaging was only done for the remaining sequences.
Table 1.
Decomposition of CD spectra of the peptide A and C
| Conformer population (%) | |||
| Water | TFE | Bound | |
| Peptide A | |||
| α-helix | 0.0 | 44.3 | 30.6 |
| β-sheet | 46.9 | 18.7 | 27.3 |
| turn | 9.6 | 0.0 | 18.5 |
| coil | 43.6 | 37.0 | 23.6 |
| Peptide C | |||
| α-helix | 0.0 | 24.0 | 28.4 |
| β-sheet | 46.3 | 41.0 | 30.5 |
| turn | 10.3 | 0.0 | 17.8 |
| coil | 43.4 | 35.0 | 23.3 |
Acknowledgments
This work was supported by grants T 32360, T 34255, and TS 040723 from OTKA, FKFP 0100/2000, 0075/2000 and NKFP 1/010 from the Ministry of Education and International Senior Research Fellowship GR067595 from the Wellcome Trust. We thank Prof. J.S. Elce (Queen’s University, Kingston, Canada) for kindly providing the expression vectors encoding for rat m-calpain.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
IUP, intrinsically unstructured protein
TFE, trifluoroethanol
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03138803.
References
- Betts, R., Weinsheimer, S., Blouse, G.E., and Anagli, J. 2003. Structural determinants of the calpain inhibitory activity of calpastatin peptide B27-WT. J. Biol. Chem. 278 7800–7809. [DOI] [PubMed] [Google Scholar]
- Bienkiewicz, E.A., Adkins, J.N., and Lumb, K.J. 2002. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1). Biochemistry 41 752–759. [DOI] [PubMed] [Google Scholar]
- Blanchard, H., Grochulski, P., Li, Y., Arthur, J.S., Davies, P.L., Elce, J.S., and Cygler, M. 1997. Structure of a calpain Ca(2+)-binding domain reveals a novel EF-hand and Ca(2+)-induced conformational changes. Nat. Struct. Biol. 4 532–538. [DOI] [PubMed] [Google Scholar]
- Daughdrill, G.W., Hanely, L.J., and Dahlquist, F.W. 1998. The C-terminal half of the anti-σ factor FlgM contains a dynamic equilibrium solution structure favoring helical conformations. Biochemistry 37 1076–1082. [DOI] [PubMed] [Google Scholar]
- Demchenko, A.P. 2001. Recognition between flexible protein molecules: Induced and assisted folding. J. Mol. Recognit. 14 42–61. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., Lawson, J.D., Iakoucheva, L.M., and Obradovic, Z. 2002. Intrinsic disorder and protein function. Biochemistry 41 6573–6582. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2002. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12 54–60. [DOI] [PubMed] [Google Scholar]
- Emori, Y., Kawasaki, H., Imajoh, S., Minami, Y., and Suzuki, K. 1988. All four repeating domains of the endogenous inhibitor for calcium-dependent protease independently retain inhibitory activity. Expression of the cDNA fragments in Escherichia coli. J. Biol. Chem. 263 2364–2370. [PubMed] [Google Scholar]
- Hackel, M., Konno, T., and Hinz, H. 2000. A new alternative method to quantify residual structure in ‘unfolded’ proteins. Biochim. Biophys. Acta 1479 155–165. [DOI] [PubMed] [Google Scholar]
- Hao, L.Y., Kameyama, A., Kuroki, S., Takano, J., Takano, E., Maki, M., and Kameyama, M. 2000. Calpastatin domain L is involved in the regulation of L-type Ca2+ channels in guinea pig cardiac myocytes. Biochem. Biophys. Res. Commun. 279 756–761. [DOI] [PubMed] [Google Scholar]
- Hosfield, C.M., Elce, J.S., Davies, P.L., and Jia, Z. 1999. Crystal structure of calpain reveals the structural basis for Ca(2+)-dependent protease activity and a novel mode of enzyme activation. EMBO J. 18 6880–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua, Q.X., Jia, W.H., Bullock, B.P., Habener, J.F., and Weiss, M.A. 1998. Transcriptional activator-coactivator recognition: Nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry 37 5858–5866. [DOI] [PubMed] [Google Scholar]
- Ishima, R., Tamura, A., Akasaka, K., Hamaguchi, K., Makino, K., Murachi, T., Hatanaka, M., and Maki, M. 1991. Structure of the active 27-residue fragment of human calpastatin. FEBS Lett. 294 64–66. [DOI] [PubMed] [Google Scholar]
- Konno, T., Tanaka, N., Kataoka, M., Takano, E., and Maki, M. 1997. A circular dichroism study of preferential hydration and alcohol effects on a denatured protein, pig calpastatin domain I. Biochim. Biophys. Acta 1342 73–82. [DOI] [PubMed] [Google Scholar]
- Lee, H., Mok, K.H., Muhandiram, R., Park, K.H., Suk, J.E., Kim, D.H., Chang, J., Sung, Y.C., Choi, K.Y., and Han, K.H. 2000. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 275 29426–29432. [DOI] [PubMed] [Google Scholar]
- Lin, G.D., Chattopadhyay, D., Maki, M., Wang, K.K., Carson, M., Jin, L., Yuen, P.W., Takano, E., Hatanaka, M., DeLucas, L.J., et al. 1997. Crystal structure of calcium bound domain VI of calpain at 1.9 Å resolution and its role in enzyme assembly, regulation, and inhibitor binding. Nat. Struct. Biol. 4 539–547. [DOI] [PubMed] [Google Scholar]
- Ma, H., Yang, H.Q., Takano, E., Lee, W.J., Hatanaka, M., and Maki, M. 1993. Requirement of different subdomains of calpastatin for calpain inhibition and for binding to calmodulin-like domains. J. Biochem. (Tokyo) 113 591–599. [DOI] [PubMed] [Google Scholar]
- Ma, H., Yang, H.Q., Takano, E., Hatanaka, M., and Maki, M. 1994. Amino-terminal conserved region in proteinase inhibitor domain of calpastatin potentiates its calpain inhibitory activity by interacting with calmodulin-like domain of the proteinase. J. Biol. Chem. 269 24430–24436. [PubMed] [Google Scholar]
- Maki, M., Takano, E., Mori, H., Sato, A., Murachi, T., and Hatanaka, M. 1987. All four internally repetitive domains of pig calpastatin possess inhibitory activities against calpains I and II. FEBS Lett. 223 174–180. [DOI] [PubMed] [Google Scholar]
- Maki, M., Takano, E., Osawa, T., Ooi, T., Murachi, T., and Hatanaka, M. 1988. Analysis of structure–function relationship of pig calpastatin by expression of mutated cDNAs in Escherichia coli. J. Biol. Chem. 263 10254–10261. [PubMed] [Google Scholar]
- Maki, M., Bagci, H., Hamaguchi, K., Ueda, M., Murachi, T., and Hatanaka, M. 1989. Inhibition of calpain by a synthetic oligopeptide corresponding to an exon of the human calpastatin gene. J. Biol. Chem. 264 18866–18869. [PubMed] [Google Scholar]
- Perczel, A. and Hollósi, M. 1996. Turns. In Circular dichroism and the conformational analysis of biomolecules (ed. G.D. Fasman), pp. 285–380. Plenum Press, New York and London.
- Pidcock, E. and Moore, G.R. 2001. Structural characteristics of protein binding sites for calcium and lanthanide ions. J. Biol. Inorg. Chem. 6 479–489. [DOI] [PubMed] [Google Scholar]
- Prevelige, P. and Fasman, G.D. 1990. Chou-Fasman prediction of the secondary structure of proteins. In Prediction of protein structure and the principles of protein conformation (ed. G.D. Fasman), pp. 391–416. Plenum Press, New York and London.
- Schád, É., Farkas, A., Jékely, G., Tompa, P., and Friedrich, P. 2002. A novel human small subunit of calpains. Biochem. J. 362 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl, S., Fernandez-Catalan, C., Braun, M., Huber, R., Masumoto, H., Nakagawa, K., Irie, A., Sorimachi, H., Bourenkow, G., Bartunik, H., et al. 2000. The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc. Natl. Acad. Sci. 97 588–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, E. and Maki, M. 1999. Structure of calpastatin and its inhibitory control of calpain. In Calpain: Pharmacology and toxicology of calcium-dependent protease (eds. K.K.W. Wang and P.-W. Yuen), pp. 25–49. Taylor and Francis, Philadelphia, PA.
- Takano, E., Maki, M., Mori, H., Hatanaka, M., Marti, T., Titani, K., Kannagi, R., Ooi, T., and Murachi, T. 1988. Pig heart calpastatin: Identification of repetitive domain structures and anomalous behavior in polyacrylamide gel electrophoresis. Biochemistry 27 1964–1972. [DOI] [PubMed] [Google Scholar]
- Takano, E., Ma, H., Yang, H.Q., Maki, M., and Hatanaka, M. 1995. Preference of calcium-dependent interactions between calmodulin-like domains of calpain and calpastatin subdomains. FEBS Lett. 362 93–97. [DOI] [PubMed] [Google Scholar]
- Todd, B., Moore, D., Deivanayagam, C.C., Lin, G.D., Chattopadhyay, D., Maki, M., Wang, K.K., and Narayana, S.V. 2003. A structural model for the inhibition of calpain by calpastatin: Crystal structures of the native domain VI of calpain and its complexes with calpastatin peptide and a small molecule inhibitor. J. Mol. Biol. 328 131–146. [DOI] [PubMed] [Google Scholar]
- Tompa, P. 2002. Intrinsically unstructured proteins. Trends Biochem. Sci. 27 527–533. [DOI] [PubMed] [Google Scholar]
- Tompa, P., Mucsi, Z., Orosz, G., and Friedrich, P. 2002. Calpastatin subdomains A and C are activators of calpain. J. Biol. Chem. 277 9022–9026. [DOI] [PubMed] [Google Scholar]
- Uemori, T., Shimojo, T., Asada, K., Asano, T., Kimizuka, F., Kato, I., Maki, M., Hatanaka, M., Murachi, T., Hanzawa, H., et al. 1990. Characterization of a functional domain of human calpastatin. Biochem. Biophys. Res. Commun. 166 1485–1493. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. 2002a. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 11 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2002b. What does it mean to be natively unfolded? Eur. J. Biochem. 269 2–12. [DOI] [PubMed] [Google Scholar]
- Wright, P.E. and Dyson, H.J. 1999. Intrinsically unstructured proteins: Re-assessing the protein structure–function paradigm. J. Mol. Biol. 293 321–331. [DOI] [PubMed] [Google Scholar]
- Yang, J.T., Wu, C.S., and Martinez, H.M. 1986. Calculation of protein conformation from circular dichroism. Methods Enzymol. 130 208–269. [DOI] [PubMed] [Google Scholar]
- Yang, H.Q., Ma, H., Takano, E., Hatanaka, M., and Maki, M. 1994. Analysis of calcium-dependent interaction between amino-terminal conserved region of calpastatin functional domain and calmodulin-like domain of μ-calpain large subunit. J. Biol. Chem. 269 18977–18984. [PubMed] [Google Scholar]