

Abstract
The Ca2+-triggered merger of two apposed membranes is the defining step of regulated exocytosis. CHOL is required at critical levels in secretory vesicle membranes to enable efficient, native membrane fusion: CHOL-sphingomyelin enriched microdomains organize the site and regulate fusion efficiency, and CHOL directly supports the capacity for membrane merger by virtue of its negative spontaneous curvature. Specific, structurally dissimilar lipids substitute for CHOL in supporting the ability of vesicles to fuse: diacylglycerol, αT, and phosphatidylethanolamine support triggered fusion in CHOL-depleted vesicles, and this correlates quantitatively with the amount of curvature each imparts to the membrane. Lipids of lesser negative curvature than cholesterol do not support fusion. The fundamental mechanism of regulated bilayer merger requires not only a defined amount of membrane-negative curvature, but this curvature must be provided by molecules having a specific, critical spontaneous curvature. Such a local lipid composition is energetically favorable, ensuring the necessary “spontaneous” lipid rearrangements that must occur during native membrane fusion—Ca2+-triggered fusion pore formation and expansion. Thus, different fusion sites or vesicle types can use specific alternate lipidic components, or combinations thereof, to facilitate and modulate the fusion pore.
INTRODUCTION
The involvement of specific membrane components in the native membrane merger mechanism, including prefusion protein functions (1,2) and lipidic functions during bilayer coalescence (3,4), has been widely discussed. The contribution of lipidic membrane components specifically to the fusion process is addressed by the stalk-pore hypothesis (3–16), which defines the underlying energetics of bilayer merger via a lipidic fusion pore. Different lipids affect the energy required to overcome the hydration layer, and affect bilayer merger in terms that include membrane curvature. Lipids with small hydrophilic headgroups relative to larger hydrophobic domains (e.g., “average molecular conformation”) are said to have negative spontaneous curvature and tend to pack in concave structures at the lipid-water interface; lipids of an opposite average molecular conformation are said to have positive curvature and form convex, micelle-like structures. The spontaneous curvature of various lipids in hydrated assemblies has been measured (17–23). Investigations into the role of lipids in the fusion process have shown that the initial formation of high energy, transient, high curvature lipidic intermediates (hemifusion) can be inhibited by addition of exogenous lipids of high positive curvature to the interacting monolayers (24,25) or by removing endogenous lipidic components of high negative curvature, such as CHOL (25). In both cases, the net negative curvature is decreased, and the positive curvature increased. Thus, a local concentration of negative curvature in the initially contacting monolayers promotes hemifusion, the necessary first transition to the fusion of two apposed bilayers; a focal enhancement of positive curvature in the distal monolayers then promotes transition to full fusion.
Liposome and viral fusion studies have highlighted the contribution of molecules with negative spontaneous curvature to the formation of the hemifusion intermediates predicted by the stalk-pore hypothesis, however, this has not previously been demonstrated in a native system capable of fast, Ca2+-triggered membrane fusion. Isolated sea urchin egg CV offer a robust, stage-specific, high purity preparation for the study of native, Ca2+-triggered membrane merger; by all available criteria, CV-CV fusion proceeds through the same molecular pathway as exocytotic CV-PM fusion (26,27). Most notably, these vesicles are amenable to rigorous biochemical manipulation and molecular quantification not possible in intact cellular systems. Here we show that Ca2+-triggered fusion of CHOL-depleted CV can be selectively rescued by the addition of structurally dissimilar lipids having negative curvature comparable to CHOL. We highlight for the first time, to our knowledge, a direct, quantitative molar correlation between the focal curvature contributions of specific, structurally unrelated membrane components—CHOL, DOG, DOPE, and αT—and the fundamental role of these specific negative curvature components in the native mechanism of Ca2+-triggered membrane fusion (Table 1, Fig. 1).
TABLE 1.
Measured spontaneous curvatures of specific lipids
FIGURE 1.

Chemical structures of specific negative curvature lipids, as in Table 1. (A) DOPC. (B) DOPA. (C) Me2-DOPE. (D) Me-DOPE. (E) DOPE. (F) DOG. (G) αT. (H) CHOL.
MATERIALS AND METHODS
Materials
DiI, mßcd, hpßcd, αT, and hexadecane were from Sigma (St. Louis, MO). CHOL-B was from Invitrogen (Carlsbad, CA). Cholesterol, DOPE, DOG, DOPC, Me-DOPE, Me2-DOPE, DOPA, and all lipid standards for HPTLC were from Avanti Polar Lipids (Alabaster, AL). All other chemicals were minimally analytical grade.
Preparations, CV treatments, and fusion assays
CV were isolated from Strongylocentrotus purpuratus (Westwind Sea Labs, Victoria, British Columbia, Canada) as previously described (26,28). All experiments were carried out in BIM (210 mM potassium glutamate, 500 mM glycine, 10 mM NaCl, 10 mM PIPES, 50 μM CaCl2, 1 mM MgCl2, 1 mM EGTA, pH 6.7) supplemented with 2.5 mM ATP and protease inhibitors (25,28). Standard end-point and kinetic fusion assays were carried out as previously described (25,26,29). CV were treated with 2 mM mßcd in BIM for 30 min (25°C). CHOL-loaded hpßcd was prepared as previously described (25), and treatments were carried out as with mßcd. DOPC, DOPA, DOPE, Me-DOPE, Me2-DOPE, αT, and DOG were delivered from 50 mM (DOPC, DOPA, DOPE) or 200 mM (αT, DOG) stocks in hexadecane to a final solvent concentration of 0.1%. CHOL-B was delivered from a DMSO stock to a final solvent concentration of 0.75%, whereas C9NBD-T was delivered from ethanol to a final solvent concentration of 0.1%. Lipids in organic solvents were either i), delivered directly to a suspension of CV (optical density 1.0) and the suspension gently mixed by inversion, or ii), diluted 10-fold into BIM and vortexed thoroughly before delivery to CV suspensions (final optical density 1.0); both delivery methods were of comparable efficacy. In each experiment, parallel solvent controls were carried out to determine the effects of organic solvent alone on CV-CV fusion. At the concentrations used, none of the organic solvents had a significant effect on any of the parameters of fusion (P ≥ 0.5). Incubation of CV with exogenous PLD (prepared as a concentrated stock in BIM) were carried out in BIM at 25°C as with previous enzyme treatments (25,29). To ensure removal of any unincorporated reagents, CV were centrifuged after all treatments and then suspended in fresh BIM. CV were then counted using a hemacytometer (25). Ca2+-activity curves were fit using the sigmoidal cumulative log-normal model (25,30,31); control conditions were fit with a two-parameter model (plateau at 100%, by definition), whereas experimental conditions were fit with a three-parameter model (TableCurve 2D) to determine upper plateau extent, Ca2+ sensitivity (EC50), and sigmoidal curve-shape parameters. Kinetic data were fit with an established model (32). The spontaneous curvature (1/R0p) for Me-DOPE and Me2-DOPE were estimated from a linear extrapolation of 1/R0p versus degree of N-methylation for DOPC (three methyl groups) and DOPE (zero methyl groups) (20,21,23).
Confocal microscopy
Eggs from S. purpuratus were isolated, washed, and dejellied as previously described (26). A dilute suspension of eggs in artificial sea water (435 mM NaCl, 40 mM MgCl, 15 mM MgSO4, 11 mM CaCl2, 10 mM KCl, 1 mM EDTA, 10 mM HEPES, pH 8.0) was incubated with 20 μM DiI (delivered from DMSO stock diluted into artificial sea water to a 1% final concentration of DMSO) for 20 min at 25°C. Labeled eggs were washed three times with sea water and stored suspended on ice until required. Cortices were isolated on glass coverslips as previously described (31). Briefly, DiI-labeled eggs were allowed to settle onto poly-L-lysine treated (1.0 mg/ml) glass bottomed culture dishes (MatTek, Ashland, MA) for 10 min, then eggs were sheared with a stream of BIM delivered from an 18-gauge syringe; shearing was forceful to produce regions of PM denuded of CV. Cortices were washed with several volumes of BIM, and subsequently incubated with 15 μM CHOL-B (delivered from DMSO diluted into BIM to a 1% final concentration) in BIM (20 min, room temperature). Images were acquired using a Zeiss LSM-510 Meta confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 40× 1.2 numerical aperture water immersion objective in the inverted configuration. Cortices were imaged using a series of Z plane optical sections to produce an image stack. The CHOL-B signal was obtained using laser excitation at 488 nm and imaged using a bandpass filter at 505–530 nm; DiI was excited at 543 nm and imaged using a 560–615 nm bandpass filter. Images were visualized and processed using ImageJ (33).
Molecular analyses
CV membranes were isolated by ultracentrifugation (90 min, 200,000 × g) after lysis (34). Total cholesterol concentrations were measured using the Amplex Red Cholesterol Assay kit (Molecular Probes, Eugene, OR) according to manufacturer instructions. Fluorescent lipid analogs were determined by fluorescence measurements on intact CV after removal of excess reagent and washing with BIM in parallel with a dilution series of the fluorescent molecule (Wallac Victor II Microplate Reader, PerkinElmer, Boston, MA). CV membrane lipids were extracted according to Bligh and Dyer (35) with some modifications. Methanol and chloroform were added sequentially to an intermediate ratio of 0.8:2:1 (H2O/CH3OH/CHCl3, v/v/v) and subsequently brought to a final ratio of 1.8:2:2 with the sequential addition of aqueous solution (0.1 M HCl, 1 M NaCl) and chloroform. The chloroform phase was recovered, and the remaining aqueous phase was rinsed with an additional 2 vol of chloroform. The organic phases were combined, dried under vacuum, and stored under N2 (−30°C) before analysis. For DOPA, DOPE, and DOG analysis, dried films were resuspended in 2:1 chloroform/methanol and loaded onto silica gel 60 HPTLC plates (EMD Chemicals, Darmstadt, Germany) previously washed with chloroform/ethyl acetate (6:4) and activated at 110°C for 30 min. HPTLC was carried out essentially as previously described (25). For αT analysis, dried films were loaded as above onto silica gel 60 preparative TLC plates (EMD Chemicals) and separated in four sequential steps using the CAMAG AMD2 (CAMAG, Wilmington, NC). Plates were developed to 30 mm with CH2Cl2/ethyl acetate/acetone (80:16:4 v/v/v), then to 50, 70, and 90 mm with 92:8, 95:5, and 98:2 v/v hexane/ethyl acetate, respectively. DOPA, DOPE, and DOG were stained on a plate with Nile Red (36) or were charred with CuSO4 (29) and the integrated fluorescence intensity was compared to a parallel dilution series of lipid standards (25). Quantification of αT was carried out on-plate using the characteristic absorbance at 288 nm, with the CAMAG TLC2 scanner (CAMAG). Integrated signal was compared to a parallel dilution series of standards on the same TLC plate. Thus, all values reported in the text are the result of direct quantification of the amount of exogenous lipid incorporated into CV membranes.
Statistical analyses
To define the minimal quantity of each lipidic species required to elicit full recovery of fusion, a linear regression was fit to the scatter data (see Fig. 3 C) sequentially, increasing the number of data points from the origin until such a time that increasing the number of points included significantly altering the slope and coefficient of determination of the line. Data points below the break point were defined as the rising phase, and those above the break as the plateau phase of the saturation curve. The X value at the point of intersection between the linear fit of the rising phase and the plateau was defined as the minimal quantity required for full recovery of fusion (see Fig. 3 D). As it was experimentally difficult to incorporate either very small or very large amounts of exogenous DOPE into the CV membrane, the minimal quantity required for recovery of fusion was calculated as the average amount of exogenous DOPE that elicited full fusion recovery in six experiments.
FIGURE 3.

Treatment of CHOL-depleted CV with negative curvature analogs rescues the extent but not the Ca2+ sensitivity or kinetics of CV-CV fusion. (A) Ca2+ activity curve of CV treated with 2 mM mßcd or sequentially with mßcd and 100 μM DOPE, 2 mM CHOL-loaded hpßcd, or 200 μM DOG or αT (n = 3–7). (B) Kinetics of CV-CV fusion in response to 157 ± 17 μM 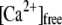 (n = 3); labels as in A. (C) Relationship between total exogenous lipid incorporated into CV membranes, as determined by quantitative analysis of isolated CV membranes, and the extent of fusion after standard mßcd treatment (red triangle). Labels as in A, except open diamond (Me-DOPE), open square (Me2-DOPE), and open circle (DOPC). (D) The amount of a given reagent incorporated into CV membranes required to fully recover fusion extent correlates linearly with the amount of negative curvature each species contributes. Labels as in A except crosses, which indicate half the minimal quantities of CHOL, DOPE, and DOG required for fusion recovery, to represent the quantity localized to the vesicle outer leaflet (blue fit y = 99.2x + 12.2, R2 = 0.92, red fit y = 195x + 25.5, R2 = 0.90). (E) Summary of CV fusion recovery after mßcd treatment and subsequent delivery of CHOL or curvature analogs in the quantities shown in D; DOPE, αT, and DOG fully support the ability to fuse, whereas DOPC, Me-DOPE, and Me2-DOPE do not, and DOPA further inhibits the extent of fusion. *(P < 0.01), **(P < 0.001) indicates difference from control and other conditions (n = 3–7).
(n = 3); labels as in A. (C) Relationship between total exogenous lipid incorporated into CV membranes, as determined by quantitative analysis of isolated CV membranes, and the extent of fusion after standard mßcd treatment (red triangle). Labels as in A, except open diamond (Me-DOPE), open square (Me2-DOPE), and open circle (DOPC). (D) The amount of a given reagent incorporated into CV membranes required to fully recover fusion extent correlates linearly with the amount of negative curvature each species contributes. Labels as in A except crosses, which indicate half the minimal quantities of CHOL, DOPE, and DOG required for fusion recovery, to represent the quantity localized to the vesicle outer leaflet (blue fit y = 99.2x + 12.2, R2 = 0.92, red fit y = 195x + 25.5, R2 = 0.90). (E) Summary of CV fusion recovery after mßcd treatment and subsequent delivery of CHOL or curvature analogs in the quantities shown in D; DOPE, αT, and DOG fully support the ability to fuse, whereas DOPC, Me-DOPE, and Me2-DOPE do not, and DOPA further inhibits the extent of fusion. *(P < 0.01), **(P < 0.001) indicates difference from control and other conditions (n = 3–7).
Two-sample two-tailed t-tests were used to determine differences (P < 0.05, unless stated otherwise) of fusion parameters between experimental conditions and parallel, internal controls. In all calculations, statistical error was propagated using standard methods. All data are reported as mean ± SE.
RESULTS
We first asked the question whether CHOL is generally enriched in areas of the membrane in which it would contribute to the exocytotic fusion mechanism (e.g., areas of CV-PM contact). CHOL-B, a probe selective for membrane regions enriched in CHOL (37,38), was used to label cortex preparations made from oocytes in which the PM had been prelabeled with the general membrane stain DiI (Fig. 2 A). Confocal fluorescence microscopy of these dual-labeled cortices consistently revealed that, in the absence of vesicles, only the PM is labeled and there is no correlation between the DiI and the CHOL-B (Fig. 2, A and C, see region in upper left of cortex versus CV-rich region in lower right of cortex). However, PM regions containing CV show a consistent correlation between DiI and CHOL-B; this correspondence in labeling is neither homogeneous nor uniform but is clearly localized to the region of the PM with docked CV (Fig. 2, B–E). The apparent heterogeneity may be a reflection of the labeling mechanism, although previous analyses do associate CHOL with the fusion machinery, which would be consistent with its nonuniform distribution across the population of CV (25). Line scans suggest that the CHOL-enriched regions labeled by CHOL-B are localized within and above the plane of the PM (Fig. 2 F). Together, these results indicate that the bulk of CHOL is associated with CV and/or the sites of vesicle attachment to the PM. Additionally, upon Ca2+-triggering, the CHOL-B label underwent rapid dispersion into the PM, consistent with exocytotic fusion and the subsequent appearance of fusion “domes” (that verify vectorial discharge of CV content across the PM (39)) as seen in the parallel bright field images (Supplementary Material, Movie S1).
FIGURE 2.

Confocal analysis of the relative CHOL distribution between the PM and CV of S. purpuratus egg cortices. (A) DiI labeling was carried out on intact oocytes before preparation of cortices, to specifically label the plasma membrane. (B) CV-PM cortex subsequent to labeling with CHOL-B to indicate regions enriched in CHOL. (C) Bright field image of the cortex preparation shown in A and B. (D) Overlay of A and B indicates only partial correlation of DiI label in the PM with the heterogeneous CHOL-B label. (E) Overlay of A, B, and C indicates that CHOL-B labeling correlates primarily with CV-rich regions; areas of the PM denuded of CV have no detectable CHOL-B label. (F) Line traces as indicated in E, reconstructed from a Z stack of A and B, indicate that CHOL-B-enriched regions are localized within and above the plane of the PM, consistent with the position of docked CV. Scale bar in A is 20 μm. Representative of 10 cortices from four separate preparations.
To couple quantitative functional and molecular assays as tightly as possible, as is required to effectively dissect molecular mechanisms, the role of CHOL and negative curvature was further probed using isolated CV that retain the minimal molecular machinery for intermembrane attachment, Ca2+ sensing, and fusion (25,26,28,29,40). Isolated, free floating CV were treated with 2 mM mßcd to selectively remove membrane CHOL (25,29). Relative to native, untreated CV, mßcd treatment resulted in a 31.4 ± 5.9% reduction in CV membrane CHOL. CHOL depletion correlated with a 26.0 ± 5.0% (n = 5) reduction in fusion extent (Fig. 3 E) and a rightward shift in Ca2+ sensitivity of 65.9 ± 15.0 μM [Ca2+]free (n = 5; Fig. 3 A), consistent with previous observations (25,29). Similar inhibitory effects were also observed for fusion kinetics (Fig. 3 B). Delivery of exogenous CHOL with hpßcd restored membrane CHOL to that of untreated CV, and the extent, Ca2+ sensitivity, and kinetics of fusion were recovered to that of control (Fig. 3, A, B, and D). In contrast, incorporation of the negative curvature analogs DOG, DOPE, or αT to CHOL-depleted CV selectively recovered the ability to fuse, but not the Ca2+ sensitivity or kinetics of fusion (Fig. 3, A and B). Similar to CHOL (25), supplementation of exogenous DOG, αT, and DOPE into native CV did not affect any fusion parameters (data not shown). As a structural analog of CHOL, CHOL-B was also delivered to the CV membrane after CHOL depletion. Although the spontaneous curvature of cholesteryl esters has not been determined experimentally, esterification of the 3′ hydroxyl group is structurally consistent with substantially decreased negative curvature (e.g., more characteristic of a neutral or even positive curvature molecule), particularly when the conjugate includes a heterocyclic fluorophore. Neither CHOL-esters (data not shown) nor CHOL-B (Fig. 3 A) were able to rescue any parameters of fusion, despite quite effective incorporation into the CV membrane (verified by fluorometry).
In a single native CV, an average of 54.5 ± 5.2 amol CHOL provide −2.20 ± 0.30 units of specific curvature (defined herein as the amol lipid per CV × spontaneous curvature, 1/R0p, Å−1). After CHOL depletion using a standardized treatment with 2 mM mßcd (25,29), the specific curvature contributed by CHOL was −1.51 ± 0.29 amol · Å−1 per CV, leaving a negative curvature deficit of 0.69 ± 0.24 amol · Å−1. Rescue of fusion by enrichment of CHOL-depleted vesicles with DOPE and DOG correlated quantitatively with the respective negative curvature contributions of these lipids rather than the actual molar incorporation (Table 1; Fig. 3, D and E), and was equivalent to the specific curvature introduced by CHOL itself for the full rescue of fusion, −0.79 ± 0.15 amol · Å−1 (P > 0.5). With DOG having the highest measured negative curvature (23), >2-fold higher than CHOL, proportionally less DOG as CHOL was required to effectively recover the extent of fusion (Fig. 3, C–E). A strong linear correlation exists between the minimal quantity of each of CHOL, DOPE, and DOG required to fully rescue the fusion extent and the respective measured spontaneous curvature of these molecules (Fig. 3 D, R2 = 0.90). Relative to CHOL, DOG, and DOPE, the required curvature contributed by αT to fully recover the fusion extent is ∼2-fold less (Fig. 3 D). This is consistent with observations that αT has effectively no transbilayer mobility over the timescale of several hours (41); thus whereas CHOL, DOG, and DOPE can rapidly equilibrate between the inner and outer leaflets of the vesicle membrane (37,42–44), αT remains almost exclusively in the outer leaflet. Since CHOL, DOG, and DOPE will effectively reach an equilibrium distribution within the time course of a single experiment (∼1 h), the resulting curvature contribution in the outer monolayer will be approximately half of the total curvature contribution (∼−0.40 ± 0.08 amol · Å−1 for CHOL). Taking transbilayer equilibration into account, there exists a strong linear correlation (coefficient of determination R2 = 0.92) between the spontaneous negative curvature of CHOL, DOPE, αT, DOG (Table 1), and the minimal quantity of each molecule incorporated into the outer monolayer of the vesicle to effect full recovery of the ability to fuse (e.g., extent, Fig. 3, C and D). Additionally, rescue of fusion with CHOL, αT, and DOG was dose-dependent, and resulted in a graded recovery of fusion extent (Fig. 3 C), confirming a relationship between the negative curvature contribution and the extent of fusion. As indicated (Table 1), the spontaneous curvature measurements of DOPE were available in both water (18–22) and high salt buffer (150 mM KCl, pH 7) (20), whereas the remaining lipids (DOG, αT, and CHOL) were each measured in water only (17,18,21,23). As DOPE is the only selected lipid of zwitterionic character (with DOG, αT, and CHOL being neutral) it is likely the only lipid that would show significant variation in the presence of aqueous ions. The measurements made in high salt buffer more appropriately approximate the aqueous environment surrounding the CV in these experiments, and as such we have used these measurements in all subsequent calculations; however, if we assume the curvature to be that measured in water (−0.035 ± 0.001 Å−1), the specific curvature contribution after DOPE recovery was still −0.51 ± 0.06 amol · Å−1.
Treatment of CV with 25.3 μM of the CHOL binding polyene antibiotic filipin, to sequester CHOL in the membrane rather than remove it (25), resulted in a selective 30.4 ± 1.8% inhibition of the extent (n = 4, P < 1 × 10−5), but not Ca2+ sensitivity of fusion (Fig. 4, A–C). This inhibition was fully reversed after delivery of 1.22 ± 0.23 amol/CV of αT. Subsequent quantitative analysis showed that the graded recovery of fusion extent correlated linearly with the amount of αT delivered to the membrane in a manner consistent with the previous results (Figs. 3 C and 4 C). The total amount of αT required to fully recover the extent of fusion was ∼3.6-fold less than was required to rescue inhibition after a comparable mßcd treatment (Fig. 3). To examine the specificity of the recovery, an identical experiment was carried out using C9NBD-T; this molecule is structurally related to αT but with a fluorophore incorporated such that the structural conformation is consistent with a substantially reduced negative curvature. C9NBD-T was unable to recover the extent of fusion, despite extensive incorporation (13.4 ± 0.5 amol/CV) in the membrane, as confirmed by washing away of excess reagent followed by fluorometric assessment (Fig. 4 A, inset).
FIGURE 4.

Inhibition of CV-CV fusion extent by filipin treatment was reversed by the addition of molecules of high negative curvature. (A) Ca2+ activity curve of CV treated with 25.3 μM filipin, or sequentially with filipin and 50, 100, or 200 μM αT or C9NBD-T (n = 4). Fluorescence measurements (inset) determined incorporation of 13.4 ± 0.5 amol/CV of C9NBD-T. (B) Kinetics of CV-CV fusion in response to 144 ± 16 μM 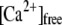 (n = 4). C. Delivery of αT to filipin-treated CV restores fusion capacity in a dose-dependent manner, correlating with the amount of αT incorporated into the membrane (P < 0.01, n = 4).
(n = 4). C. Delivery of αT to filipin-treated CV restores fusion capacity in a dose-dependent manner, correlating with the amount of αT incorporated into the membrane (P < 0.01, n = 4).
Previous studies have detailed the potent inhibitory effects on fusion of adding high positive curvature molecules, such as LPC, to secretory vesicles (24,45), including addition of LPC to CHOL-depleted CV (25). At molar quantities comparable to CHOL, delivery of DOPC (20.8 ± 7.1 amol/CV), a lipid with effectively neutral curvature (23), to CHOL-depleted vesicles, had no effect on any of the fusion parameters (n = 4, Fig. 3 C). Similarly, additions of 24.5 ± 3.6 and 22.3 ± 4.0 amol/CV of Me-DOPE and Me2-DOPE, respectively, two derivatives of DOPE with spontaneous curvature predicted to be intermediate to DOPE and DOPC (46), also failed to recover the extent of fusion (n = 4, Fig. 3 C). Based on an estimated spontaneous curvature of −0.0313 Å−1, Me-DOPE incorporation amounts to −0.767 ± 0.113 amol · Å−1, equivalent to the curvature deficit after CHOL depletion (Fig. 3). This suggests that total negative curvature alone is insufficient to enable fusion, but rather that only molecules of a specific negative spontaneous curvature can support the molecular rearrangements leading to pore formation.
Similarly, and in contrast to the effects of DOG, DOPE, and αT, addition of another negative curvature analog, DOPA, was also unable to recover the ability of CHOL-depleted CV to undergo Ca2+-triggered fusion (Fig. 5 A). Treatment of native CV with DOPA also resulted in a potent inhibition of the extent and Ca2+ sensitivity of fusion, with a corresponding decrease in kinetics (Fig. 5 A). The measured spontaneous curvature of DOPA is −0.022 Å−1 (20). Thus delivery of 6.4 ± 0.9 amol of DOPA per CV correlates to a specific curvature contribution of −0.145 ± 0.020 amol · Å −1, which, although less net curvature than required for complete recovery of fusion extent after CHOL depletion, is comparable to the amounts of CHOL, αT, and DOG delivered to CV to effect partial recovery (Fig. 3 C). As with DOPE, the spontaneous curvature of DOPA becomes more negative in the presence of aqueous salts (Table 1). The spontaneous curvature of DOPA in water is −0.0077 Å−1 (20), similar to that of DOPC. Additionally, enzymatic generation of endogenous PA in CV membranes via PLD activity showed comparable inhibitory effects on fusion extent (Fig. 5 B); however, effects on Ca2+ sensitivity and kinetics were less substantial than with exogenous DOPA (Fig. 5 B, inset). Quantitative analysis of PLD-treated CV membranes indicated that endogenously generated PA was required at levels substantially less than the exogenous DOPA to yield comparable inhibitory effects. This likely reflects the localized production of a particular range of PA species, as opposed to exogenous supplementation with only DOPA.
FIGURE 5.

Phosphatidic acid inhibits fusion. Treatment of CHOL-depleted CV with the curvature analog DOPA did not recover but rather further inhibited the extent of CV-CV fusion (purple). Similarly, supplementing native CV with DOPA (green) inhibited fusion to approximately the same extent as did depletion of CHOL. (A) Ca2+ activity curve of CV treated with 200 μM DOPA, 2 mM mßcd or sequentially with mßcd and DOPA (n = 4). (Inset) Kinetics of CV-CV fusion in response to 236 ± 34 μM 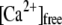 (n = 4). In a comparable fashion, the generation of endogenous PA in native CV also inhibits the extent, Ca2+ sensitivity, and kinetics of CV-CV fusion. (B) Ca2+ activity curve of CV treated with increasing concentrations of exogenous PLD as indicated. (Inset) Kinetics of CV-CV fusion in response to 91 ± 15 μM
(n = 4). In a comparable fashion, the generation of endogenous PA in native CV also inhibits the extent, Ca2+ sensitivity, and kinetics of CV-CV fusion. (B) Ca2+ activity curve of CV treated with increasing concentrations of exogenous PLD as indicated. (Inset) Kinetics of CV-CV fusion in response to 91 ± 15 μM  (n = 5).
(n = 5).
DISCUSSION
The ability of vesicles to undergo Ca2+-triggered fusion is dependent on the localized presence of CHOL as a molecule of high negative spontaneous curvature that supports the formation and transition of transient fusion intermediates (25,47,48). Imaging confirms that CHOL is predominantly localized to CV-enriched regions of the PM. The fundamental ability of CHOL-depleted CV to fuse, as assessed by the total extent of fusion, is rescued by the quantitative recovery of membrane negative curvature using specific, structurally dissimilar lipidic curvature analogs, each with a spontaneous negative curvature (1/R0p) equivalent to or more negative than CHOL.
Specific effects of negative curvature lipids
All Ca2+-activity curves, including the fusion of CHOL-depleted and lipid-recovered samples, were translationally invariant. Thus, despite the different treatments used, such conservation of the underlying curve-shape parameters of each of these Ca2+-activity curves is minimally consistent with a single common fusion mechanism (25,26,29,31). Nonetheless, rescue of fusion capacity with DOPE, αT, and DOG does not correlate with rescue of Ca2+ sensitivity or kinetics of fusion, indicating that these parameters can be effectively separated experimentally (25,29). Although the ability of vesicles to undergo fusion is dependent in part on the focal curvature of the membrane, the efficiency (Ca2+ sensitivity and kinetics) of fusion is defined by membrane components associated with critical CHOL levels, most probably via organization of proteins and lipids within CHOL and sphingomyelin-enriched microdomains (25,29). The activity or interactions of various protein components of the fusion machinery may be differentially modulated by CHOL levels in CV, either through direct interaction with CHOL or through general changes in the lipid environment. Such effects of CHOL on the activities of different membrane proteins are quite well documented (49–53). Thus it is not inconceivable that the localization or sensitivity of Ca2+-sensing molecules may be differently affected by local alterations in CHOL density relative to the effects of these alterations on the protein machinery that promotes rapid fusion kinetics.
Lipidic curvature analogs, although likely able to associate with CHOL-rich microdomains, do not actively contribute to the formation or stability of such microdomains (54,55). As DOPE-, αT-, and DOG-recovered CV are able to undergo the native Ca2+-triggered fusion reaction without stabilizing microdomains or reorganizing efficiency factors at the fusion site, these results are consistent with the concept of a conserved minimal fusion machine of lower inherent Ca2+ sensitivity, with the associated protein and lipid components modulating the efficiency of triggering, pore formation, and pore expansion (25,28,29,40). This selective ability of specific negative curvature molecules to recover the inherent ability to fuse demonstrates that negative curvature itself is one necessary component of the fast native Ca2+-triggered fusion mechanism. This quantitative finding appears most consistent with the stalk-pore model as a mechanism for native membrane fusion, with negative curvature components contributing to initial membrane dimpling or formation of point-like protrusion (7) and the formation and expansion of the hemifusion diaphragm (3,5,6,56–58).
Negative curvature acts only in the contacting monolayers
The formation of a stalk intermediate occurs exclusively between the outer monolayer of a single CV and the cytoplasmic leaflet of the PM (in CV-PM fusion) or between the outer monolayers of two adjacent CV (in homotypic CV-CV fusion). As αT has effectively no transbilayer mobility (41) within the timescale of a single experiment (∼1 h), the finding that the curvature contributed by αT to fully recover fusion is approximately half that required of CHOL, DOG, or DOPE demonstrates that the negative curvature contribution is via the outer monolayer of the CV, again consistent with the stalk-pore hypothesis. Thus negative curvature is required specifically for the formation of a stalk intermediate between the contacting monolayers of two adjacent CV. This is consistent with observations that high positive curvature molecules such as LPC reversibly arrest fusion at a stage that follows Ca2+ triggering but before monolayer merger (24,45). After sequestration of CHOL by filipin treatment, αT is required at doses even lower than those for recovery of fusion after CHOL depletion with mßcd. Clearly, whereas both mßcd and filipin functionally “remove” CHOL from the membrane, the filipin treatment requires less curvature contribution for recovery, since this has only to effect rescue of the ability to fuse; this is consistent with suboptimal doses of CHOL effecting only partial rescue of the ability to fuse, with the full native complement of CHOL required to completely rescue both fusion and efficiency (25). In effect, this is most likely due to differences in the modes of action of mßcd and filipin, in particular the fact that only mßcd is highly disruptive to CHOL enriched microdomains (25,59).
PA is unlikely to act directly in the fusion step of exocytosis
The role of PA in fusion via PLD activity has been discussed both indirectly in vesicle trafficking and exocytosis (60) and directly in the aggregation and fusion of liposomes (61). As the incorporation of exogenous DOPA into CV membranes, or the formation of endogenous PA did not enhance the Ca2+ sensitivity, and potently inhibited fusion kinetics (Fig. 5), it appears unlikely that intermembrane Ca2+ binding to PA headgroups plays any significant role in the native fusion mechanism. Indeed our results suggest that the positive role PA may play in the native exocytotic pathway is not through a direct role in Ca2+ triggered membrane merger, but likely upstream in vesicle attachment and kinase-related modulatory functions (62); in terms of fusion, PA may thus actually function as a local negative regulator. It is not clear if the transbilayer distribution of PA might have differential effects; given the relatively slow transbilayer mobility of PA the treatments used in this study (e.g., exogenous PLD or DOPA) are likely only to affect PA levels in the outer leaflet of the CV membrane. Previous studies implicating PA in the fusion process have typically involved manipulations of endogenous PLD, and as such may have a more symmetric effect on the distribution of PA (63,64). As with Me-DOPE, Me2-DOPE, and DOPC, the spontaneous negative curvature of DOPA is substantially less than that of CHOL, consistent with the finding that lipids with spontaneous curvature equivalent to or more negative than CHOL are able to substitute for CHOL in the fusion process, whereas lipids of lesser negative curvature cannot. PA may also associate with different regions of the CV membrane due to its inherent charge, whereas CHOL, DOPE, αT, and DOG are able to spatially and/or functionally associate with the docking and calcium-sensing machinery. The observation that exogenous PLD potently inhibits fusion extent at lower membrane concentrations of PA, relative to the delivery of exogenous DOPA, implies that endogenous PA may be generated at specific sites, or from specific molecular species that associate with different regions of the CV membrane than does DOPA. Additionally, as PLD treatments less potently inhibit the Ca2+ sensitivity and kinetics of fusion, relative to exogenous DOPA, PA may directly and potently inhibit the formation of high curvature fusion intermediates, whereas the inhibitory effects on efficiency may be related to less specific charge effects. As an anionic lipid, PA can create defects in lipid packing due to lateral phase separation driven by the binding of divalent ions; this would also result in the aggregation of proteins with a high affinity for PA (65).
Membrane lipids define the initial stalk formation
Of the lipids examined in this study, three have measured spontaneous curvature that is equivalent to or more negative than CHOL (DOPE, αT, and DOG) whereas six have measured or predicted spontaneous curvature that is less negative than CHOL (DOPC, Me-DOPE, Me2-DOPE, DOPA, CHOL-B, and C9NBD-T). The functional response of CHOL-depleted CV to the delivery of each of these lipid species parallels this division: whereas a lesser quantity of a lipid having greater spontaneous negative curvature than CHOL (αT or DOG), or a similar amount of an equivalent curvature molecule (DOPE), both rescue fusion (Fig. 3), a lipid of less spontaneous negative curvature is unable to substitute for CHOL in the fusion process despite providing the same net negative curvature contribution (Figs. 3 C and 5). This implies not only that a total defined quantity of membrane negative curvature is required to enable fusion, but also that the components that contribute the curvature must have a specific, critical spontaneous curvature value to effectively support fusion pore formation.
Extending these observations, including the enrichment of CHOL in CV and at docking/fusion sites on the PM (Fig. 2), we can predict that the curvature provided by CHOL is required to support the energetically favorable formation of a high curvature stalk intermediate. As the monolayer spontaneous curvature is the weighted average of the spontaneous curvature of all of the membrane components forming the stalk, the local lipid composition will define the specific size of the initial stalk. If, in addition to CHOL, the local membrane is also enriched in other negative curvature molecules such as αT or DOG, the total curvature of the stalk intermediate would be less still, and thus more favorable for rapid pore formation (12,13).
Cholesterol at the site of fusion
The data presented here quantitatively extend our understanding of the fusion process. Our previous studies indicate that the fusion site is organized by microdomains enriched in CHOL and sphingomyelin (25,29), enabling spatial coordination of protein and lipid components essential to membrane deformation/bending, Ca2+-sensing, triggering, and modulation. In light of these findings, we hypothesize that the lipidic fusion site (the site of initial formation of point-like protrusion (7) and subsequent stalk formation) is enriched in CHOL and other specific negative curvature components (e.g., phosphatidylethanolamine, diaglycerol, αT). We further suggest that this lipidic fusion site may exist in one of three possible states. First, the fusion site may exist in a liquid-disordered state capable of rapidly forming high curvature structures (66) as a region of varied fluidity within a single intact microdomain. Second, the fusion site may be organized as a discrete (liquid-disordered) lipid phase bounded by multiple liquid-ordered microdomains (67). Finally, the fusion site may exist as a CHOL-rich, liquid-ordered domain in resting CV, which rapidly transitions to a liquid-disordered phase as a result of the physiological triggering event, likely mediated by protein conformational changes upon Ca2+-activation (67). The challenge is now to experimentally test these possibilities.
Although the necessary negative curvature is largely contributed by CHOL in CV membranes, these findings imply that different fusion sites, vesicles, or secretory cell types could use other lipidic components, in addition to CHOL or similar endogenous sterols, to provide optimal local negative curvature to initiate and modulate the fusion process. CHOL is also known to promote the membrane active effects of such negative curvature lipids (47). An optimized lipid composition may serve to further reduce energy constraints arising from the molecular reorganization required for stalk formation and subsequent pore opening and expansion (1,68,69). As an essential component of the native fusion mechanism, effects on membrane negative curvature must be assessed in any manipulations used to probe the native fusion mechanism (70).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
J.R.C. acknowledges support of the Natural Sciences and Engineering Research Council of Canada, the Canadian Institutes of Health Research, the Alberta Heritage Foundation for Medical Research, and the Heart and Stroke Foundation of Canada. M.A.C. is the recipient of a Postgraduate Scholarship Award from the Natural Sciences and Engineering Research Council of Canada.
The authors thank Dr. G. Zamponi for kindly providing access to the confocal microscope; F. Rhemtulla, R. H. Butt, and K. Furber for helpful discussions; and D. Bininda and C. Skolseg for assistance with aquatics.
Abbreviations used: CHOL, cholesterol; DOPE, dioleoylphosphatidylethanolamine; αT, α-tocopherol; DOG, dioleoylglycerol; DOPC, dioleoylphosphatidylcholine; DOPA, dioleoylphosphatidic acid; Me-DOPE, N-methyl dioleoylphosphatidylethanolamine; Me2-DOPE, N,N-dimethyl dioleoylphosphatidylethanolamine; CV, cortical vesicles; CHOL-B, cholesteryl BODIPY FL-C12; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; PLD, phospholipase D; PM, plasma membrane; HPTLC, high-performance thin layer chromatography; BIM, baseline intracellular media; DMSO, dimethylsulfoxide; C9NBD-T, C9-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-tocopherol; mßcd, methyl-β-cyclodextrin; hpßcd, hydroxypropyl-β-cyclodextrin; LPC, lysophosphatidylcholine; PA, phosphatidic acid.
Editor: Joshua Zimmerberg.
References
- 1.Arac, D., X. Chen, H. A. Khant, J. Ubach, S. J. Ludtke, M. Kikkawa, A. E. Johnson, W. Chiu, T. C. Sudhof, and J. Rizo. 2006. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 13:209–217. [DOI] [PubMed] [Google Scholar]
- 2.Chen, X., D. Arac, T. M. Wang, C. J. Gilpin, J. Zimmerberg, and J. Rizo. 2006. SNARE-mediated lipid mixing depends on the physical state of the vesicles. Biophys. J. 90:2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozlovsky, Y., L. V. Chernomordik, and M. M. Kozlov. 2002. Lipid intermediates in membrane fusion: formation, structure, and decay of hemifusion diaphragm. Biophys. J. 83:2634–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel, D. P. 1999. The modified stalk mechanism of lamellar/inverted phase transitions and its implications for membrane fusion. Biophys. J. 76:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozlov, M. M., and V. S. Markin. 1983. Biofizika. 28:242–247. Possible mechanism of membrane fusion. [PubMed] [Google Scholar]
- 6.Chernomordik, L., M. M. Kozlov, and J. Zimmerberg. 1995. Lipids in biological membrane fusion. J. Membr. Biol. 146:1–14. [DOI] [PubMed] [Google Scholar]
- 7.Efrat, A., L. V. Chernomordik, and M. M. Kozlov. 2007. Point-like protrusion as a prestalk intermediate in membrane fusion pathway. Biophys. J. 92:L61–L63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozlovsky, Y., and M. M. Kozlov. 2002. Stalk model of membrane fusion: solution of energy crisis. Biophys. J. 82:882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markin, V. S., M. M. Kozlov, and V. L. Borovjagin. 1984. On the theory of membrane fusion. The stalk mechanism. Gen. Physiol. Biophys. 3:361–377. [PubMed] [Google Scholar]
- 10.Chernomordik, L. V., M. M. Kozlov, G. B. Melikyan, I. G. Abidor, V. S. Markin, and Y. Chizmadzhev. 1985. The shape of lipid molecules and monolayer membrane fusion. Biochimica et Biophysica Acta. 812:643–655. [Google Scholar]
- 11.Leikin, S. L., M. M. Kozlov, L. V. Chernomordik, V. S. Markin, and Y. A. Chizmadzhev. 1987. Membrane fusion: overcoming of the hydration barrier and local restructuring. J. Theor. Biol. 129:411–425. [DOI] [PubMed] [Google Scholar]
- 12.Kozlov, M. M., S. L. Leikin, L. V. Chernomordik, V. S. Markin, and Y. A. Chizmadzhev. 1989. Stalk mechanism of vesicle fusion. Intermixing of aqueous contents. Eur. Biophys. J. 17:121–129. [DOI] [PubMed] [Google Scholar]
- 13.Siegel, D. P. 1993. Energetics of intermediates in membrane fusion: comparison of stalk and inverted micellar intermediate mechanisms. Biophys. J. 65:2124–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markin, V. S., and J. P. Albanesi. 2002. Membrane fusion: stalk model revisited. Biophys. J. 82:693–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozlovsky, Y., A. Efrat, D. P. Siegel, and M. M. Kozlov. 2004. Stalk phase formation: effects of dehydration and saddle splay modulus. Biophys. J. 87:2508–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuzmin, P. I., J. Zimmerberg, Y. A. Chizmadzhev, and F. S. Cohen. 2001. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. USA. 98:7235–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradford, A., J. Atkinson, N. Fuller, and R. P. Rand. 2003. The effect of vitamin E on the structure of membrane lipid assemblies. J. Lipid Res. 44:1940–1945. [DOI] [PubMed] [Google Scholar]
- 18.Chen, Z., and R. P. Rand. 1997. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 73:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epand, R. M., N. Fuller, and R. P. Rand. 1996. Role of the position of unsaturation on the phase behavior and intrinsic curvature of phosphatidylethanolamines. Biophys. J. 71:1806–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kooijman, E. E., V. Chupin, N. L. Fuller, M. M. Kozlov, B. de Kruijff, K. N. J. Burger, and P. R. Rand. 2005. Spontaneous curvature of phosphatidic acid and lysophosphatidic acid. Biochemistry. 44:2097–2102. [DOI] [PubMed] [Google Scholar]
- 21.Leikin, S., M. M. Kozlov, N. L. Fuller, and R. P. Rand. 1996. Measured effects of diacylglycerol on structural and elastic properties of phospholipid membranes. Biophys. J. 71:2623–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rand, R. P., N. L. Fuller, S. M. Gruner, and V. A. Parsegian. 1990. Membrane curvature, lipid segregation, and structural transitions for phospholipids under dual-solvent stress. Biochemistry. 29:76–87. [DOI] [PubMed] [Google Scholar]
- 23.Szule, J. A., N. L. Fuller, and R. P. Rand. 2002. The effects of acyl chain length and saturation of diacylglycerols and phosphatidylcholines on membrane monolayer curvature. Biophys. J. 83:977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel, S. S., E. A. Leikina, and L. V. Chernomordik. 1993. Lysophosphatidylcholine reversibly arrests exocytosis and viral fusion at a stage between triggering and membrane merger. J. Biol. Chem. 268:25764–25768. [PubMed] [Google Scholar]
- 25.Churchward, M. A., T. Rogasevskaia, J. Hofgen, J. Bau, and J. R. Coorssen. 2005. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci. 118:4833–4848. [DOI] [PubMed] [Google Scholar]
- 26.Coorssen, J. R., P. S. Blank, M. Tahara, and J. Zimmerberg. 1998. Biochemical and functional studies of cortical vesicle fusion: the SNARE complex and Ca2+ sensitivity. J. Cell Biol. 143:1845–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmerberg, J., P. S. Blank, I. Kolosova, M. S. Cho, M. Tahara, and J. R. Coorssen. 2000. A stage-specific preparation to study the Ca(2+)-triggered fusion steps of exocytosis: rationale and perspectives. Biochimie. 82:303–314. [DOI] [PubMed] [Google Scholar]
- 28.Coorssen, J. R., P. S. Blank, F. Albertorio, L. Bezrukov, I. Kolosova, X. Chen, P. S. Backlund Jr., and J. Zimmerberg. 2003. Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell Sci. 116:2087–2097. [DOI] [PubMed] [Google Scholar]
- 29.Rogasevskaia, T., and J. R. Coorssen. 2006. Sphingomyelin-enriched microdomains define the efficiency of native Ca2+-triggered membrane fusion. J. Cell Sci. 119:2688–2694. [DOI] [PubMed] [Google Scholar]
- 30.Vogel, S. S., P. S. Blank, and J. Zimmerberg. 1996. Poisson-distributed active fusion complexes underlie the control of the rate and extent of exocytosis by calcium. J. Cell Biol. 134:329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blank, P. S., M. S. Cho, S. S. Vogel, D. Kaplan, A. Kang, J. Malley, and J. Zimmerberg. 1998. Submaximal responses in calcium-triggered exocytosis are explained by differences in the calcium sensitivity of individual secretory vesicles. J. Gen. Physiol. 112:559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blank, P. S., S. S. Vogel, J. D. Malley, and J. Zimmerberg. 2001. A kinetic analysis of calcium-triggered exocytosis. J. Gen. Physiol. 118:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abramoff, M. D., P. J. Magelhaes, and S. J. Ram. 2004. Image processing with ImageJ. Biophotonics Int. 11:36–42. [Google Scholar]
- 34.Hibbert, J. E., R. H. Butt, and J. R. Coorssen. 2006. Actin is not an essential component in the mechanism of calcium-triggered vesicle fusion. Int. J. Biochem. Cell Biol. 38:461–471. [DOI] [PubMed] [Google Scholar]
- 35.Bligh, E., and W. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917. [DOI] [PubMed] [Google Scholar]
- 36.Fowler, S. D., W. J. Brown, J. Warfel, and P. Greenspan. 1987. Use of Nile Red for the rapid in situ quantitation of lipids on thin-layer chromatograms. J. Lipid Res. 28:1225–1232. [PubMed] [Google Scholar]
- 37.Bai, J., and R. E. Pagano. 1997. Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry. 36:8840–8848. [DOI] [PubMed] [Google Scholar]
- 38.Reaven, E., L. Tsai, and S. Azhar. 1995. Cholesterol uptake by the ‘selective’ pathway of ovarian granulosa cells: early intracellular events. J. Lipid Res. 36:1602–1617. [PubMed] [Google Scholar]
- 39.Zimmerberg, J., and J. Liu. 1988. Ionic and permeability requirements for exocytosis in vitro in sea urchin eggs. J. Membr. Biol. 101:199–207. [DOI] [PubMed] [Google Scholar]
- 40.Szule, J. A., S. E. Jarvis, J. E. Hibbert, J. D. Spafford, J. E. Braun, G. W. Zamponi, G. M. Wessel, and J. R. Coorssen. 2003. Calcium-triggered membrane fusion proceeds independently of specific presynaptic proteins. J. Biol. Chem. 278:24251–24254. [DOI] [PubMed] [Google Scholar]
- 41.Tiurin, V. A., V. E. Kagan, E. A. Serbinova, N. V. Gorbunov, and A. N. Erin. 1986. The interaction of alpha-tocopherol with phospholipid liposomes: the absence of transbilayer mobility. Biull. Eksp. Biol. Med. 102:689–692. [PubMed] [Google Scholar]
- 42.Leventis, R., and J. R. Silvius. 2001. Use of cyclodextrins to monitor transbilayer movement and differential lipid affinities of cholesterol. Biophys. J. 81:2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganong, B. R., and R. M. Bell. 1984. Transmembrane movement of phosphatidylglycerol and diacylglycerol sulfhydryl analogues. Biochemistry. 23:4977–4983. [DOI] [PubMed] [Google Scholar]
- 44.Marx, U., G. Lassmann, H. G. Holzhutter, D. Wustner, P. Muller, A. Hohlig, J. Kubelt, and A. Herrmann. 2000. Rapid flip-flop of phospholipids in endoplasmic reticulum membranes studied by a stopped-flow approach. Biophys. J. 78:2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chernomordik, L. V., S. S. Vogel, A. Sokoloff, H. O. Onaran, E. A. Leikina, and J. Zimmerberg. 1993. Lysolipids reversibly inhibit Ca2+-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 318:71–76. [DOI] [PubMed] [Google Scholar]
- 46.Hamai, C., T. Yang, S. Kataoka, P. S. Cremer, and S. M. Musser. 2006. Effect of average phospholipid curvature on supported bilayer formation on glass by vesicle fusion. Biophys. J. 90:1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coorssen, J. R., and R. P. Rand. 1990. Effects of cholesterol on the structural transitions induced by diacylglycerol in phosphatidylcholine and phosphatidylethanolamine bilayer systems. Biochem. Cell Biol. 68:65–69. [DOI] [PubMed] [Google Scholar]
- 48.Vogel, S. S., L. V. Chernomordik, and J. Zimmerberg. 1992. Calcium-triggered fusion of exocytotic granules requires proteins in only one membrane. J. Biol. Chem. 267:25640–25643. [PubMed] [Google Scholar]
- 49.Barrantes, F. J. 2007. Cholesterol effects on nicotinic acetylcholine receptor. J. Neurochem. 103(Suppl. 1):72–80. [DOI] [PubMed] [Google Scholar]
- 50.Chang, T. Y., C. C. Chang, N. Ohgami, and Y. Yamauchi. 2006. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22:129–157. [DOI] [PubMed] [Google Scholar]
- 51.Epand, R. F., A. Thomas, R. Brasseur, S. A. Vishwanathan, E. Hunter, and R. M. Epand. 2006. Juxtamembrane protein segments that contribute to recruitment of cholesterol into domains. Biochemistry. 45:6105–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Epand, R. M. 2006. Cholesterol and the interaction of proteins with membrane domains. Prog. Lipid Res. 45:279–294. [DOI] [PubMed] [Google Scholar]
- 53.Soltani, C. E., E. M. Hotze, A. E. Johnson, and R. K. Tweten. 2007. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA. 104:20226–20231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samsonov, A. V., I. Mihalyov, and F. S. Cohen. 2001. Characterization of cholesterol-sphingomyelin domains and their dynamics in bilayer membranes. Biophys. J. 81:1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaikh, S. R., A. C. Dumaual, A. Castillo, D. LoCascio, R. A. Siddiqui, W. Stillwell, and S. R. Wassall. 2004. Oleic and docosahexaenoic acid differentially phase separate from lipid raft molecules: a comparative NMR, DSC, AFM, and detergent extraction study. Biophys. J. 87:1752–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chanturiya, A., L. V. Chernomordik, and J. Zimmerberg. 1997. Flickering fusion pores comparable with initial exocytotic pores occur in protein-free phospholipid bilayers. Proc. Natl. Acad. Sci. USA. 94:14423–14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chernomordik, L., A. Chanturiya, J. Green, and J. Zimmerberg. 1995. The hemifusion intermediate and its conversion to complete fusion: regulation by membrane composition. Biophys. J. 69:922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chernomordik, L. V., and J. Zimmerberg. 1995. Bending membranes to the task: structural intermediates in bilayer fusion. Curr. Opin. Struct. Biol. 5:541–547. [DOI] [PubMed] [Google Scholar]
- 59.Awasthi-Kalia, M., P. P. Schnetkamp, and J. P. Deans. 2001. Differential effects of filipin and methyl-beta-cyclodextrin on B cell receptor signaling. Biochem. Biophys. Res. Commun. 287:77–82. [DOI] [PubMed] [Google Scholar]
- 60.Roth, M. G., K. Bi, N. T. Ktistakis, and S. Yu. 1999. Phospholipase D as an effector for ADP-ribosylation factor in the regulation of vesicular traffic. Chem. Phys. Lipids. 98:141–152. [DOI] [PubMed] [Google Scholar]
- 61.Blackwood, R. A., J. E. Smolen, A. Transue, R. J. Hessler, D. M. Harsh, R. C. Brower, and S. French. 1997. Phospholipase D activity facilitates Ca2+-induced aggregation and fusion of complex liposomes. Am. J. Physiol. 272:C1279–C1285. [DOI] [PubMed] [Google Scholar]
- 62.Coorssen, J. R. 1996. Phospholipase activation and secretion: evidence that PLA2, PLC, and PLD are not essential to exocytosis. Am. J. Physiol. 270:C1153–C1163. [DOI] [PubMed] [Google Scholar]
- 63.Humeau, Y., N. Vitale, S. Chasserot-Golaz, J. L. Dupont, G. Du, M. A. Frohman, M. F. Bader, and B. Poulain. 2001. A role for phospholipase D1 in neurotransmitter release. Proc. Natl. Acad. Sci. USA. 98:15300–15305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zeniou-Meyer, M., N. Zabari, U. Ashery, S. Chasserot-Golaz, A. M. Haeberle, V. Demais, Y. Bailly, I. Gottfried, H. Nakanishi, A. M. Neiman, G. Du, M. A. Frohman, M. F. Bader, and N. Vitale. 2007. Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J. Biol. Chem. 282:21746–21757. [DOI] [PubMed] [Google Scholar]
- 65.Faraudo, J., and A. Travesset. 2007. Phosphatidic acid domains in membranes: effect of divalent counterions. Biophys. J. 92:2806–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tenchov, B. G., R. C. MacDonald, and D. P. Siegel. 2006. Cubic phases in phosphatidylcholine-cholesterol mixtures: cholesterol as membrane “fusogen”. Biophys. J. 91:2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.London, E. 2005. How principles of domain formation in model membranes may explain ambiguities concerning lipid raft formation in cells. Biochim. Biophys. Acta. 1746:203–220. [DOI] [PubMed] [Google Scholar]
- 68.Martens, S., M. M. Kozlov, and H. T. McMahon. 2007. How synaptotagmin promotes membrane fusion. Science. 316:1205–1208. [DOI] [PubMed] [Google Scholar]
- 69.Rizo, J., X. Chen, and D. Arac. 2006. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 16:339–350. [DOI] [PubMed] [Google Scholar]
- 70.Szule, J. A., and J. R. Coorssen. 2004. Comment on “Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis”. Science. 306:813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.