

Abstract
We have used a sequence prediction algorithm and a novel sampling method to design protein sequences for the WW domain, a small β-sheet motif. The procedure, referred to as SPANS, designs sequences to be compatible with an ensemble of closely related polypeptide backbones, mimicking the inherent flexibility of proteins. Two designed sequences (termed SPANS-WW1 and SPANS-WW2), using only naturally occurring l-amino acids, were selected for study and the corresponding polypeptides were prepared in Escherichia coli. Circular dichroism data suggested that both purified polypeptides adopted secondary structure features related to those of the target without the aid of disulfide bridges or bound cofactors. The structure exhibited by SPANS-WW2 melted cooperatively by raising the temperature of the solution. Further analysis of this polypeptide by proton nuclear magnetic resonance spectroscopy demonstrated that at 5°C, it folds into a structure closely resembling a natural WW domain. This achievement constitutes one of a small number of successful de novo protein designs through fully automated computational methods and highlights the feasibility of including backbone flexibility in the design strategy.
Keywords: Protein structure, protein folding, stability and mutagenesis, protein design, WW domain
A common goal of many protein design efforts is the creation of computer-automated algorithms that generate amino acid sequences compatible with a target three-dimensional structure. Dahiyat and Mayo (1997) utilized this approach for the first time to design FSD-1, a protein that adopts the zinc finger fold and a number of other examples have followed (Regan and DeGrado 1988; Zhou et al. 1994; Betz et al. 1997; Bryson et al. 1998; Harbury et al. 1998; Kortemme et al. 1998; Walsh et al. 1999; Lopez de la Paz et al. 2001. For a recent review, see Kraemer-Pecore et al. 2001). Most of the full sequence design achievements involve proteins that are entirely or largely α-helical, structures that exhibit a prevalence of short-range interactions. In contrast, β-sheet proteins depend on long-range H-bonding interactions that are a challenge for computational design (Ottesen and Imperiali 2001; Karanicolas and Brooks III 2003). Early efforts recognized the importance of stabilizing the topology, and resorted to metal ions and disulfide bridges to encode the specificity of the fold (Pessi et al. 1993; Quinn et al. 1994; Yan and Erickson 1994). Work toward the de novo design of simple β-hairpin peptides (de Alba et al. 1996; Ramirez-Alvarado et al. 1996) and the iterative development of a small β-sheet protein (Kortemme et al. 1998; Lopez de la Paz et al. 2001) later met with mixed results. We report here the automated design of a β-sheet protein devoid of ligands and cross-links through the implementation of a sequence prediction algorithm, or SPA (Raha et al. 2000), coupled with a novel sampling procedure that integrates information from an ensemble of backbone structures. This algorithm can be used to select one or more sequences that have a high preference for a specified fold.
We have chosen to design sequences for a naturally occurring structural motif, the WW domain, using a backbone structure derived from the Pin1 protein. The use of a detailed backbone structure from an existing protein has many advantages. First, the existence of the fold in nature guarantees that design success is indeed possible. Second, the sequence and thermodynamic characteristics of the wild-type sequence provide useful reference points for the extent of success of the design procedure. Finally, when the target motif is a member of a large natural family, statistical information derived from the family members can be used to discover weaknesses in the design process.
The WW domain is a small natural module that is in many cases capable of folding autonomously. It is thought to be primarily responsible for binding to proline-containing regions of partner proteins (Otte et al. 2003). The domain is comprised of only 34–40 amino acids and adopts a three-stranded antiparallel β-sheet structure (Macias et al. 1996; Koepf et al. 1999b; Deechongkit and Kelly 2002). We have used the backbone structure of the WW domain from human peptidyl-prolyl cis–trans isomerase (hPin1; Ranganathan et al. 1997) as our framework. The advantages of using the WW domain from hPin1 are its two-state folding kinetics and its ability to fold into the desired structure after single-site mutagenesis at most positions (Jäger et al. 2001). The primary structure and fold of the hPin1 WW domain are given in Table 1 and Figure 1 ▶, respectively. We report the characterization of two polypeptides resulting from the implementation of our algorithm on the WW backbone scaffold. Thermodynamic measurements and structural inspection demonstrated that at least one of these two candidate sequences adopted preferentially the target WW fold.
Table 1.
The primary structures of the WW domain from hPin1 and the designed WW domains SPANS–WW1 and SPANS–WW2
| Amino acid sequence | Protein |
| KLPPGWEKRM SRSSGRVYYF NHITNASQWE RPSG | hPin1 WW |
| N . . S . . TP . T KSGASEN . . Y . KE .NEVTNT . . TD | SPANS-WW1 (35%) |
| S . . S . . TQLT KA DDTT . . Y . KT . DVVTNT . . TD | SPANS-WW2 (32%) |
The sheet regions in the wild-type WW domain are underlined. The two residues that were mutated in the wild-type sequence (Lys8 and Trp29, see text) are identified in bold face. Numbers in parentheses indicate the percent identity with the wild-type hPin1 sequence. A dot indicates a residue identical to that in wild-type hPin1 WW domain.
Figure 1.

Ribbon diagram of the WW domain found in complex with hPin1 (PDB ID code 1PIN [Ranganathan et al. 1997], residues 6 to 39, with numbering adjusted for consistency with this work). (A) The protein contains two Trp residues; Trp6 is part of a hydrophobic cluster, and Trp29 is exposed to solvent. (B) Jäger et al. (2001) have identified two hydrophobic clusters; the first contains Leu2, Trp6, Tyr19, and Pro32, appearing on the left of the sheet in this orientation; the second contains Arg9, Tyr18, and Phe20 on the opposite side of the sheet. Asn21 belongs to a stabilizing H-bonded network.
Results and Discussion
Protein design and the backbone ensemble approach
For practical reasons, most protein design attempts have used a fixed or stationary backbone. However, proteins exhibit some level of vibrational dynamics under native conditions, and it has been firmly established that they respond to changes in sequence through adjustments of their backbone geometry (Alber et al. 1988). Freezing the coordinates of the backbone is thus overly restrictive, and the relaxation of this unrealistic constraint is expected to provide a broader range of solutions to the sequence-structure problem (Harbury et al. 1995, 1998; Wollacott and Desjarlais 2001; Larson et al. 2003). To overcome the limitations imposed in SPA by a fixed backbone, we have developed an approach that mimics the natural flexibility of proteins through incorporation of an ensemble of closely related backbone structures. This modified version of SPA is referred to as sequence prediction algorithm for numerous states (SPANS). This algorithm has a variety of appropriately weighted scoring functions that represent energetic or free energetic factors that stabilize or destabilize protein folding, including van der Waals interactions, hydrogen bonding, electrostatics, nonpolar and polar desolvation, and entropic effects. Coupled with these is a novel sampling procedure that integrates information from a large set of backbone structures and simulation results.
The backbone ensemble used in the design process was derived from the WW domain backbone coordinates contained in 1PIN. To begin the creation of each new backbone, a Monte Carlo expansion and contraction algorithm was applied by which initial perturbation by ±5° of φ and Ψ backbone angles at all positions in the structure was followed by refinement of the angles until the backbone reached a preset maximal root mean squared deviation (r.m.s.d.) value relative to the reference backbone. The refinement procedure involved a perturbation of a single, randomly chosen backbone angle by ±1°. If the r.m.s.d. decreased, the move was accepted; if the r.m.s.d. increased, the move was rejected. For the ensemble used herein, the maximal r.m.s.d. value was set at 0.30 Å; when the total r.m.s.d. reached this cutoff, the refinement procedure was stopped for that backbone model and the procedure was begun again with the wild-type backbone to obtain a different perturbed backbone model. Thirty nondegenerate backbones, all related to the initial experimentally determined backbone structure by less than 0.30 Å, were thus created. Figure 2A ▶ depicts a typical backbone ensemble derived from hPin1 and obtained by this procedure.
Figure 2.

(A) Backbone ensemble used in SPANS. All backbones have an r.m.s.d. lower than 0.3 Å compared to the wild-type backbone and are developed through Monte Carlo moves of the (ϕ,Ψ) wild-type backbone angles. (B) The sequences of lowest free energy as selected by SPANS on each of the 30 backbone structures are placed on the wild-type backbone. The thickness of the amino acids correlates directly to their probabilities of occurrence in the final sequence according to the selections made by SPANS. SPANS–WW1 was generated from this composite structure.
A sequence design simulation using SPA (Raha et al. 2000) was then performed separately on each member of the backbone ensemble. An initial genetic algorithm (GA) gave a sequence of reasonable energy for further calculation purposes. This sequence was used as follows to estimate the context-dependent fitness of every allowed amino acid and its possible rotamers at every position on the protein backbone under consideration. First, the side chain identities and rotamers at all other positions not immediately under inspection were frozen to the sequence selected in the first GA. The rotamers of all amino acid types (except for cysteine, histidine, and methionine) were then sampled exhaustively for the position of interest, and the energy of each corresponding model was evaluated as described previously (Raha et al. 2000). The Boltzmann weight of each sampled side chain and rotamer combination was added to an ongoing partition function in one of two ways. In the design procedure used for SPANS–WW1, multiple “sub-rotamer” states were stochastically sampled about each canonical rotamer (typically, 15 sub-rotamer states within 20° of the central rotamer state are sampled randomly), and the Boltzmann weights of all of these states were combined into one “super-rotamer” and included in the partition function. In an alternative method (used for SPANS–WW2), each canonical rotamer was optimized by a torsion-space steepest descent minimization; this involved moving each of the side chain dihedral angles separately in each direction by 5° and evaluating the effect on the energy. The procedure was applied to all dihedral angles, and those that led to a decrease in energy were combined at the end.
A set of partition functions {Qx,r,i} for all amino acid rotamers at all positions in the protein defined a probabilistic fitness matrix:
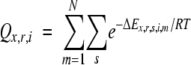 |
(1) |
where x is the amino acid type, s is a sub-rotamer state of rotamer r of amino acid x, i is the position in the structure, m is the model under evaluation, and N is the total number of models used. Ex,r,s,i,m is the total calculated energy, according to the SPA scoring function, of the specific model under evaluation given the current sub-rotamer of amino acid type x at position i. The partition function for each amino acid and rotamer combination was continually updated as more backbone structures and models under evaluation were added to the simulation. Because each model contained a unique configuration of backbone structure, side chain identities, and rotamers, each rotamer state was exposed to a wide range of environments.
To ensure self-consistency in the final probability matrix and free energy values, three cycles of design and sampling were performed over the backbone ensemble. The Qx,r,i values from each cycle, representing the cumulative probability of each rotamer state, were used to guide the next cycle of design simulations by serving as a probabilistic selection matrix for amino acids and rotamers. Figure 2B ▶ illustrates the probabilities of all allowed amino acid rotamer states as calculated using the SPANS method on the hPin1 WW backbone. The single sequences designed herein were based directly on the amino acid with the highest calculated probability at each position.
The results from the SPANS simulations are matrices detailing mean field-free energies for every residue and rotamer combination. Although the Qx,r,i values represent probabilities useful for designing individual sequences, such matrices can also readily be used to generate libraries of protein sequences consistent with the fold.
Designed WW domains
The application of SPANS to backbone ensembles derived from the hPin1 WW domain led to two amino acid sequences, SPANS–WW1 and SPANS–WW2 (Table 1). Theoretical pIs were calculated to be 6.3 (SPANS–WW1) and 4.8 (SPANS–WW2), compared to the hPin1 WW value of 10.4. As is typical for a well-parameterized protein design algorithm (Kuhlman and Baker 2000; Raha et al. 2000), the designed sequences have notable similarities to natural WW domains and similar secondary structure propensities as calculated by PsiPred (Jones 1999). The levels of identity, however, are at most 35% with the hPin1 sequence.
Homology between the designed proteins and the WW family occurred primarily in and around the first hydrophobic cluster (Fig. 1B ▶). Two of the three conserved prolines (3 and 32) were consistently selected. This was most likely due to the key role of prolines in forming a hydrophobic core without burying an unprotected backbone nitrogen atom and in fixing the backbone at those positions. In addition, the common WW pattern of three consecutive aromatics was preserved in the designed proteins (YYY versus YYF). Also conserved in the designed sequences is the Asn at position 21, which makes several hydrogen bonding interactions with surrounding backbone atoms and is important for stability of the domain (Jäger et al. 2001). Interestingly, the free energy matrices rank Asp as the next most probable residue type at this position, consistent with its prevalence in natural WW domains. The preservation of most of the highly conserved WW domain residues in the designed sequences suggests that the subtle balance among factors that stabilize or destabilize folding is reasonably well approximated by the scoring functions used in SPA. Although each designed protein maintained the structural Trp participating in the hydrophobic core (Trp6), neither sequence contained the functional Trp (Trp29). In the wild-type protein, Trp29 is significantly solvent-exposed (Fig. 1A ▶) and is involved directly in binding to the peptide substrate. Because substrate binding was not a constraint of the simulations, there was no particular driving force for the selection of a Trp at position 29; interestingly, the placement of a Trp at this location in the simulations yielded a low theoretical calculated energy within the WW matrix. The absence of Trp29 from the designed sequences has implications for the interpretation of the spectroscopic data, as explained later.
The WW domain from human Pin1
The hPin1 WW and, to a lesser extent, the WW domain from Yes-associated protein (hYap), exhibit a strong positive peak in their circular dichroism spectra (Koepf et al. 1999b; Macias et al. 2000; Deechongkit and Kelly 2002; Nguyen et al. 2003). For the hPin1 WW domain, the maximum occurs at 226 nm, as shown in Figure 3 ▶. This distinctive feature is useful as a qualitative signature of the WW fold. However, mutation of the functional Trp to Ala (W29A) in the hPin1 sequence yielded a significant decrease in the intensity of this positive band under conditions strongly favoring the folded (N) state. Thus, the main portion of the 226-nm signal in the native protein was attributed to the presence of Trp29, and variations in the spectrum were expected depending on the residue occupying position 29. The WW domain from hPin1 exhibited a cooperative thermal denaturation when studied by circular dichroism (Fig. 4 ▶). The midpoint of the transition (Tm) was approximately 55°C. The W29A replacement resulted in a decrease in Tm of ~4°C.
Figure 3.

CD spectra of the WW proteins at 2°C. Molar residual ellipticities are given in mdeg cm2/dmole. (□) Wild-type WW from hPin1; (⋄) W29A hPin1; (▵) SPANS–WW1; (○) SPANS–WW2. Buffer conditions were 10 mM MOPS at pH 7.2 (10 mM phosphate at pH 7.0 for SPANS–WW2), and 100 mM NaCl; protein concentrations were ~50 μM.
Figure 4.

Thermal denaturation profile of the WW domain from hPin1. The molar residual ellipticity at 230 nm was followed as a function of temperature. Buffer conditions were 10 mM MOPS at pH 7.2, 100 mM NaCl; protein concentration was 50 μM. CD acquisition conditions were as described in the Materials and Methods section.
SPANS–WW1
The CD spectrum of SPANS-WW1 at pH 7 contained only a weak feature at ~230 nm (Fig. 3 ▶). The ellipticity at this wavelength decreased reversibly but not cooperatively with increasing temperature (Fig. 5A ▶). Addition of 40% glycerol to SPANS–WW1 induced a slight increase in ellipticity near 230 nm, and rendered the spectrum similar to that of the native WW domain from hYap (Koepf et al. 1999b; Kraemer-Pecore 2002). This was taken as an indication for the correct structural propensity.
Figure 5.
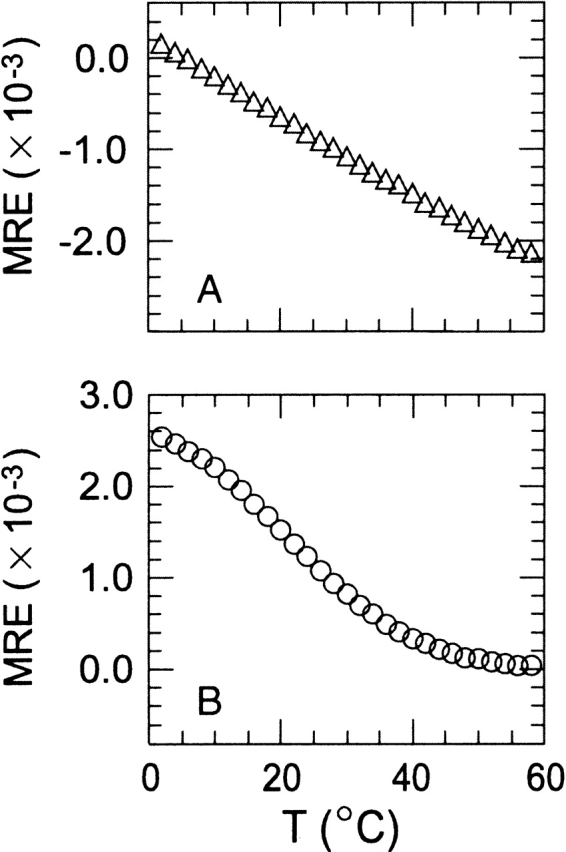
Thermal denaturation profiles of (A) SPANS–WW1 and (B) SPANS–WW2. The molar residual ellipticity at 230 nm was followed as a function of temperature. Buffer conditions were 10 mM MOPS at pH 7.2, 100 mM NaCl for SPANS–WW1 and 10 mM phosphate at pH 7.0, 100 mM NaCl for SPANS–WW2; protein concentrations were 50–60 μM. CD acquisition conditions were as described in the Materials and Methods section.
The selection of a proline at position 8 in the first β-strand of WW1 (Table 1 and Fig. 1 ▶) was initially a cause for concern because a Pro in that region is not found in natural WW domains and may introduce detrimental steric constraints on the sheet. To inspect this possibility, the mutation Lys8 → Pro (K8P) was introduced in the wild-type hPin1 protein. The CD data collected on the mutant revealed the 230-nm feature typical of wild-type hPin1 WW domain (shown in Supplemental Material). The K8P replacement led to a destabilization comparable to that of the W29A replacement mentioned earlier, causing a drop in Tm of ~2°C. These results suggested that the presence of Pro8 could be tolerated in a WW-type fold and that its choice by SPANS in the designed protein was reasonable. Further biophysical characterization of SPANS–WW1 was not attempted because of the low-intensity 230-nm signal, the shallow thermal denaturation, and the poor solubility, all of which supported that the target fold, although likely sampled, was not highly populated.
SPANS–WW2
SPANS–WW2 (Fig. 6 ▶) exhibited a maximum in ellipticity at ~230 nm, similar in intensity to the W29A hPin1 mutant signal but shifted to the red by 5 nm (Fig. 3 ▶). The thermal denaturation monitored by CD indicated a reversible and moderately sigmoidal transition with a Tm near 20°C (Fig. 5B ▶), in a marked improvement over SPANS–WW1. The curve paralleled that of the target WW domain (Fig. 4 ▶) and further structural characterization was therefore attempted by NMR spectroscopy.
Figure 6.

SPANS–WW2 designed amino acids and rotamers on the wild-type WW backbone. (A) A ribbon diagram with explicit aromatic residues. (B) The Cα trace in the same orientation. In addition to the aromatic residues, four side chains discussed in the text are included.
At 35°C, the 1D 1H spectrum of SPANS–WW2 exhibited little chemical shift dispersion (Fig. 7a ▶). This was expected of the ensemble of exchanging conformations present above Tm. Lowering the temperature led to the gradual appearance of resolved signals. The constant position of these resonances indicated slow exchange on the chemical shift time scale between a folded polypeptide and the disordered ensemble observed at higher temperatures. To characterize the three-dimensional features of the folded state, homonuclear 2D data (NOESY, DQF-COSY, and TOCSY) were collected in H2O and D2O at 5°C, a temperature at which the proportion of folded polypeptide exceeded that of the other species in solution (>70% by integration). These spectra allowed for the assignment of several spin systems.
Figure 7.
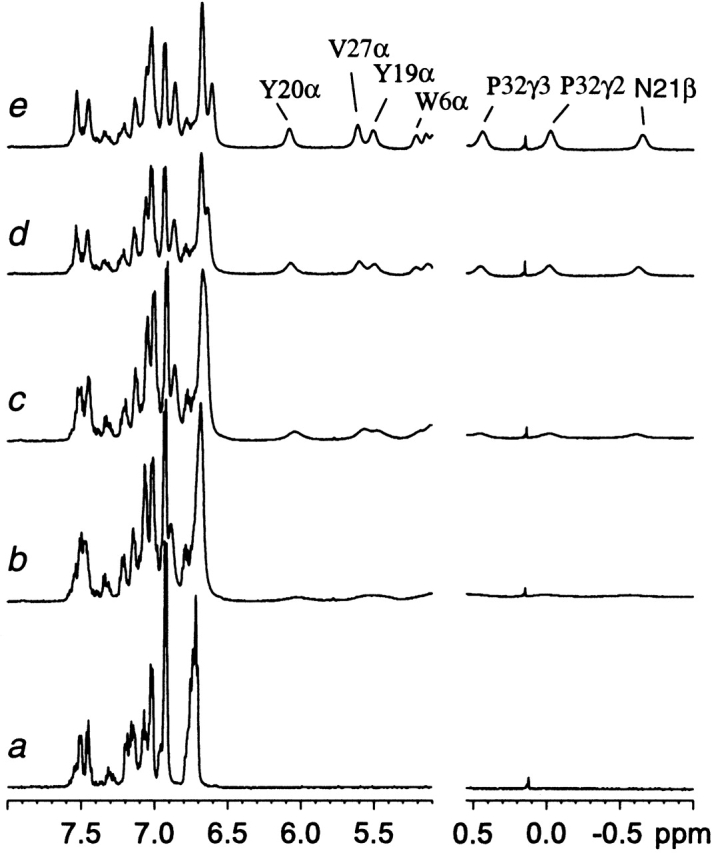
1H NMR data collected on SPANS–WW2 (900 μM) in D2O at pH 7 at 35°C (a), 20°C (b), 15°C (c), 10°C (d), and 5°C (e). Signals from the folded state are not detectable above 25°C. The intensities are scaled arbitrarily.
The aromatic resonances of Trp6, Tyr18, Tyr19, and Tyr20 in the folded conformer provided convenient starting points to analyze the spectra. The NH-CαH-CβH2 moieties of these residues were identified through NOEs to the ring CδH’s. In a hint of a hydrophobic cluster stabilized by long-range interactions, dipolar contacts were observed between one of the tyrosine rings and Trp6. Sequential effects in the 18–20 stretch identified this residue as the central tyrosine of the YYY triad. From these ring assignments and the model of SPANS–WW2 (Fig. 6 ▶), other side chains could be found. Of particular structural interest are the aliphatic residues experiencing a large upfield ring current shift (Fig. 7e ▶). For example, the two shifted protons at 0.4 ppm and 0 ppm had NOEs to Trp6 and Tyr19 and could be attributed to a proline ring using correlation data. The NOEs pointed to Pro32 (Fig. 6 ▶). The third resolved signal (−0.7 ppm) was traced to a methylene proton in a CαH-CβH2 spin system. NOEs between this side chain and Trp6, but not Tyr19 or the shifted proline, were consistent with assignment to Asn21. This was confirmed through sequential effects to Tyr20. One methyl group of a valine residue was found in contact with the shifted β proton of Asn21; this valine was the first of a sequential VV pair and therefore, identified as Val26. Additional assignments followed by a nearest neighbor approach (a list is provided in the Electronic Supplemental Material). Two networks of contacts were identified: centered around Trp6, including Tyr19, Asn21, Val26, Thr28, Pro32, and centered around Tyr18, including Leu9, Lys11, Val27, and Tyr20. It is important to note that the NOEs and chemical shifts were consistent with a specific and long-lived arrangement of the side chains, indicating the preponderance of certain rotameric states. For several resolved core residues, and as far as could be determined at this level of analysis, the preferred state appeared to agree with that targeted by the algorithm. The formation of distinct hydrophobic clusters in SPANS–WW2 is in contrast with the molten-globular species often obtained for designed proteins when side chain packing is not optimal.
The secondary structure of folded SPANS–WW2 could also be characterized with the NMR data. More than a third of the Cα protons were found downfield from 4.7 ppm, reflecting the presence of β secondary structure. Accordingly, CαHi-NHi+1 NOEs were readily followed in three extended strands, encompassing residues 6 to 12, 18 to 22, and 26 to 29. A portion of the NOESY fingerprint region is shown in Figure 8 ▶ to illustrate the connectivities involving several of these protons. Furthermore, CαH-CαH NOEs, such as those shown in Figure 9 ▶, determined the register of the three strands and confirmed the formation of the WW sheet. The strongest observed dipolar connectivities are schematized in Figure 10 ▶. NHi-NHi+1 NOEs were detected from residue 23 to residue 26, in the turn between the second and third strands. The conformational bias for the intended structure is further supported by a remarkable correspondence of CαH chemical shifts for similar residues in SPANS–WW2 and the apo hPin1 WW domain (Kowalski et al. 2002). Examples include Trp6 (5.21 ppm in SPANS–WW2 and 5.27 ppm in apo hPin1 WW domain); Tyr19 (5.51 ppm and 5.30 ppm); and Tyr/Phe20 (6.08 ppm and 5.69 ppm). For each of these CαH protons, the random coil value is ~4.42 ppm. Figure 11 ▶ summarizes the differences between observed and random coil CαH shifts in both peptides.
Figure 8.

Portion of a SPANS–WW2 NOESY spectrum. Protein concentration was 900 μM in 90% H2O/10% D2O at pH 7 and 5°C. Some of the cross-peaks indicating the formation of a β sheet are labeled i/i-1, where i represents NHi and i-1 represents CαHi-1.
Figure 9.
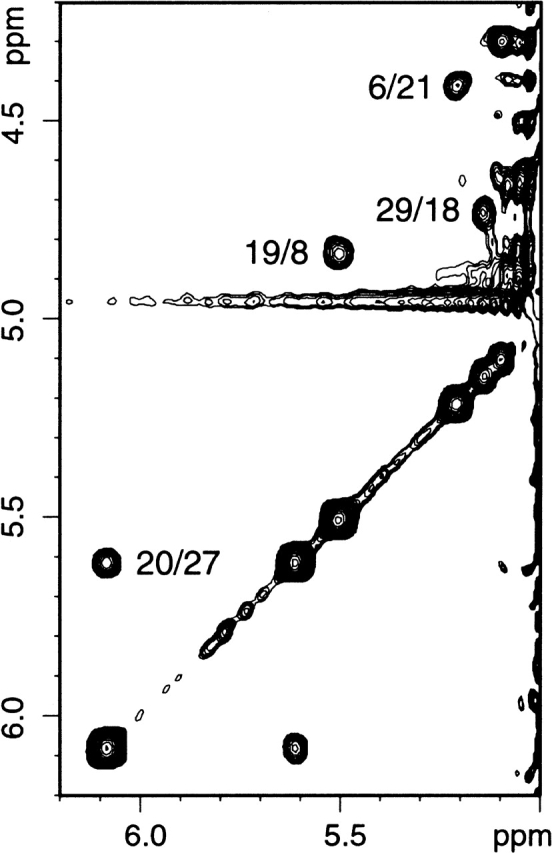
Portion of a SPANS–WW2 NOESY spectrum. Protein concentration was 700 μM in D2O at pH 7 and 5°C. The CαH-CαH NOEs determine the register of the three-stranded β sheet. Cross-peaks are labeled i/j; the chemical shift of i is read in the direct dimension.
Figure 10.

The β structure of the hPin1 WW domain. The NOEs observed in SPANS–WW2 are marked with arrows. These contacts indicate population of a WW-like topology.
Figure 11.

Plot of (δobs–δrc) versus residue number for assigned CαH resonances in SPANS–WW2 (this work, gray bars) and apo hPin1 WW domain (Kowalski et al. 2002; BMRB Accession 5248, open bars). The random coil shifts were taken from the BMRB site (http://www.bmrb.wisc.edu/ref_info/csishift.txt). Numerical values are provided in the Electronic Supplemental Material.
The NOEs between Leu2 and Pro32 as well as Thr33 demonstrated that the termini of the chain were in close proximity as in WW domains (Fig. 1 ▶). Nevertheless, some extent of reorganization was expected in this region. Four of the first 10 residues in the wild-type primary structure are ionizable, whereas none are in the same region of the SPANS polypeptides. This is likely to affect the details of the structure and the balance of conformations through altered electrostatic interactions. It is also possible that the replacement Pro4 → Ser weakens the hydrogen bond between the backbone carbonyl oxygen of residue 4 and the side chain of Asn21. Although Pro32 was found in the proximity of Trp6, the ring current shifts did not match the predicted values for all protons. These and other peculiarities of the sequence may be responsible for differences in the terminal region. The calculation of a high-resolution structure with proton data was not feasible, in part because of the persistence of slow conformational exchange, and in part because of overlap from residues in the loops; however, the sequential and interresidue NOEs and the shifts demonstrated that the majority of the polypeptide molecules adopted on average a secondary and tertiary structure similar to those of the WW target.
In evaluating the SPANS–WW2 results, it is worth pointing out that not all naturally occurring WW domains are stable. A recent survey reports that at 12°C, only 30 of 42 naturally occurring WW domains were fully folded in the absence of ligand (Otte et al. 2003). Slow conformational exchange on the chemical shift time scale was observed for the Trp side chains of some unstable WW domain peptides (Ferguson et al. 2001; Otte et al. 2003), as was detected here for Trp6 in SPANS–WW2 in TOCSY and NOESY data. Thus, the algorithm identified a sequence coding for both realistic thermodynamic stability and activation energy barrier to conformational changes disrupting the folded state. Some natural WW domains are known for the complexity of their folding transition (Koepf et al. 1999b; Crane et al. 2000; Karanicolas and Brooks III 2003; Kuznetsov et al. 2003). The sigmoidal unfolding profile observed for SPANS– WW2 (Fig. 5B ▶) is suggestive of a cooperative process. However, additional characterization is necessary for a more complete interpretation of this behavior and to determine the statedness of the transition.
SPANS–WW2 and SPANS–WW1 versus the hPin1 WW domain
Both designed proteins exhibited decreased stability compared to the WW domain from hPin1. Interestingly, they contained none of the severely destabilizing wild-type mutations identified by Gruebele and co-workers (Jäger et al. 2001). Two types of interactions appear crucial in stabilizing the native state of the hPin1 WW domain—hydrophobic, in two segregated clusters, and H-bonding, in an extended network. Remarkably, almost all of the residues involved in the two hydrophobic clusters (Leu2, Trp6, Tyr19, Pro32 in cluster 1, and Tyr18, Phe20, and the n-propyl group of Arg9 in cluster 2; Fig. 1 ▶) were present in the designed sequences. SPANS–WW1 had one mutation (F20Y) to this hydrophobic cluster; SPANS–WW2 had this mutation and another (R9L). The F20Y mutation is conservative and may exert little effect on the stability of the fold. The R9L mutation in SPANS–WW2 is less conservative; although the hydrophobicity of this side chain is similar to that of Arg. The van der Waals interactions between residue 9 and the rest of cluster 2 may be weakened because of the Leu9 rotameric state selected in the SPANS–WW2 matrix. Both of the designed proteins exhibited similar basic hydrogen-bonded interactions detailed to be important for structural stability in the wild-type polypeptide; the largest deviation involved the loss of a postulated interaction between Glu7 and Arg9 (Jäger et al. 2001). The designed proteins also had a mutation of the functional, solvent-exposed Trp (W29N in both); a W29A mutation in the wild type was shown to be moderately destabilizing (3 kJ mole−1 < ΔΔG° < 6 kJ mole−1), possibly because of the loss of a cation–aromatic interaction with Arg16 or an aromatic–aromatic interaction with Tyr18 (Jäger et al. 2001). Asn in this position may have the same effect, as it cannot participate in these interactions.
It remains unclear why SPANS–WW2 populated a typical WW-like fold, whereas SPANS–WW1 did not to any great extent. The side chain hydrogen bonding networks predicted for WW2 were more dispersed throughout the protein structure than those for WW1; perhaps these additional hydrogen bonds available to WW2 served to select for the target structure and maintain it at higher temperatures. This may indicate that the treatment of rotamers used in the design procedure for WW2 was more effective than that used for WW1.
Relationship to other β protein design
It is instructive to compare our WW domain results with other successful designs and the properties of “mini-proteins.” Andersen and co-workers prepared a 20-residue polypeptide capable of folding into a stable “Trp-cage” motif (Neidigh et al. 2002). Unlike the WW domain, this small motif contains an α structure, but it has in common with it essential hydrophobic interactions between Trp and Pro residues. This type of interaction may play an important role in stabilizing the folded state of several small proteins (Gellman and Woolfson 2002; Neidigh et al. 2002).
Production of small β proteins has been demonstrated in several laboratories (de Alba et al. 1996; Ramirez-Alvarado et al. 1996; Schenk and Gellman 1998; Lopez de la Paz et al. 2001; Ottesen and Imperiali 2001). Key to the stabilization of the fold is the engineering of proper reverse turns; the use of d-amino acids has been particularly helpful to this end (Schenk and Gellman 1998; Ottesen and Imperiali 2001), as has the placement of disulfide bridges at or near turns to increase the propensity of the desired fold (Ottesen and Imperiali 2001). In addition, this body of work indicates the importance of cross-strand interactions and judicious exposure of hydrophobic side chains to solvent to define a single conformation by limiting the ability of the strands to alter register.
The procedure applied here demonstrates a simple means to include backbone flexibility in protein design. With this method, residues found to be key to the fold are still retrieved but excessive steric constraints are relieved and the peptide can adjust to satisfy nonbonding interactions. The cumulative backbone displacements are likely to distort the structure, at least locally, compared to the target, but the desired fold is attained. Although we have not demonstrated that the use of backbone flexibility is essential for successful full sequence design, it appears to be an appropriate method for mitigating the errors inherent in the more frequently used fixed backbone assumption. It is also arguably more realistic. In addition, the sampling methods we describe provide a new avenue for treatment of more sophisticated design problems. Examples include the design of proteins with specifically tuned dynamic properties, and the design of sequences that are compatible with a number of discrete conformational states.
Full sequence design remains a considerable challenge for the protein design community. The primary difficulty is the identification of one or more suitable sequences out of the manifold possible sequences for a given protein length. For the design problems treated herein, there were 2034 possible sequences from which to choose. A suitable sequence must preferentially adopt the desired fold versus myriad alternative folded, unfolded, and aggregated states. The data strongly suggest that our SPANS algorithm and backbone sampling methods have generated at least one novel WW domain, constituting a successful search through sequence space. This novel sequence folds autonomously into a state that is similar to structures adopted by wild-type WW domains. Despite this success, several issues remain for our design algorithm. First, only one of the two designed sequences described herein convincingly adopts the target WW domain structure. Second, although this sequence is more stable than many naturally occurring WW domains, it is considerably less stable than the wild-type WW domain from which the target structure was derived. This result indicates that deficiencies remain in the scoring functions or sampling methods, or both, used to generate the sequences. It is hoped that future developments focusing on the quality of scoring functions and a systematic analysis of the backbone degrees of freedom will lead to further improvements in predictive capacity.
Materials and methods
Synthesis of the designed WW domains
Unless otherwise mentioned, DNA oligomers were purchased from Invitrogen Life Technologies (Carlsbad, CA) at a 50-nmole scale with standard desalting purification; enzymes and enzymatic buffers were purchased from New England BioLabs (Beverly, MA); competent cells were purchased as a glycerol stock from Novagen, except for DH5α competent cells, which were originally purchased from Invitrogen Life Technologies as a glycerol stock; and chemicals were purchased from J.T. Baker (Philipsburg, NJ) and used without further purification.
A novel fusion system was developed for the expression and purification of the WW peptides (WW from hPin1 as well as SPANS–WW1 and -WW2), which used the high expression of the amino terminus of a D78Y mutant of calmodulin from Homo sapiens (N-Cam.Y) in Escherichia coli and the ease of purification of N-Cam systems through Ca2+-dependent binding to phenyl Sepharose. A linker was placed in a position amino-terminal to the WW gene product for proteolytic purposes; its sequence was ENLYFQ/GS, where / indicates the site of protease cleavage by the mutant (His)6-tagged NIa-Pro tobacco etch virus protease. The NIa cleavage system was chosen because of its cleavage to ~95% completion in most non-SDS buffers in 48 h at room temperature. This fusion system led to the ready expression and purification of all of the WW designs discussed herein.
Synthetic oligonucleotides were used to construct inserts that coded for the proteins of interest. The assembled genes were then cloned into a pAED4-derived fusion system 3′ to the gene coding for N-Cam.Y and the NIa linker region. Proteins were expressed in BL21(DE3)pLysS E. coli cells or HMS174(DE3) pLysS E. coli cells to the same approximate level. All were localized in the soluble portion of the cell. Expressed fusion proteins were purified using calcium-dependent affinity of N-Cam.Y for phenyl Sepharose resin; the supernatant after cell lysis was applied at low pressure using a BioRad LP system over a phenyl Sepharose column in the presence of 5 mM Ca2+ and 500 mM Na+ at pH 7.5. Once the background cellular proteins had been washed from the column, the fusion protein was eluted with a 5 mM EDTA and 500 mM Na+ solution at pH 7.5. The NIa protease was then added in a 1:100 or 2:100 protease-to-protein solution volume ratio in the EDTA buffer; this reaction was allowed to proceed for 2 to 3 days at room temperature. Cleavage of the proteins with the NIa protease was estimated to be ~90%–95% complete as determined by SDS-PAGE.
The cleaved WW domains were isolated using reversed phase HPLC or further phenyl Sepharose purification (where in this instance the WW design was contained in the eluent from the first wash). In the case of the former purification scheme, the column was washed with 100% HPLC-grade water with 0.1% TFA. A gradient of 90% acetonitrile (Burdick and Jackson)/10% HPLC-grade water with 0.1% TFA was initialized. The fractions of interest were lyophilized and the solid material resuspended in distilled water. The identity and integrity of the designed proteins were qualitatively analyzed by SDS-PAGE and confirmed by electrospray mass spectrometry. Concentrations of the proteins were calculated using the absorbance at 280 nm in nondenaturing conditions and the absorbance coefficient as calculated from the primary structure.
Mutations were made as follows. Primers were designed with high complementarity to the surrounding regions of the gene but alterations (to reflect the desired mutation) in the region of the codon to be mutated. The primers were designed to maximize Tm (by increasing the GC content) and minimize the amount of noncomplementarity of the primer with the template. Reaction mixtures were prepared of 35.7 μL of distilled water, 5 μL of 10× ThermoPol buffer, 5 μL of 10 ng/μL plasmid template, 1.4 μL of each primer at 10 μM, and 0.5 μL of Vent DNA polymerase, 2 U/μL. The PCR program consisted of 2 min at 95°C, and 16 cycles of 30 sec at 95°C followed by 55°C for 8.5 min. The reaction mixtures were cooled to 4°C. This mixture was digested with 1 μL of 20 U/μL DpnI and incubated at 37°C for 1–2 h. Two microliters of this reaction mixture was transformed into DH5α and plated. Several of the colonies were sequenced for the presence or absence of the mutation in question.
Spectroscopic characterization
The purified protein was characterized by CD on an Aviv CD model 62 DS (Lakewood, NJ). A positive peak near 230 nm is typical of the WW-type fold (Koepf et al. 1999a; Deechongkit and Kelly 2002). Circular dichroism spectra were, therefore, used to probe for the presence of the WW fold and the ~230 nm peak was followed to assess its thermal stability. The buffer conditions were 10 mM phosphate at pH 7.0 (or 10 mM MOPS at pH 7.2), and 100 mM NaCl. The protein concentrations were between 50 and 60 μM. The temperature of acquisition was 2°C to populate the native state. For the thermal denaturation experiments, the temperature was raised from 2°C to 98°C in 1.5°–2°C steps. Equilibration time at each temperature was 0.1–1.5-min and averaging time was 7–90 sec depending on the protein. Reversibility was tested by recording data from 98°C to 2°C in the same stepwise fashion. The thermal denaturation profiles were fitted to a Gibbs-Helmholtz free-energy equation using a nonlinear least-squares routine (Nfit, University of Texas, Galveston, TX), as given in equation 2:
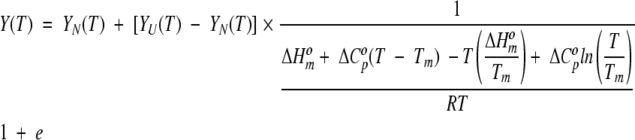 |
(2) |
In this equation, Y(T) represents the observed signal at any temperature; YN(T) represents the signal of the native state; YU(T) represents the signal of the unfolded state; and the thermodynamic quantities are for the N → U reaction. The subscript m indicates quantities at the midpoint of the transition. The absence of a well-defined folded state baseline prevented a determination of thermodynamic quantities and only estimates of Tm, a relatively robust parameter, are reported. Because no conditions were found under which the folded state was fully populated, chemical denaturation was expected to show an equally ill-defined native baseline and was not attempted to obtain a standard free energy of unfolding.
SPANS–WW2 exhibited an intriguing pH-dependent behavior at high concentration of protein. When it was resuspended after lyophilization and the pH was raised above the pI, reduced positive ellipticity at 230 nm and greatly increased turbidity and viscosity were observed. To break the aggregates, the samples were briefly heated to 95°C and allowed to cool gradually to room temperature. This annealing process restored a clear solution and the typical 230-nm peak. The trapping of the peptide in multimeric structure is reminiscent of the metastable β network observed in amyloid peptides and in some proteins under non-native conditions (Dobson 1999).
For 1H NMR spectroscopy, solutions of SPANS–WW2 (700–900 μM) were prepared in 90% H2O/10% D2O or 100% D2O by resuspension of the lyophilized powder. Several annealing steps were necessary when raising the pH above the theoretical pI of the protein. One annealing cycle was comprised of placing the sample on a 90°C heat block for 5 min, allowing the system to cool to room temperature, and adjusting the pH again. The pH(*) was measured with a Beckman φ71 pH meter equipped with a small bore probe (Mettler Toledo, Columbus, OH) and adjusted to 6.8–7.2 with HCl (DCl) or NaOH (NaOD). Sodium azide (Sigma, ~3 mM) was added for inhibition of bacterial growth and the buffer was adjusted to 20 mM phosphate for the D2O experiments. Data were collected at 600 MHz on a Bruker DRX spectrometer (Billerica, MA) with probe temperature set between 5° and 35°C. All homonuclear 2D data were obtained at 5°C for optimal population of the folded state. NOESY (Kumar et al. 1980), DQF-COSY (Rance et al. 1983) and relaxation-compensated TOCSY (Cavanagh and Rance 1992) data were collected with TPPI quadrature detection in the indirect dimension (10 kHz spectral width, 4096* × 512 points, 64 transients). The mixing time was 120 msec for the NOESY data, and 45 msec for the TOCSY data, with a DIPSI-2 spin locking pulse train (Shaka et al. 1988). Suppression of the water signal was achieved with a 1.2-sec low-power saturation pulse or a modified WATERGATE sequence (Piotto et al. 1992; Sklenář et al. 1993). Time–domain data were subjected to multiplication with a 45°-shifted squared sine bell function before transformation. Final matrix size was 2048 × 2048 real. For the 1D variable temperature study in D2O, the temperature of the probe was decreased from 35°C to 5°C in 5°C increments. A total of 256 to 4000 transients were collected per temperature with other parameters as above. Chemical shifts were referenced indirectly to DSS through water after correction for temperature (Wishart et al. 1995).
Electronic supplemental material
The supplementary information (five pages) contains: (1) A table of secondary structure prediction for wild-type hPin1 WW domain, SPANS–WW1 and SPANS–WW2; (2) the CD spectrum and thermal denaturation of K8P hPin1; (3) the CD spectrum of SPANS–WW1 with osmolyte; (4) a 1D NMR spectrum of SPANS–WW2; (5) a list of SPANS–WW2 chemical shifts; (6) a list of CαH shifts in SPANS–WW2 and apo hPin1 WW domain. The information is within a single pdf file (cmkp-esm.pdf).
Acknowledgments
We are grateful to Bellamarie Bregar for her assistance in preparing and characterizing W29A variant of the hPin1 WW domain, to B. Christie Vu for her assistance with obtaining the NMR data for SPANS–WW2, to Dr. Song Tan from the Biochemistry and Molecular Biology Department in the Pennsylvania State University for a sample of the NIa cleavage protease, to Dr. Jennifer Doudna (Yale University) for the plasmid coding the (His)6-tagged NIa cleavage protease mutant, to the members of the Lecomte and Matthews groups for their assistance, and to Dr. Christopher Falzone for comments. Figures 1 ▶ and 6 ▶ were generated with MOLSCRIPT (Kraulis 1991). This work was supported in part by NIH grant GM-54217 (JTJL) and a National Science Foundation Career Award CHE 9876234 (JRD).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BMRB, BioMagResBank
bp, base pair
CD, circular dichroism
DQF-COSY, double-quantum-filtered correlated spectroscopy
DSS, 2,2-dimethyl-2-silapentane-5-sulfonate
EDTA, ethylenediaminetetraacetic acid
GA, genetic algorithm
MALDI, matrix-assisted laser desorption ionization
MOPS, 3–[N-morpholino] propanesulfonic acid
MRE, molar residual ellipticity
NIa, nuclear inclusion a
NOE, nuclear Overhauser effect
NOESY, two-dimensional nuclear Overhauser effect spectroscopy
PAGE, polyacrylamide gel electrophoresis
PCR, polymerase chain reaction
ppm, parts per million
r.m.s.d., root-mean-square deviation
SDS, sodium dodecylsulfate
SPA, sequence prediction algorithm
SPANS, sequence prediction algorithm for numerous states
TFA, trifluoroacetic acid
TOCSY, totally correlated two-dimensional spectroscopy
TPPI, time-proportional phase incrementation
Tris, tris(hydroxymethyl) aminomethane
Supplemental material: See www.proteinscience.org
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03190903.
References
- Alber, T., Bell, J.A., Sun, D.P., Nicholson, H., Wozniak, J.A., Cook, S., and Matthews, B.W. 1988. Replacements of Pro86 in phage T4 lysozyme extend an α-helix but do not alter protein stability. Science 239 631– 635. [DOI] [PubMed] [Google Scholar]
- Betz, S.F., Liebman, P.A., and DeGrado, W.F. 1997. De novo design of native proteins: Characterization of proteins intended to fold into antiparallel, rop-like, four-helix bundles. Biochemistry 36 2450–2458. [DOI] [PubMed] [Google Scholar]
- Bryson, J.W., Desjarlais, J.R., Handel, T.M., and DeGrado, W.F. 1998. From coiled coils to small globular proteins: Design of a native-like three-helix bundle. Protein Sci. 7 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh, J. and Rance, M. 1992. Suppression of cross-relaxation effects in TOCSY spectra via a modified DIPSI-2 mixing sequence. J. Magn. Reson. 96 670–678. [Google Scholar]
- Crane, J.C., Koepf, E.K., Kelly, J.W., and Gruebele, M. 2000. Mapping the transition state of the WW domain β-sheet. J. Mol. Biol. 298 283– 292. [DOI] [PubMed] [Google Scholar]
- Dahiyat, B.I., and Mayo, S.L. 1997. De novo protein design: fully automated sequence selection. Science 278 82–87. [DOI] [PubMed] [Google Scholar]
- de Alba, E., Jimenez, M.A., Rico, M., and Nieto, J.L. 1996. Conformational investigation of designed short linear peptides able to fold into β-hairpin structures in aqueous solution. Fold. Des. 1 133–144. [DOI] [PubMed] [Google Scholar]
- Deechongkit, S. and Kelly, J.W. 2002. The effect of backbone cyclization on the thermodynamics of β-sheet unfolding: stability optimization of the PIN WW domain. J. Am. Chem. Soc. 124 4980–4986. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24 329–332. [DOI] [PubMed] [Google Scholar]
- Ferguson, N., Pires, J.R., Toepert, F., Johnson, C.M., Pan, Y.P., Volkmer-Engert, R., Schneider-Mergener, J., Daggett, V., Oschkinat, H., and Fersht, A. 2001. Using flexible loop mimetics to extend φ-value analysis to secondary structure interactions. Proc. Natl. Acad. Sci. 98 13008– 13013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellman, S.H. and Woolfson, D.N. 2002. Mini-proteins Trp the light fantastic. Nat. Struct. Biol. 9 408–410. [DOI] [PubMed] [Google Scholar]
- Harbury, P.B., Tidor, B., and Kim, P.S. 1995. Repacking protein cores with backbone freedom: Structure prediction for coiled coils. Proc. Natl. Acad. Sci. 92 8408–8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury, P.B., Plecs, J.J., Tidor, B., Alber, T., and Kim, P.S. 1998. High-resolution protein design with backbone freedom. Science 282 1462– 1467. [DOI] [PubMed] [Google Scholar]
- Jäger, M., Nguyen, H., Crane, J.C., Kelly, J.W., and Gruebele, M. 2001. The folding mechanism of a β-sheet: the WW domain. J. Mol. Biol. 311 373–393. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Karanicolas, J. and Brooks III, C.L. 2003. The structural basis for biphasic kinetics in the folding of the WW domain from a formin-binding protein: Lessons for protein design? Proc. Natl. Acad. Sci. 100 3954–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepf, E.K., Petrassi, H.M., Ratnaswamy, G., Huff, M.E., Sudol, M., and Kelly, J.W. 1999a. Characterization of the structure and function of W → F WW domain variants: Identification of a natively unfolded protein that folds upon ligand binding. Biochemistry 38 14338–14351. [DOI] [PubMed] [Google Scholar]
- Koepf, E.K., Petrassi, H.M., Sudol, M., and Kelly, J.W. 1999b. WW: An isolated three-stranded antiparallel β-sheet domain that unfolds and refolds reversibly; evidence for a structured hydrophobic cluster in urea and GdnHCl and a disordered thermal unfolded state. Protein Sci. 8 841– 853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme, T., Ramirez-Alvarado, M., and Serrano, L. 1998. Design of a 20-amino acid, three-stranded β-sheet protein. Science 281 253–256. [DOI] [PubMed] [Google Scholar]
- Kowalski, J.A., Liu, K., and Kelly, J.W. 2002. NMR solution structure of the isolated Apo Pin1 WW domain: Comparison to the x-ray crystal structures of Pin1. Biopolymers 63 111–121. [DOI] [PubMed] [Google Scholar]
- Kraemer-Pecore, C.M. 2002. “Computational design and experimental verification of protein domains.” Ph.D. thesis. The Pennsylvania State University, University Park, PA.
- Kraemer-Pecore, C.M., Wollacott, A.M., and Desjarlais, J.R. 2001. Computational protein design. Curr. Opin. Chem. Biol. 5 690–695. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kuhlman, B. and Baker, D. 2000. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. 97 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A., Ernst, R.R., and Wüthrich, K. 1980. A 2D NOE experiment for the elucidation of complete proton–proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 95 1–6. [DOI] [PubMed] [Google Scholar]
- Kuznetsov, S.V., Hilario, J., Keiderling, T.A., and Ansari, A. 2003. Spectroscopic studies of structural changes in two β-sheet-forming peptides show an ensemble of structures that unfold noncooperatively. Biochemistry 42 4321–4332. [DOI] [PubMed] [Google Scholar]
- Larson, S.M., Garg, A., Desjarlais, J.R., and Pande, V.S. 2003. Increased detection of structural templates using alignments of designed sequences. Proteins 51 390–396. [DOI] [PubMed] [Google Scholar]
- Lopez de la Paz, M., Lacroix, E., Ramirez-Alvarado, M., and Serrano, L. 2001. Computer-aided design of β-sheet peptides. J. Mol. Biol. 312 229– 246. [DOI] [PubMed] [Google Scholar]
- Macias, M.J., Hyvonen, M., Baraldi, E., Schultz, J., Sudol, M., Saraste, M., and Oschkinat, H. 1996. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature 382 646–649. [DOI] [PubMed] [Google Scholar]
- Macias, M.J., Gervais, V., Civera, C., and Oschkinat, H. 2000. Structural analysis of WW domains and design of a WW prototype. Nat. Struct. Biol. 7 375–379. [DOI] [PubMed] [Google Scholar]
- Neidigh, J.W., Fesinmeyer, R.M., and Andersen, N.H. 2002. Designing a 20-residue protein. Nat. Struct. Biol. 9 425–430. [DOI] [PubMed] [Google Scholar]
- Nguyen, H., Jäger, M., Moretto, A., Gruebele, M., and Kelly, J.W. 2003. Tuning the free-energy landscape of a WW domain by temperature, mutation, and truncation. Proc. Natl. Acad. Sci. 100 3948–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte, L., Wiedemann, U., Schlegel, B., Pires, J.R., Beyermann, M., Schmieder, P., Krause, G., Volkmer-Engert, R., Schneider-Mergener, J., and Oschkinat, H. 2003. WW domain sequence activity relationships identified using ligand recognition propensities of 42 WW domains. Protein Sci. 12 491– 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottesen, J.J. and Imperiali, B. 2001. Design of a discretely folded mini-protein motif with predominantly β-structure. Nat. Struct. Biol. 8 535–539. [DOI] [PubMed] [Google Scholar]
- Pessi, A., Bianchi, E., Crameri, A., Venturini, S., Tramontano, A., and Sollazzo, M. 1993. A designed metal-binding protein with a novel fold. Nature 362 367–369. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenár̂, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Quinn, T.P., Tweedy, N.B., Williams, R.W., Richardson, J.S., and Richardson, D.C. 1994. βdoublet: De novo design, synthesis, and characterization of a β-sandwich protein. Proc. Natl. Acad. Sci. 91 8747–8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha, K., Wollacott, A.M., Italia, M.J., and Desjarlais, J.R. 2000. Prediction of amino acid sequence from structure. Protein Sci. 9 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Alvarado, M., Blanco, F.J., and Serrano, L. 1996. De novo design and structural analysis of a model β-hairpin peptide system. Nat. Struct. Biol. 3 604–612. [DOI] [PubMed] [Google Scholar]
- Rance, M., Sørensen, O.W., Bodenhausen, G., Wagner, G., Ernst, R.R., and Wüthrich, K. 1983. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 117 479–485. [DOI] [PubMed] [Google Scholar]
- Ranganathan, R., Lu, K.P., Hunter, T., and Noel, J.P. 1997. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89 875–886. [DOI] [PubMed] [Google Scholar]
- Regan, L. and DeGrado, W.F. 1988. Characterization of a helical protein designed from first principles. Science 241 976–978. [DOI] [PubMed] [Google Scholar]
- Schenk, H.L. and Gellman, S.H. 1998. Use of a designed triple-stranded antiparallel β-sheet to probe β-sheet cooperativity in aqueous solution. J. Am. Chem. Soc. 120 4869–4870. [Google Scholar]
- Shaka, A.J., Lee, C., and Pines, A. 1988. Iterative schemes for bilinear operators; application to spin decoupling. J. Magn. Reson. 77 274–293. [Google Scholar]
- Sklenář, V., Piotto, M., Leppik, R., and Saudek, V. 1993. Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. 102 241–245. [Google Scholar]
- Walsh, S.T., Cheng, H., Bryson, J.W., Roder, H., and DeGrado, W.F. 1999. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. 96 5486–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6 135–140. [DOI] [PubMed] [Google Scholar]
- Wollacott, A.M. and Desjarlais, J.R. 2001. Virtual interaction profiles of proteins. J. Mol. Biol. 313 317–342. [DOI] [PubMed] [Google Scholar]
- Yan, Y. and Erickson, B.W. 1994. Engineering of betabellin 14D: Disulfide-induced folding of a β-sheet protein. Protein Sci. 3 1069–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, N.E., Kay, C.M., and Hodges, R.S. 1994. The role of interhelical ionic interactions in controlling protein folding and stability. De novo designed synthetic two-stranded α-helical coiled-coils. J. Mol. Biol. 237 500–512. [DOI] [PubMed] [Google Scholar]