

Abstract
Chemical modification of Torpedo californica acetylcholinesterase by the natural thiosulfinate allicin produces an inactive enzyme through reaction with the buried cysteine Cys 231. Optical spectroscopy shows that the modified enzyme is “native-like,” and inactivation can be reversed by exposure to reduced glutathione. The allicin-modified enzyme is, however, metastable, and is converted spontaneously and irreversibly, at room temperature, with t1/2 ≃ 100 min, to a stable, partially unfolded state with the physicochemical characteristics of a molten globule. Osmolytes, including trimethylamine-N-oxide, glycerol, and sucrose, and the divalent cations, Ca2+, Mg2+, and Mn2+ can prevent this transition of the native-like state for >24 h at room temperature. Trimethylamine-N-oxide and Mg2+ can also stabilize the native enzyme, with only slight inactivation being observed over several hours at 39°C, whereas in their absence it is totally inactivated within 5 min. The stabilizing effects of the osmolytes can be explained by their differential interaction with the native and native-like states, resulting in a shift of equilibrium toward the native state. The stabilizing effects of the divalent cations can be ascribed to direct stabilization of the native state, as supported by differential scanning calorimetry.
Keywords: Acetylcholinesterase, calorimetry, chemical chaperone, conformational change, forces and stability, molten globule, protein folding
In recent years, experimental evidence has accumulated indicating that certain globular proteins may assume metastable conformations (Carr et al. 1997; Sohl et al. 1998). In some cases, proteins in such metastable states display exposed hydrophobic surfaces, which may lead to aggregation, as appears to be the case for the prion protein (Safar et al. 1994). Similar metastable states may occur on the folding pathway, both in vitro and in vivo, and, under conditions of overexpression in prokaryotic systems, may result in sequestration of the expressed protein in inclusion bodies (Speed et al. 1996). Physicochemical characterization of such metastable states is thus not only of fundamental value, but also of interest in a biotechnological context and in relation to the so-called conformational diseases (Carrell and Lomas 1997; Wanker 2000; Solomon et al. 2001).
The discovery and characterization of several classes of chaperone proteins has shown that nature copes with the problem of aggregation of folding intermediates by using special classes of proteins whose role is to protect them from aggregation. These chaperone proteins may also actively assist the folding process. The best known of them are the prokaryotic GroEL/GroES system, and its eukaryotic counterpart hsp60, both of which have been studied intensively (Thirumalai and Lorimer 2001). Obviously, low-molecular-weight counterparts of these proteins would be valuable both in preventing aggregation and in facilitating folding, especially in a biotechnological context, and might also offer an approach to the treatment of conformational diseases (Tatzelt et al. 1996). The term “chemical chaperone” has been used to describe such compounds, but, in fact, they are a chemically diverse set of compounds, and their common feature is their osmolyte activity (Yancey et al. 1982). They include sugars (e.g., trehalose), methylammonium derivatives such as trimethylamine N-oxide (TMAO), polyols (e.g., glycerol and polethyleneglycol), and amino acids and their derivatives (for reviews, see Bolen and Baskakov 2001; Davis-Searles et al. 2001). Because of their osmolyte activity, they can replace much of the solvent water adhering to proteins, and can shift the equilibrium state for an unfolded protein toward the folded state (Qu et al. 1998; Timasheff 1998).
Torpedo californica acetylcholinesterase (TcAChE) is a homodimer whose two catalytic subunits are linked by an interchain disulfide bridge close to its C terminus (MacPhee-Quigley et al. 1986; Gibney et al. 1988). It contains, in addition, three intrachain disulfide bonds, and one free cysteine, Cys 231. Chemical modification of by various sulfhydryl reagents results in its inactivation. This is accomplished by chemical modification of the single free sulfhydryl group, on Cys 231, which is buried within the hydrophobic core of the protein, ~7 Å from Oγ of the active-site serine, S200 (Sussman et al. 1991; Kreimer et al. 1994). Mutation of Cys 231 produces an enzyme insensitive to sulfhydryl reagents (Morel et al. 1999).
Inactivation is accompanied by conversion to one of two principal unfolded states. One of these states, produced by modification with either bulky and/or charged disulfides and alkylating agents (Dolginova et al. 1992; Kreimer et al. 1994), is stable under physiological conditions, and displays the characteristics of a molten globule (MG) state (Fink 1995; Arai and Kuwajima 2000). The second state, produced by mercury derivatives, is metastable, and transforms spontaneously to the MG state, with a half-life of ~1 h at room temperature and physiological pH (Kreimer et al. 1994). The physicochemical features of this state are close to those of the native state, and it can revert to the native state upon demodification with a sulfhydryl reagent. We thus named this state quasinative, N*. In contrast, demodification of the MG state does not regenerate the native state. No recovery of enzymic activity can be detected, and spectroscopic measurements show that it fully retains its MG characteristics. The relationship among the three states can be schematically represented as shown in Scheme 1 ▶ (Shin et al. 1997):
Scheme 1.

Thus TcAChE provides an attractive experimental model for studying the intraconversion of partially unfolded states and their possible stabilization. In the following, a novel variant of the N* state was obtained by chemical modification with the natural small noncharged reactive compound, allicin (diallylthiosulfinate; Cavallito and Bailey 1944). We show that this state can be stabilized and reactivated not only by some typical osmolytes, but also by certain divalent cations.
Results
Inhibition of enzymic activity by allicin
Allicin is a natural product, present at high levels in fresh crushed garlic, which acts as a potent sulfhydryl reagent because it possesses an activated disulfide bond (Rabinkov et al. 1998):
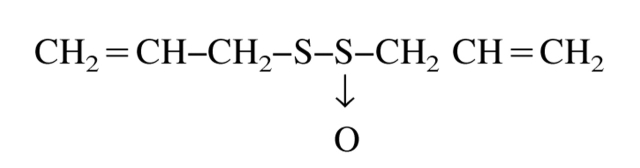
We chose to investigate its effect on TcAChE not only because of its intrinsically high reactivity, but also because it is very hydrophobic and small, and thus might be expected to react rapidly with the buried sulfhydryl, Cys 231, of TcAChE (Miron et al. 2000), and because modification can readily be reversed by suitable sulfhydryl reagents (Rabinkov et al. 1998).
Incubation of TcAChE with allicin produced rapid inactivation, which was time- and concentration-dependent (Fig. 1 ▶). Dilution 1000-fold did not result in any recovery of enzymic activity, indicating that inhibition is indeed caused by covalent modification, as is the case for other sulfhydryl reagents (Dolginova et al. 1992; Kreimer et al. 1994). Exposure to allicin of the C231S mutant (Morel et al. 1999), under similar experimental conditions, produced no detectable inactivation (data not shown). Kinetic analysis showed that allicin inhibited the enzyme with pseudo-first-order kinetics. The reaction order with respect to the allicin concentration was 0.94, consistent with inactivation by one molecule of allicin per subunit. GSH (5 mM) restored ≥80% of the original enzymic activity if added shortly after inhibition by allicin.
Figure 1.

Kinetics of inhibition of TcAChE by allicin. The enzyme (3.6 μM) was incubated with the appropriate concentration of allicin at 23°C. The decrease in enzymic activity as a function of time is shown at three allicin concentrations: (•) 0.5 mM; (○) 3 mM; and (▴) 8 mM.
The irreversible inhibition by allicin, its restoration by GSH, and the absence of any effect of allicin on the activity of the C231S mutant, all indicate that inhibition was due to modification of the single free sulfhydryl of TcAChE by a thiol-disulfide exchange reaction (Scheme 2 ▶ below).
Scheme 2.

As shown in Figure 2 ▶, the extent of recovery of enzymic activity upon treatment of the allicin-modified enzyme with GSH decreases as a function of time, with a t1/2 of 100 min under the experimental conditions used. Thus, the allicin-modified enzyme is transformed spontaneously to a nonreactivatable form, displaying behavior similar to that of TcAChE modified by mercurials (Kreimer et al. 1994).
Figure 2.

Time-dependent loss by allicin-modified TcAChE of capacity for reactivation by GSH. TcAChE (1 μM) was inhibited >80% by incubation with 1 mM allicin at room temperature for 25 min. The sample was maintained at room temperature for >24 h; at appropriate intervals, 10-μL aliquots were withdrawn, mixed with 5 μL of freshly reduced GSH, and incubated at 23°C for 30 min, prior to assay using 2 mM PNPA in Sorenson buffer.
Inactivation by allicin was not accompanied by aggregation of TcAChE, as demonstrated by sucrose gradient centrifugation (data not shown).
Spectroscopic properties of allicin-modified TcAChE
CD, intrinsic fluorescence, and ANS binding were used for spectroscopic characterization of the allicin-modified enzyme. Figure 3 ▶ compares the CD spectra, in the near and far UV, of native TcAChE, freshly prepared allicin-modified TcAChE, and MG-TcAChE produced by thermal denaturation as described previously (Kreimer et al. 1995, 1996). The CD spectrum of the allicin-modified enzyme, in both the near and far UV, is very close to that of the unmodified enzyme. Thus, changes in tertiary and secondary structure are very small, compared with the substantial changes produced upon conversion to the MG state.
Figure 3.

Effect of modification by allicin on the CD spectra of TcAChE in the near and far UV. (1) Native TcAChE; (2) allicin-modified TcAChE after 22°C for 1 h, subsequent to modification as described in Materials and Methods; (3) as in 2, but 24 h after modification; (4) MG produced by heating of TcAChE at 39°C for 1 h.
Figure 4 ▶ displays the normalized emission spectra for native and allicin-modified TcAChE, and for TcAChE modified by 5,5′-dithiodipyridine (DTP), which displays the features of an MG state (Dolginova et al. 1992). Modification by allicin results in a 1–2-nm red shift in the emission maximum, much smaller than the ~8-nm red shift produced by modification with DTP. Modification with allicin produced, however, a pronounced enhancement in ANS fluorescence, to values 6–10-fold higher than for native TcAChE (Fig. 5 ▶), similar to those seen for the DTP-modified enzyme (Dolginova et al. 1992), indicating a pronounced increase in exposed hydrophobic surface. This increase in ANS fluorescence occurred with a t1/2 of ~4 min. This half-life is very similar to the t1/2 values for inactivation under similar experimental conditions (Fig. 1 ▶). Thus, the rate of transition to the conformational state, which binds ANS, is very similar to the rate of chemical modification.
Figure 4.

Effects of modification by allicin or by DTP on the intrinsic fluorescence spectrum of TcAChE. (Solid line) Native TcAChE; (dashed line) allicin-modified TcAChE; (dotted/dashed line) DTP-modified TcAChE.
Figure 5.

Allicin-induced increase in ANS fluorescence of TcAChE. The enzyme (2.5 μM), was incubated with 3 mM allicin in the presence of 0.1 mM ANS. The points show the averages of triplicate values. In some cases, the error bars are smaller than the symbols. The t1/2 for transition from the state that binds ANS poorly, the N state, to the state that binds ANS maximally, the quasinative state, N*, is 3.9 min.
The spectral characteristics of the freshly prepared allicin-modified enzyme, taken together with the fact that catalytic activity could be recovered upon demodification with GSH, show that it closely resembles the quasinative N* state of TcAChE produced upon modification with mercurials (Kreimer et al. 1994; Shin et al. 1997). Figure 6 ▶, A and B, shows that concomitantly with the recovery of catalytic activity upon demodification, ANS fluorescence decreases substantially, providing evidence that the exposed hydrophobic surfaces have again become, at least partially, sequestered.
Figure 6.
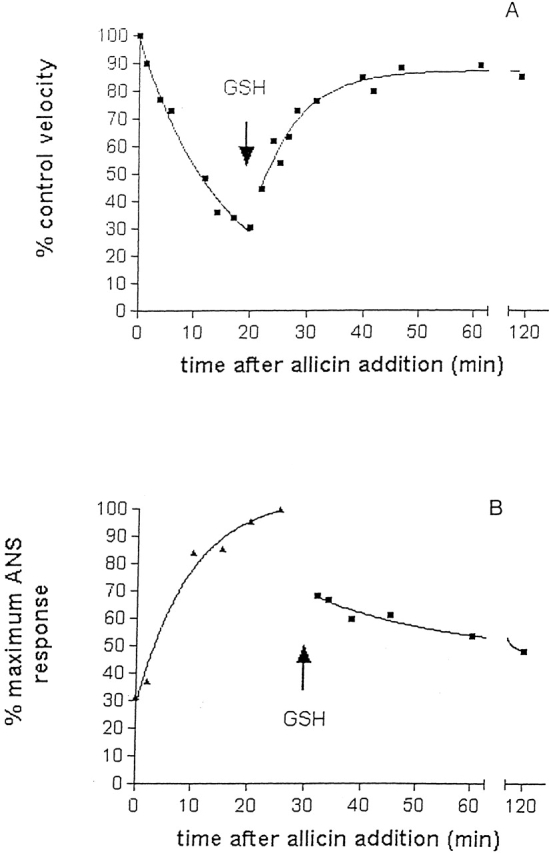
Reversal by GSH of inactivation of TcAChE, and concomitant capacity to bind ANS, produced by modification with allicin. Native enzyme (2.5 μM) was incubated at 23°C for 20 min with 1 mM allicin, in the absence (A) and presence (B) of 0.1 mM ANS. Aliquots from A were taken for enzymic assay, and from B for estimation of ANS binding by fluorescence measurements. After 20 min (A) and 30 min (B), 11 mM GSH (final concentration) was added, and recovery of activity and decrease in ANS binding in A and B, respectively, were monitored as prior to addition of GSH.
Trapping of short-lived aggregates by cross-linking with hypericin
We earlier showed that the photosensitive dye hypericin (Miskovsky 2002) could cross-link partially unfolded species of TcAChE, but not the native enzyme (Weiner et al. 1999). This approach could further be used to trap transiently formed oligomers or aggregates not readily detectable by routinely used techniques such as SDS-PAGE or sucrose gradient centrifugation (Shin et al. 2002). Figure 7 ▶ shows that TcAChE freshly modified with allicin produces oligomeric species when irradiated in the presence of hypericin. The pattern produced upon SDS-PAGE under reducing conditions is not very different from that obtained upon similar treatment of an MG species of the enzyme produced by treatment with 1.2 M Gdn.HCl for 2 h. This provides additional evidence that modification by allicin generated exposed hydrophobic surfaces.
Figure 7.
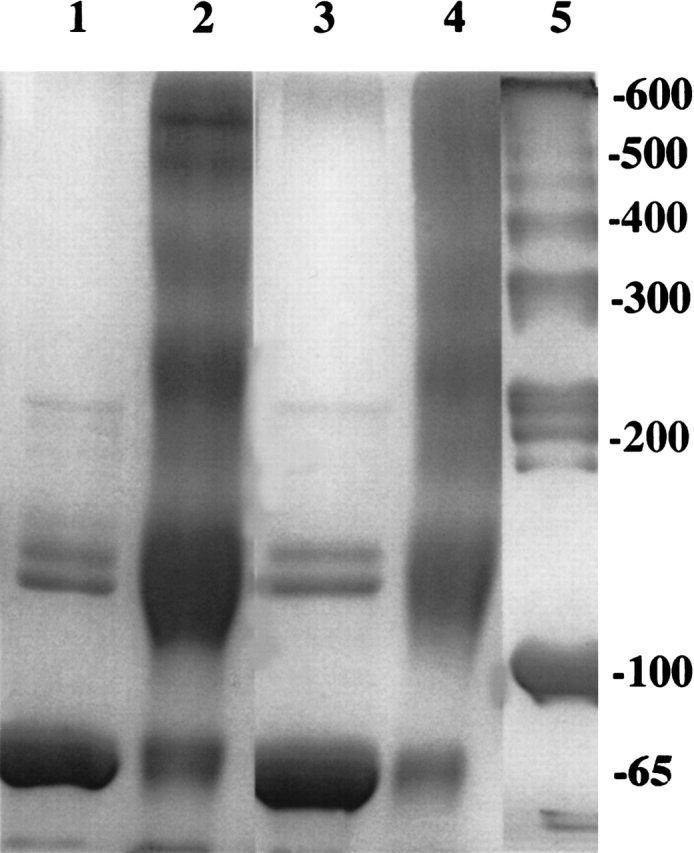
Cross-linking by hypericin of N* TcAChE, produced by modification with allicin, and of MG TcAChE, produced by treatment with 1.2 M Gdn.HCl. Allicin modification was carried out with 1 mM allicin at 23°C for 30 min. Treatment with 1.2 M Gdn.HCl was in Sorenson buffer at 22°C for 2 h, followed by removal of the Gdn.HCl by washing three times in a YM-30 Centricon centrifugal filter. Irradiation in the presence of 1 mM hypericin for 1 h was performed as described in Materials and Methods. SDS-PAGE was as described previously (Weiner et al. 1999). (Lane 1) Allicin-modified (N*); (lane 2) allicin-modified enzyme after irradiation for 1 h in the presence of hypericin; (lane 3) Gdn.HCl-treated (MG) TcAChE; (lane 4) Gdn.HCl-treated enzyme after irradiation for 1 h in the presence of hypericin; (lane 5) molecular weight markers.
Stability of allicin-modified TcAChE
As mentioned above, whereas native TcAChE is stable for prolonged periods in aqueous solution at room temperature, the allicin-modified enzyme is metastable, converting spontaneously to a stable MG state similar to that produced by modification with disulfides. Curve 3 in Figure 3 ▶ shows that the CD spectrum of allicin-modified TcAChE, after 24 h at room temperature, has changed dramatically, in both the near and far UV, compared with both curves 1 (native TcAChE) and 2 (allicin-modified enzyme after 1 h). It possesses the spectral characteristics of an MG state, as can be seen by comparison with curve 4 in both the near and far UV. Demodification with GSH has no effect on these spectra, and no recovery of enzymic activity can be detected. Thus, whereas the state produced initially by allicin is quasinative, N*, that produced after prolonged incubation, like that produced immediately by modification with bulky and/or charged disulfides and alkylating agents (Dolginova et al. 1992; Kreimer et al. 1994), is partially unfolded, displaying many of the features of the MG state (Fink 1995; Arai and Kuwajima 2000). Thus, allicin modification produces states very similar to those depicted in Scheme 1 ▶.
Our earlier studies showed that the MG state of TcAChE is very stable, most likely thermodynamically more stable than both the native state and the quasinative, N*, state (Kreimer et al. 1994, 1995). It can also be reached from the unfolded (U) state (Eichler et al. 1994). We therefore sought means of stabilizing the allicin-modified enzyme, thereby retarding the N* → MG transition. One obvious approach involved the use of osmolytes (Tatzelt et al. 1996; Timasheff 1998; Bolen and Baskakov 2001). As shown in Figure 8 ▶, two commonly used osmolytes, TMAO and glycerol, both strongly stabilize allicin-modified TcAChE. Under the experimental conditions used, the modified enzyme alone loses the capacity to be reactivated by GSH, with a t1/2 of 1–2 h, whereas, in the presence of 3 M TMAO, for example, >75% reactivation can be achieved, even after 23 h.
Figure 8.

Effects of glycerol, TMAO, and Mg2+ on the degree of reactivation by GSH of N* TcAChE produced by allicin. The native enzyme (1.5 μM) was inhibited by incubation with 1 mM allicin at 23°C for 25 min, yielding 90% inhibition. Samples were then treated with (♦) 1.1 M glycerol, (▾) 3 M TMAO, (▴) 10 mM MgCl2, or (•) an equivalent volume of the incubation buffer. At appropriate times, 10-μL aliquots were mixed with 5 μL of fresh 30 mM aqueous GSH and incubated at 23°C for 30 min. The amount of reactivation was assayed spectrophotometrically on 2 mM PNPA in Sorenson buffer. The velocity measured was corrected for GSH-catalyzed hydrolysis of PNPA.
It is well known that divalent cations often stabilize proteins (Uversky et al. 2000; Xu et al. 2002). In particular, we were aware that cations, for example, Mg2+, have long been used both for extraction of AChE from electric organ tissue and as activators of the enzyme (Changeux 1966). We decided, therefore, to test their effect as stabilizers of allicin-modified TcAChE. As seen in Figure 8 ▶, in the presence of 10 mM MgCl2, the allicin-modified enzyme retains at least 80% of its capacity to be reactivated by GSH, and the same is true for MgSO4 (data not shown). Similar stabilization can be achieved using either 0.1–1 mM MnCl2 or 2–10 mM CaCl2 (data not shown).
Figure 9 ▶ shows the effect of Mg2+ on the CD spectra of allicin-modified TcAChE in the near and far UV. By this spectroscopic criterion, too, Mg2+ largely prevents transition to the MG state of the allicin-modified enzyme, even after 24 h.
Figure 9.

Effect of Mg2+ on the CD spectra of allicin-modified TcAChE in the near and far UV. (1) Native TcAChE + 10 mM MgCl2; (2) allicin-modified TcAChE at 23°C for 1 h, after modification as described in Materials and Methods; (3) as in 2, but 24 h after modification; (4) allicin-modified TcAChE + 10 mM MgCl2 at 22°C for 24 h, after modification.
Because, as shown above, the spectroscopic characteristics of TcAChE freshly modified with allicin closely resemble those of the native enzyme (Figs. 3 ▶ and 4 ▶), it is not possible to use these parameters to assess the effects of the stabilizing agents on the conformation of the modified enzyme. However, as already pointed out, the allicin-modified enzyme binds ANS much more than does native TcAChE (Fig. 5 ▶), binding being at levels similar to those displayed by the MG state. The high concentrations of osmolytes required to achieve substantial stabilization made this approach questionable for assessing their effect. However, the fact that almost complete stabilization by Mg2+ could be achieved at a concentration of 10 mM permitted us to assess its effect on ANS binding (Fig. 10 ▶). It can be seen that a reduction in ANS fluorescence was obtained in the presence of 10 mM MgCl2, although not as large as produced concomitantly with reactivation by GSH (see above).
Figure 10.

Effect of Mg2+ on the ANS-binding capacity of native and allicin-modified TcAChE. ANS, at a final concentration of 0.2 mM, was added to either native or freshly prepared allicin-modified TcAChE (1.5 μM), in the presence or absence of 10 mM MgCl2.
The fact that the allyl group is a small nonpolar moiety led us to consider the possibility that we might be able to reactivate the allicin-modified TcAChE without demodification. Figure 11 ▶ presents data showing that this can, indeed, be achieved. Thus, if the allicin-modified enzyme is exposed to either TMAO or Mg2+, or to both together, immediately after modification, substantial activity is recovered.
Figure 11.

Effects of TMAO and Mg2+ on the activity of N* TcAChE. The enzyme (4 μM) was incubated with 1 mM allicin at 23°C for 20 min. The inhibited enzyme was either demodified by addition of GSH to a final concentration of 2 mM, to determine maximum reactivation capacity; or incubated with TMAO, at a final concentration of 4 M; or of MgCl2, at a final concentration of 10 mM; or both together, before dilution 102–103-fold into the assay mixture, followed by immediate assay. (1) Control; (2) allicin treated; (3) allicin treatment followed by GSH; (4) allicin treatment followed by TMAO; (5) allicin treatment followed by Mg2+; (6) allicin treatment followed by TMAO + Mg2+.
Our working hypothesis for explaining the stabilizing effect of the divalent ion is that, by binding to the N state of the enzyme, it shifts the equilibrium from the N* state toward the N state (Scheme 1 ▶).
To test the effects of the stabilizing agents on native TcAChE, we carried out thermal inactivation studies. Figure 12 ▶ shows that both Mg2+ and TMAO have a strong stabilizing effect. Thus, in their absence, the t1/2 for inactivation of TcAChE at 39°C is <1 min. In the presence of 5 mM MgCl2, it loses <5% activity over a period of 2 h, and in the presence of 3 M TMAO, it loses <10% activity.
Figure 12.

Effects of Mg2+ and TMAO on thermal denaturation of TcAChE. The native enzyme was incubated at 39°C in the presence and absence of MgCl2. At appropriate times, aliquots were withdrawn and assayed for enzymic activity. (•) No MgCl2; (▵) 0.1 mM MgCl2; (♦) 1 mM MgCl2; (○) 5 mM MgCl2; (▴) 3 M TMAO.
Stabilization by MgCl2 was further characterized by high-sensitivity DSC. Figure 13 ▶ shows the excess molar heat capacities obtained at different scan rates in the presence and absence of 10 mM MgCl2. The heat absorption curve apparent Tm (temperature at the maximum of the heat capacity profile) was found to be dependent on the scan rate, and denaturation was always calorimetrically irreversible, because no thermal effect was observed on heating the enzyme solution a second time. The effect of scan rate on the calorimetric profiles clearly indicates that they correspond to irreversible, kinetically controlled transitions. For this reason, analysis of DSC transitions on the basis of equilibrium thermodynamics was ruled out (Freire et al. 1990), and was performed as described earlier (Kreimer et al. 1995), using the simple two-state irreversible model (see Materials and Methods). The effect of MgCl2 was to increase the thermostability of TcAChE. Thus, the Tm at a scan rate of 1 K/min in the presence of 10 mM MgCl2 is 47.3° ± 0.2°C, whereas the value in the absence of MgCl2 is 41.7° ± 0.2°C. Quite similar values are obtained for T*, that is, the temperature at which k = 1 min−1 (47.4° ± 0.2°C in the presence of MgCl2, and 42.7° ± 0.2°C in its absence). The Arrhenius activation energy for TcAChE in the presence of MgCl2, 197.5 ± 1.8 kcal/mole, is much higher than in its absence, 112.9 ± 1.6 kcal/mole.
Figure 13.
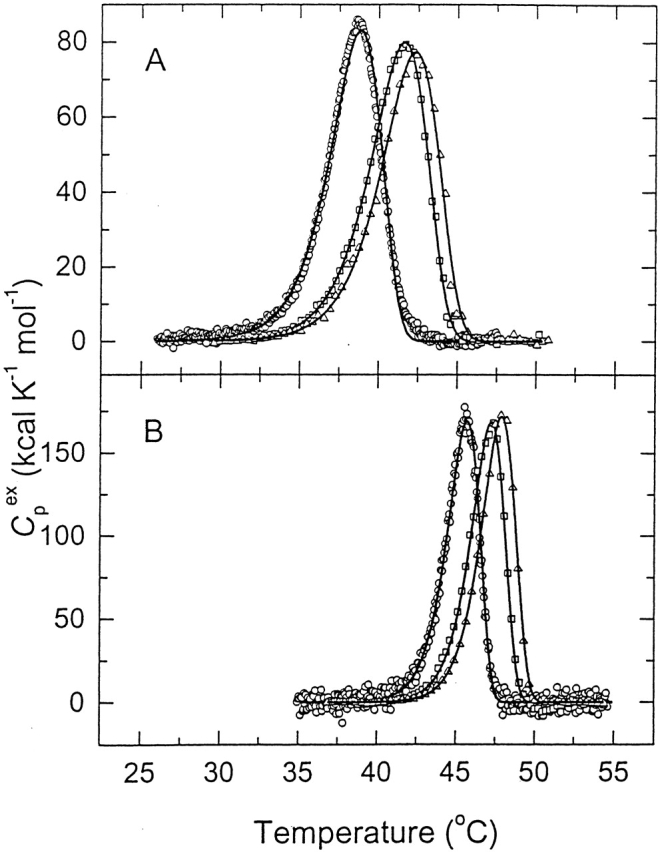
Differential scanning calorimetry of native TcAChE in the absence and presence of Mg2+. The curves show the temperature dependence of the excess molar heat capacity at three scan rates: (○) 0.21 K/min; (□) 1.0 K/min; (▵) 1.45 K/min. The buffer was 0.1 M NaCl/10 mM HEPES (pH 7.5), and the concentration of enzyme was 9.2 μM for the scans at 0.21 K/min; 6.2 μM for those at 1.0 K/min, and 5.6 μM for those at 1.45 K/min. (A) Control; (B) in the presence of 10 mM MgCl2. The solid lines represent the best fits of the experimental data to equation 1.
Another criterion used to differentiate between native and partially unfolded states is sensitivity to proteolysis. We earlier showed that the MG state of TcAChE produced by modification with the disulfide, DTP, is highly susceptible to tryptic digestion under conditions in which the native enzyme is totally resistant (Dolginova et al. 1992). The N* state produced by allicin modification, even though much more similar to native TcAChE, is also very sensitive to trypsin, as seen by comparing lanes 2 and 4 in the SDS-PAGE experiment displayed in Figure 14 ▶. Lane 5 shows that by this criterion, too, Mg2+ produces considerable stabilization, a finding best explained by assuming that, in binding to the enzyme, it produces a more compact conformation resembling that of the native enzyme.
Figure 14.

SDS-PAGE analysis of susceptibility to tryptic digestion of native and allicin-modified TcAChE. SDS-PAGE was carried out under nonreducing conditions on 5%–15% acrylamide gradient gels. Enzyme samples (1 mg/mL) were digested with trypsin (1% [w/w]) at 23°C for 1 h. (Lane 1) Control, native TcAChE; (lane 2) trypsin-digested native TcAChE; (lane 3) TcAChE modified by treatment with 1 mM allicin at 22°C for 30 min; (lane 4) allicin-modified enzyme digested with trypsin; (lane 5) allicin-modified enzyme digested with trypsin in the presence of 1 mM MgCl2; (lane 6) molecular weight markers.
Discussion
We earlier showed that chemical modification of the buried free cysteine, Cys 231, in TcAChE inhibits enzymic activity. Inhibition by alkylating agents, bulky disulfides, and charged thiosulfonates occurred, in all cases, concomitantly with irreversible transformation to an MG species (Dolginova et al. 1992; Kreimer et al. 1994, 1996). However, mercuric ions and organomercurials generated a catalytically inactive species, the quasinative, N* state, from which activity could be recovered by demodification. We presented crystallographic evidence that the N* state was transiently stabilized by interactions of the mercury ion not only with Cys 231 but also with Ser 229O and Ser 229Oγ (Kreimer et al. 1994). The natural thiosulfinate, allicin, which is small and hydrophobic, is the first sulfhydryl reagent, apart from mercurials, to generate an N* form of TcAChE, and the molecular basis for the transient stabilization in this case remains to be clarified. Our spectroscopic data show that the structure of the N* state is very similar to that of the native state. However, both ANS binding and the existence of short-lived complexes trapped by cross-linking with hypericin reveal the presence of exposed hydrophobic surfaces. In any event, the N* state produced by allicin decays spontaneously and irreversibly to an MG state unless demodified shortly after formation.
The central observations made in the present study are that the N* → MG transition of TcAChE can be retarded by two families of compounds, osmolytes and divalent cations.
It is commonly considered that osmolytes exert their refolding effect on proteins by raising the chemical potential of an unfolded or partially unfolded state more than that of the native state, thereby increasing the (positive) Gibbs free energy (ΔG) difference between them (Timasheff 1998; Bolen and Baskakov 2001). If this type of analysis is applied to our experimental system (Scheme 3 ▶), our data can be explained by assuming that the osmolyte substantially raises the potentials of both the N* and MG states, as well as of the activation energy barrier between the two states, while raising the potential of the N state much less. This should shift the equilibrium in Scheme 1 ▶ to the left, decreasing the concentration of the metastable N* state, and thus retarding the irreversible N* → MG transition.
Scheme 3.
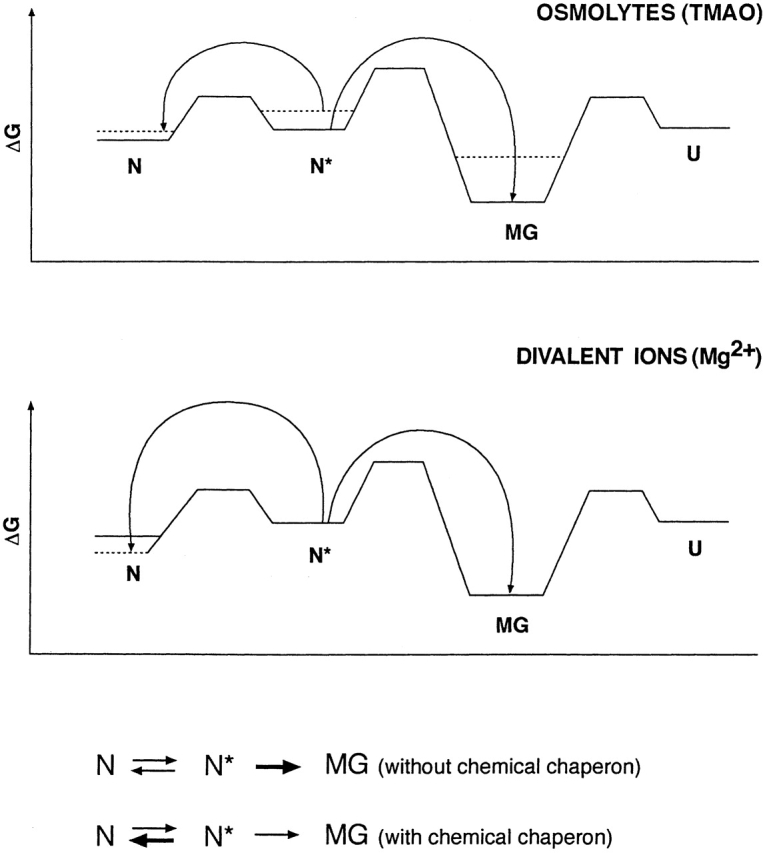
Proposed profile of relative free energy levels of conformational states of TcAChE in the presence and absence of chemical chaperones. The solid lines show energy levels in the absence of osmolytes and Mg2+, and the dashed lines, the levels in their presence.
A plausible explanation for the effect of divalent cations assumes that, by binding tightly to the N state, they decrease its free energy, and, thereby, ΔG between the N and N* states, thus shifting the N ― N* transition toward N (Scheme 3 ▶). The observed decrease in ANS fluorescence of N* in the presence of Mg2+ provides experimental support for this explanation. Independent evidence for stabilization of the N state by divalent cations comes from their effect on thermal inactivation and from the DSC studies presented above (Figs. 12 ▶ and 13 ▶). The DSC data show that binding of Mg2+ raises the transition temperature by ~5°C, increases the sharpness of the transition, and significantly increases its activation energy.
The occurrence of long-lived partially unfolded states, which may reside in either a kinetic or a thermodynamic trap, has been the subject of considerable interest in recent years (for a recent study, see Jaswal et al. 2002). For several proteins it has been shown that partially unfolded states are more stable thermodynamically than the native state, and thus provide examples of kinetic traps. These proteins include serpins (Huber and Carrell 1989), influenza hemagglutinin (Carr et al. 1997), α-lytic protease (Eder et al. 1993; Sohl et al. 1998), human insulin (Hua et al. 1995), and the native prion protein (Baskakov et al. 2001). We earlier presented evidence that partially unfolded states of TcAChE with features of an MG state are examples of thermodynamic traps (Kreimer et al. 1994, 1996), and we are not able to recover any enzymic activity even using either biological or chemical chaperones. A similar MG state is arrived at whether the starting point is the native (N) or unfolded (U) state (Eichler et al. 1994). This state appears to be stabilized by interactions between the two subunits of the TcAChE dimer, as shown by sucrose gradient centrifugation under reducing conditions (Kreimer et al. 1996).
The N* state produced by modification with allicin, as described above, can be stabilized, and even refolded, by treatment with either osmolytes or divalent cations. Thus, the N* state occupies a local energy minimum, separated by a low energy barrier from the N state. The osmolyte, by raising the free energy of N* more than that of N, shifts the equilibrium further toward N, and may also lower the activation energy barrier. With respect to the effect of divalent cations, we have clear evidence that the native enzyme is stabilized by them. This may serve to drive the equilibrium in its direction, and provide an adequate explanation for our observations (see Scheme 3 ▶).
Stabilization of proteins by ligands, whether inorganic cations or others, is well documented (see, e.g., Uversky et al. 2000; Xu et al. 2002). The data that we have presented show that, for TcAChE, divalent cations lower the energy of the N state and thereby both protect against unfolding and shift the equilibrium between the N and N* states. They can thus be regarded as chemical chaperones in the same sense as the term has been used for osmolytes (see, e.g., Tatzelt et al. 1996), even though they act by a different mechanism, as shown by the comparison in Scheme 3 ▶. Moreover, as in the present case, they have the potential to be effective at much lower concentrations, in the millimolar range, which may reflect specific interactions.
Evidence has been presented that partially unfolded proteins, produced by heat shock, oxidative stress, or chemical modification, in which exposure of hydrophobic surfaces has been increased, can trigger the heat shock response (Ananthan et al. 1986; Morimoto 1993; Freeman et al. 1999; Gosslau et al. 2001). Such treatments might be expected to produce irreversible damage, and termination of the signal would thus only occur via degradation of the unfolded protein (Goldberg 1992; Weiner et al. 1994). The N* states that we describe, whether produced by chemical modification or by physicochemical perturbation, are separated by a low energy barrier from the native protein, and thus could provide an “on/off switch” for a stress response.
Materials and methods
Materials
TcAChE was the dimeric (G2) glycosylphosphatidylinositol-anchored form purified from electric organ tissue of Torpedo californica by affinity chromatography subsequent to solubilization with phosphatidylinositol-specific phospholipase C (Sussman et al. 1988). The C231S mutant of TcAChE, expressed in COS cells (Morel et al. 1999), was given to us by Suzanne Bon and Jean Massoulié (Ecole Normale Supérieure, Paris).
Allicin and hypericin were gifts from Talia Miron and Aharon Rabinkow and from Yehuda Mazur, respectively (Weizmann Institute of Science). Reduced glutathione (GSH), 1-anilino-8-naphthalenesulfonic acid (ANS, magnesium salt), and TMAO were from Sigma; glycerol (J.T. Baker) and sucrose (BDH) were both analytical grade. Other salts and buffers were all analytical grade.
Assay methods
Concentrations and activities of AChE were determined as described previously (Kreimer et al. 1994).
Buffers
Unless stated otherwise, experiments were carried out in Sorenson buffer (namely, 0.067 M Na/K phosphate at pH 7.5) at 23°C.
Modification of TcAChE by allicin
This was performed essentially as described previously for other sulfhydryl reagents (Dolginova et al. 1992; Kreimer et al. 1994), except that Sorenson buffer at 23°C was used. Demodification used GSH at a final concentration of 5 mM (Kreimer et al. 1994).
Sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE)
SDS/PAGE was performed according to Shin et al. (2002).
Tryptic digestion
This was done using 1% (v/v) trypsin at room temperature, as described (Dolginova et al. 1992), except that Sorenson buffer was used.
Cross-linking of TcAChE
Cross-linking of partially unfolded states of TcAChE was performed using the photosensitive compound hypericin (Miskovsky 2002), as described earlier (Weiner et al. 1999).
Differential Scanning Calorimetry (DSC)
DSC measurements were performed on a MicroCal MC-2D differential scanning microcalorimeter (MicroCal Inc.) using a 1.22-mL cell, as described previously (Kreimer et al. 1996; Marcos et al. 1999). Protein solutions were dialyzed against the desired buffer, and the dialysate was used as a reference. Solutions were degassed by stirring under vacuum prior to scanning. Experimental traces were corrected for the calorimeter baseline by scanning the appropriate buffer solution in both cells of the calorimeter (Ruiz-Arribas et al. 1994). The reversibility of the thermal transitions was checked by examining the reproducibility of the calorimetric trace in a second heating of the sample immediately after cooling subsequent to the first scan. The experimental calorimetric traces were corrected for the effect of instrument response time according to Lopez Mayorga and Freire (1987). The molar excess heat capacity curves were smoothed and plotted using the Windows-based software package Origin, supplied by MicroCal. Data were analyzed by nonlinear least squares fitting as described earlier (Kurganov et al. 1997).
In accordance with our earlier study (Kreimer et al. 1995), only one model was considered in analyzing the process of denaturation of TcAChE. This is a two-state model, with only two significantly populated macroscopic states, the initial or native (N) state, and the final or denatured (D) state, in which the transition is governed by a first-order kinetic constant, k, that changes with temperature, according to the Arrhenius equation (Sanchez-Ruiz 1992). In this case, the excess heat capacity, Cpex, is given by the following equation (Kurganov et al. 1997):
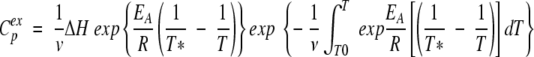 |
where ν = dT/dt is a scan rate value, ΔH the enthalpy difference between denatured and native states, R the gas constant, EA the activation energy of the denaturation process, and T* the temperature at which k = 1 min−1.
Spectroscopic measurements
Intrinsic fluorescence and binding of ANS used a Shimadzu RF-540 fluorometer, at 22°C, as described (Kreimer et al. 1994). CD spectra measurements used an Aviv Model 202 circular dichroism spectrometer.
Acknowledgments
This study was supported by a grant from the Israel Science Foundation and by the Benoziyo Center for Neuroscience. V.L.S. acknowledges receipt of a travel grant from the Kimmelman Center for Biomolecular Structure and Assembly (Rehovot). I.S. is Bernstein-Mason Professor of Neurochemistry. We thank Talia Miron and Aharon Rabinkow, and Yehuda Mazur for their generous gifts of allicin and hypericin, respectively, and Suzanne Bon and Jean Massoulié for the C231S TcAChE mutant.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03110703.
References
- Ananthan, J., Goldberg, A.L., and Voellmy, R. 1986. Abnormal proteins serve as eukaryotic stress signals and trigger the activation of heat shock genes. Science 232 522–524. [DOI] [PubMed] [Google Scholar]
- Arai, M. and Kuwajima, K. 2000. Role of the molten globule state in protein folding. Adv. Protein Chem. 53 209–282. [DOI] [PubMed] [Google Scholar]
- Baskakov, I.V., Legname, G., Prusiner, S.B., and Cohen, F.E. 2001. Folding of prion protein to its native α-helical conformation is under kinetic control. J. Biol. Chem. 276 19687–19690. [DOI] [PubMed] [Google Scholar]
- Bolen, D.W. and Baskakov, I.V. 2001. The osmophobic effect: Natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 310 955–963. [DOI] [PubMed] [Google Scholar]
- Carr, C.M., Chaudhry, C., and Kim, P.S. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. 94 14306–14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell, R.W. and Lomas, D.A. 1997. Conformational diseases. Lancet 350 134–138. [DOI] [PubMed] [Google Scholar]
- Cavallito, C.J. and Bailey, J.H. 1944. Allicin, the antibacterial principle of Allium sativum. I. Isolation, physical properties and antibacterial action. J. Am. Chem. Soc. 66 1950–1951. [Google Scholar]
- Changeux, J.-P. 1966. Responses of acetylcholinesterase from Torpedo marmorata to salts and curarizing drugs. Mol. Pharmacol. 2 369–392. [PubMed] [Google Scholar]
- Davis-Searles, P.R., Saunders, A.J., Erie, D.A., Winzor, D.J., and Pielak, G.J. 2001. Interpreting the effects of small uncharged solutes on protein-folding equilibria. Annu. Rev. Biophys. Biomol. Struct. 30 271–306. [DOI] [PubMed] [Google Scholar]
- Dolginova, E.A., Roth, E., Silman, I., and Weiner, L.M. 1992. Chemical modification of Torpedo acetylcholinesterase by disulfides: Appearance of a “molten globule” state. Biochemistry 31 12248–12254. [DOI] [PubMed] [Google Scholar]
- Eder, J., Rheinnecker, M., and Fersht, A.R. 1993. Folding of subtilisin BPN′: Role of the pro-sequence. J. Mol. Biol. 233 293–304. [DOI] [PubMed] [Google Scholar]
- Eichler, J., Kreimer, D.I., Varon, L., Silman, I., and Weiner, L. 1994. A ‘molten globule’ of Torpedo acetylcholinesterase undergoes thiol-disulfide exchange. J. Biol. Chem. 269 30093–30096. [PubMed] [Google Scholar]
- Fink, A.L. 1995. Compact intermediate states in protein folding. Annu. Rev. Biophys. Biomol. Struct. 24 495–522. [DOI] [PubMed] [Google Scholar]
- Freeman, M.L., Borrelli, M.J., Meredith, M.J., and Lepock, J.R. 1999. On the path to the heat shock response: Destabilization and formation of partially unfolded protein intermediates. A consequence of protein thiol modification. Free Radical Biol. Med. 26 737–745. [DOI] [PubMed] [Google Scholar]
- Freire, E., van Osdol, W.W., Mayorga, O.L., and Sanchez-Ruiz, J.M. 1990. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Annu. Rev. Biophys. Biophys. Chem. 19 159–188. [DOI] [PubMed] [Google Scholar]
- Gibney, G., MacPhee-Quigley, K., Thompson, B., Vedvick, T., Low, M.G., Taylor, S.S., and Taylor, P. 1988. Divergence in primary structure between the molecular forms of acetylcholinesterase. J. Biol. Chem. 263 1140–1145. [PubMed] [Google Scholar]
- Goldberg, A.L. 1992. The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. Eur. J. Biochem. 203 9–23. [DOI] [PubMed] [Google Scholar]
- Gosslau, A., Ruoff, P., Mohsenzadeh, S., Hobohm, U., and Rensing, L. 2001. Heat shock and oxidative stress-induced exposure of hydrophobic protein domains as common signal in the induction of hsp68. J. Biol. Chem. 276 1814–1821. [DOI] [PubMed] [Google Scholar]
- Hua, Q.X., Gozani, S.N., Chance, R.E., Hoffmann, J.A., Frank, B.H., and Weiss, M.A. 1995. Structure of a protein in a kinetic trap. Nat. Struct. Biol. 2 129–138. [DOI] [PubMed] [Google Scholar]
- Huber, R. and Carrell, R.W. 1989. Implications of the three-dimensional structure of α 1-antitrypsin for structure and function of serpins. Biochemistry 28 8951–8966. [DOI] [PubMed] [Google Scholar]
- Jaswal, S.S., Sohl, J.L., Davis, J.H., and Agard, D.A. 2002. Energetic landscape of α-lytic protease optimizes longevity through kinetic stability. Nature 415 343–346. [DOI] [PubMed] [Google Scholar]
- Kreimer, D.I., Dolginova, E.A., Raves, M., Sussman, J.L., Silman, I., and Weiner, L. 1994. A metastable state of Torpedo californica acetylcholinesterase generated by modification with organomercurials. Biochemistry 33 14407–14418. [DOI] [PubMed] [Google Scholar]
- Kreimer, D.I., Shnyrov, V.L., Villar, E., Silman, I., and Weiner, L. 1995. Irreversible thermal denaturation of Torpedo californica acetylcholinesterase. Protein Sci. 4 2349–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreimer, D.I., Shin, I., Shnyrov, V.L., Villar, E., Silman, I., and Weiner, L. 1996. Two partially unfolded states of Torpedo acetylcholinesterase. Protein Sci. 5 1852–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurganov, B.I., Lyubarev, A.E., Sanchez-Ruiz, J.M., and Shnyrov, V.L. 1997. Analysis of differential scanning calorimetry data for proteins. Criteria of validity of one-step mechanism of irreversible protein denaturation. Biophys. Chem. 69 125–135. [DOI] [PubMed] [Google Scholar]
- Lopez Mayorga, O. and Freire, E. 1987. Dynamic analysis of differential scanning calorimetry data. Biophys. Chem. 87 87–96. [DOI] [PubMed] [Google Scholar]
- MacPhee-Quigley, K., Vedvick, T.S., Taylor, P., and Taylor, S.S. 1986. Profile of the disulfide bonds in acetylcholinesterase. J. Biol. Chem. 261 13565–13570. [PubMed] [Google Scholar]
- Marcos, M.J., Chehín, R., Arrondo, J.L., Zhadan, G.G., Villar, E., and Shnyrov, V.L. 1999. pH-dependent thermal transitions of lentil lectin. FEBS Lett. 443 192–196. [DOI] [PubMed] [Google Scholar]
- Miron, T., Rabinkov, A., Mirelman, D., Wilchek, M., and Weiner, L. 2000. The mode of action of allicin: Its ready permeability through phospholipid membranes may contribute to its biological activity. Biochim. Biophys. Acta 1463 20–30. [DOI] [PubMed] [Google Scholar]
- Miskovsky, P. 2002. Hypericin—A new antiviral and antitumor photosensitizer: Mechanism of action and interaction with biological macromolecules. Curr. Drug Targets 3 55–84. [DOI] [PubMed] [Google Scholar]
- Morel, N., Bon, S., Greenblatt, H.M., Van Belle, D., Wodak, S.J., Sussman, J.L., Massoulié, J., and Silman, I. 1999. Effect of mutations within the peripheral anionic site on the stability of acetylcholinesterase. Mol. Pharmacol. 55 982–992. [DOI] [PubMed] [Google Scholar]
- Morimoto, R.I. 1993. Cells in stress: Transcriptional activation of heat shock genes. Science 259 1409–1410. [DOI] [PubMed] [Google Scholar]
- Qu, Y.X., Bolen, C.L., and Bolen, D.W. 1998. Osmolyte-driven contraction of a random coil protein. Proc. Natl. Acad. Sci. 95 9268–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinkov, A., Miron, T., Konstantinovski, L., Wilchek, M., Mirelman, D., and Weiner, L. 1998. The mode of action of allicin: Trapping of radicals and interaction with thiol containing proteins. Biochim. Biophys. Acta 1379 233–244. [DOI] [PubMed] [Google Scholar]
- Ruiz-Arribas, A., Santamaria, R.I., Zhadan, G.G., Villar, E., and Shnyrov, V.L. 1994. Differential scanning calorimetry study of the thermal stability of xylanase from Streptomyces halstedii JM8. Biochemistry 33 13787–13791. [DOI] [PubMed] [Google Scholar]
- Safar, J., Roller, P.P., Gajdusek, D.C., and Gibbs Jr., C.J. 1994. Scrapie amyloid (prion) protein has the conformational characteristics of an aggregated molten globule folding intermediate. Biochemistry 33 8375–8383. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ruiz, J.M. 1992. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry. Biophys. J. 61 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, I., Kreimer, D., Silman, I., and Weiner, L. 1997. Membrane-promoted unfolding of acetylcholinesterase: A possible mechanism for insertion into the lipid bilayer. Proc. Natl. Acad. Sci. 94 2848–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, I., Wachtel, E., Roth, E., Bon, C., Silman, I., and Weiner, L. 2002. Thermal denaturation of Bungarus fasciatus acetylcholinesterase: Is aggregation a driving force in protein unfolding? Protein Sci. 11 2022–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohl, J.L., Jaswal, S.S., and Agard, D.A. 1998. Unfolded conformations of α-lytic protease are more stable than its native state. Nature 395 817–819. [DOI] [PubMed] [Google Scholar]
- Solomon, B., Taraboulos, A., and Katchalski-Katzir, E. 2001. Conformational diseases—A compendium. The Center for the Study of Emerging Diseases, Jerusalem. [DOI] [PubMed]
- Speed, M.A., Wang, D.I., and King, J. 1996. Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition. Nat. Biotechnol. 14 1283–1287. [DOI] [PubMed] [Google Scholar]
- Sussman, J.L., Harel, M., Frolow, F., Varon, L., Toker, L., Futerman, A.H., and Silman, I. 1988. Purification and crystallization of a dimeric form of acetylcholinesterase from Torpedo californica subsequent to solubilization with phosphatidylinositol-specific phospholipase C. J. Mol. Biol. 203 821–823. [DOI] [PubMed] [Google Scholar]
- Sussman, J.L., Harel, M., Frolow, F., Oefner, C., Goldman, A., Toker, L., and Silman, I. 1991. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 253 872–879. [DOI] [PubMed] [Google Scholar]
- Tatzelt, J., Prusiner, S.B., and Welch, W.J. 1996. Chemical chaperones interfere with the formation of scrapie prion protein. EMBO J. 15 6363–6373. [PMC free article] [PubMed] [Google Scholar]
- Thirumalai, D. and Lorimer, G.H. 2001. Chaperonin-mediated protein folding. Annu. Rev. Biophys. Biomol. Struct. 30 245–269. [DOI] [PubMed] [Google Scholar]
- Timasheff, S.N. 1998. Control of protein stability and reactions by weakly interacting cosolvents: The simplicity of the complicated. Adv. Protein Chem. 51 355–432. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Gillespie, J.R., and Fink, A.L. 2000. Why are “natively unfolded” proteins unstructured under physiologic conditions? Protein 41 415–427. [DOI] [PubMed] [Google Scholar]
- Wanker, E.E. 2000. Protein aggregation in Huntington’s and Parkinson’s disease: Implications for therapy. Mol. Med. Today 6 387–391. [DOI] [PubMed] [Google Scholar]
- Weiner, L., Kreimer, D., Roth, E., and Silman, I. 1994. Oxidative stress transforms acetylcholinesterase to a molten-globule like state. Biochem. Biophys. Res. Comm. 198 915–922. [DOI] [PubMed] [Google Scholar]
- Weiner, L., Roth, E., Mazur, Y., and Silman, I. 1999. Targeted cross-linking of a molten globule form of acetylcholinesterase. Biochemistry 38 11401–11405. [DOI] [PubMed] [Google Scholar]
- Xu, X., Liu, Q., and Xie, Y. 2002. Metal ion-induced stabilization and refolding of anticoagulation factor II from the venom of Agkistrodon acutus. Biochemistry 41 3546–3554. [DOI] [PubMed] [Google Scholar]
- Yancey, P.H., Clark, M.E., Hand, S.C., Bowlus, R.D., and Somero, G.N. 1982. Living with water stress: Evolution of osmolyte systems. Science 217 1214–1222. [DOI] [PubMed] [Google Scholar]