

Abstract
Bidirectional signaling between neurons and glial cells has been demonstrated in brain slices and is believed to mediate glial modulation of synaptic transmission in the CNS. Our laboratory has characterized similar neuron–glia signaling in the mammalian retina. We find that light-evoked neuronal activity elicits Ca2+ increases in Müller cells, which are specialized retinal glial cells. Neuron to glia signaling is likely mediated by the release of ATP from neurons and is potentiated by adenosine. Glia to neuron signaling has also been observed and is mediated by several mechanisms. Stimulation of glial cells can result in either facilitation or depression of synaptic transmission. Release of D-serine from Müller cells might also potentiate NMDA receptor transmission. Müller cells directly inhibit ganglion cells by releasing ATP, which, following hydrolysis to adenosine, activates neuronal A1 receptors. The existence of bidirectional signaling mechanisms indicates that glial cells participate in information processing in the retina.
Keywords: Calcium, astrocyte, Müller cell, glial cell, retina, ATP, adenosine, light response, ganglion cell
INTRODUCTION
In recent years, glial modulation of synaptic transmission and neuronal excitability has been recognized as an important function of glial cells (Volterra et al., 2002; Newman, 2003b). First demonstrated in astrocyte/neuron co-cultures (Parpura et al., 1994; Araque et al., 1998a; Araque et al., 1998b) and later in brain slices (Pasti et al., 1997; Kang et al., 1998), the release of transmitters from glial cells can have a significant influence on neuronal activity. Glial cells have also been shown to modulate neuronal function in the retina (Newman, 2004), which is a useful preparation for studying glia–neuron signaling.
Two types of macroglial cells are present in the retinae of most mammals; astrocytes and Müller cells (Fig. 1) (Newman, 2001a). Astrocytes are confined largely to the nerve fiber layer at the inner border of the retina. They interact primarily with the axons of ganglion cells and do not contact neuronal synapses. In contrast, Müller cells span the entire neural retina from the inner retinal border to the photoreceptor layer. The processes of Müllers cell ramify within the two synaptic layers of the retina, the inner and outer plexiform layers, and surround and sometimes contact neuronal synapses. Throughout most of the retina, Müller cells are the only macroglial cells present and they function as specialized astrocytes.
Fig. 1. Glial cells of the mammalian retina.
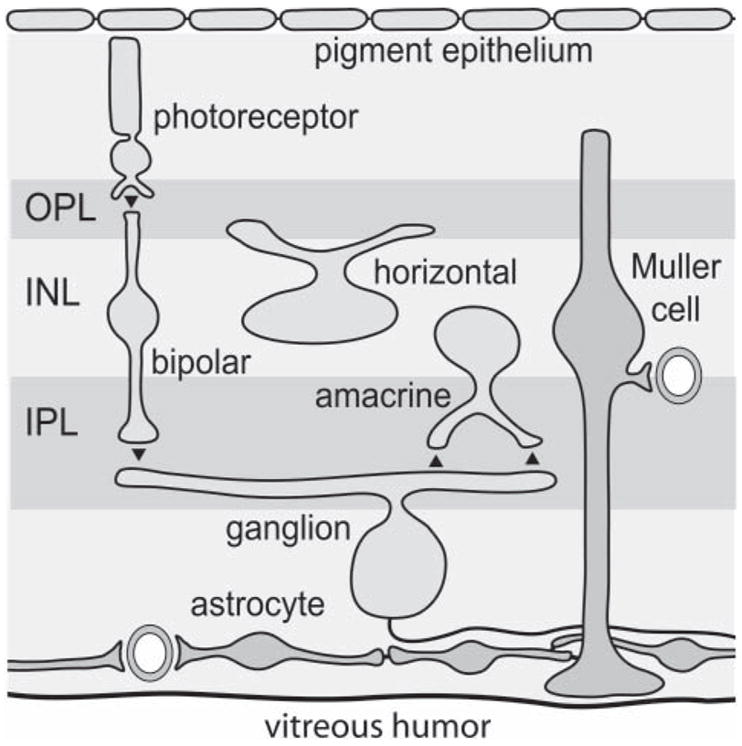
Astrocytes, ubiquitous CNS glial cells, are confined primarily to the nerve-fiber layer adjacent to the inner (vitreal) surface of the retina. Müller cells, which are specialized radial glial cells found only in the retina, extend from the vitreal surface to the photoreceptor layer. Müller cells, but not astrocytes, span the two synaptic layers of the retina, the inner plexiform layer (IPL) and the outer plexiform layer (OPL). Their somata lie in the inner nuclear layer (INL). Astrocytes are coupled to each other and to Müller cells by gap junctions. The major classes of retinal neurons are also illustrated. Synapses are indicated by filled triangles.
Glial cells can influence neuronal excitability by several mechanisms (Newman, 2005), including the uptake of glutamate and GABA following release at the synapse, and the regulation of extracellular K+ and pH. These processes are considered indirect mechanisms of neuronal modulation (Newman, 2005). Glial cells also modulate neuronal excitability directly, by releasing either transmitters or co-agonists onto neurons (Volterra et al., 2002). Similarly, neurons can modulate glial-cell function by the release of neurotransmitters (Schipke and Kettenmann, 2004). In this review, I focus on work from our laboratory that demonstrates direct communication between neurons and glial cells in the retina through the release of chemical messengers.
Neuron to glia signaling
Active neurons can evoke increases in Ca2+ in glial cells. In brain slices, electrical stimulation of neurons increases Ca2+ concentrations in neighboring astrocytes (Schipke and Kettenmann, 2004). This neuron to glia signaling is mediated by the release of transmitters from neurons. Release of glutamate (Dani et al., 1992; Porter and McCarthy, 1996; Pasti et al., 1997), GABA (Kang et al., 1998), acetylcholine (Araque et al., 2002) and ATP (Bowser and Khakh, 2004) can evoke Ca2+ increases by activating glial metabotropic receptors. The release of nitric oxide from neurons can also produce glial Ca2+ increases (Matyash et al., 2001).
We have investigated whether similar neuron to glia signaling occurs in the retina. Calcium levels in retinal Müller cells and astrocytes have been monitored with the Ca2+-indicator dye Fluo-4 when the retina is stimulated with light flashes. Characterizing neuron to glia signaling in the retina has the advantage over similar studies in brain slices in that a natural stimulus, light, rather than electrical stimuli, can be used to excite neurons.
Müller cell Ca2+ transients
Previously unpublished experiments from our laboratory demonstrate that transient increases in Ca2+ are generated in Müller cells (Fig. 2). These transients occur at low frequency (~10 transients per Müller cell per 1000 seconds) and do not propagate between cells. Flickering light stimulation increases the occurrence of these Ca2+ transients. Averaging the Ca2+ transients in Müller cells over many trials reveals that Ca2+ rises rapidly at the onset of a flickering stimulus and remains elevated for the duration of the stimulus. Ca2+ transients are not observed in astrocytes.
Fig. 2. Calcium transients in retinal glial cells.
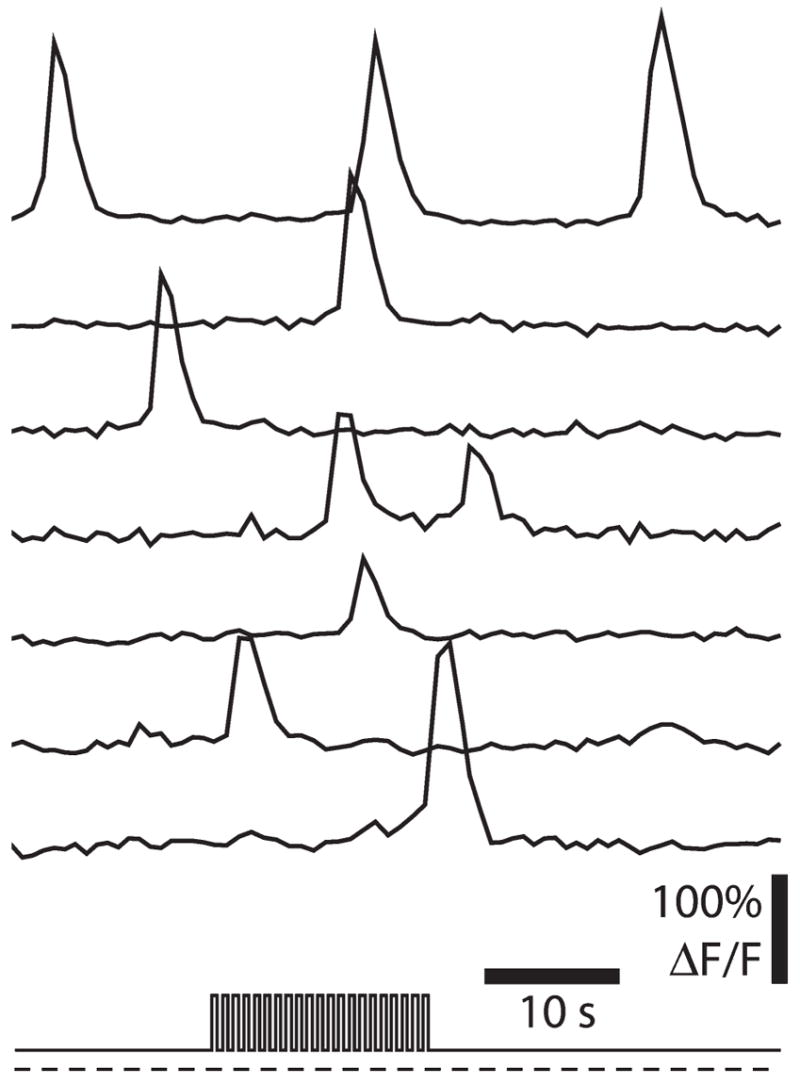
Calcium fluorescence is measured simultaneously in seven Müller cells at the vitreal surface of the retina. The light-stimulus protocol, a 16-second episode of flickering light superimposed on a dim background light, is illustrated at the bottom. Calcium transients are generated in each of the Müller cells. On average, Ca2+ transients occur at a higher rate during the flickering stimulus than during the dim background illumination. Unpublished results.
The light-evoked increase in Ca2+ transients in Müller cells is largely blocked by the purinergic receptor antagonist suramin, indicating that neuronal release of ATP is responsible for the increase in glial Ca2+. The light-evoked increase in Ca2+ transients is also blocked by tetrodotoxin, which indicates that neuron to glia signaling is mediated by neurons that generate action potentials. In the retina, only amacrine cells and ganglion cells generate action potentials and these two types of inner retinal neurons are likely to signal to glia. Cholinergic starburst amacrine cells, which are believed to co-release ATP, could mediate neuron to glia signaling. Ganglion cells might also contribute to light-evoked increases in Ca2+ in glial cells. Antidromic stimulation of ganglion cell axons evokes a substantial increase in Müller cell Ca2+ transients. This increase is also blocked by suramin, indicating that ganglion cells might release ATP in the retina.
Adenosine potentiation of Müller cell Ca2+ responses
Adenosine agonists [either 100 μM adenosine or 2 μM 1-(6-Amino-9H-purin-9-yl)-1-deoxy-N-ethyl-b-D-ribofura-nuronamide 5′-N-Ethylcarboxamidoadenosine (NECA)] potentiate light-evoked Müller cell Ca2+ increases. In the presence of an adenosine agonist, light ON elicits a large increase in Ca2+ in all Müller cells. Adenosine-potentiated increases are ~100-fold larger than the increase observed in the absence of an adenosine agonist. The adenosine-potentiated, light-evoked Ca2+ increase begins in Müller cell processes in the inner plexiform layer and spreads proximally into the endfeet of Müller cells at the inner surface of the retina.
In the presence of adenosine, addition of ATP evokes large increases in Müller cell Ca2+. The light-evoked Ca2+ increase is nearly abolished by suramin and by apyrase, an ectoenzyme that hydrolyzes ATP. By contrast, the light-evoked increase is not reduced substantially by antagonists of glutamate, GABA and acetylcholine receptors. In addition, the metabotropic glutamate agonist trans-ACPD does not evoke Müller cell Ca2+ increases. Together, these results indicate that neuron to glia signaling in the retina is mediated by the release of ATP from neurons.
Glia to neuron signaling
We have characterized glial modulation of neuronal activity in the retina in a number of studies. Initially, the effect of glial cells on light-evoked spike activity in ganglion cells was characterized. Subsequent studies have determined the mechanisms by which glial cells modulate neuronal activity.
Glial modulation of light-evoked activity in ganglion cells
Mechanical stimulation of single astrocytes evokes Ca2+ increases that spread up to 200 μm from the stimulated cell to neighboring astrocytes and Müller cells (Newman and Zahs, 1997). The effect of these activated glial cells on neurons has been assessed by recording the light-evoked spike activity of a nearby ganglion cell with an extracellular microelectrode (Newman and Zahs, 1998). We found that stimulated glial cells either enhance or depress light-evoked neuronal activity (Fig. 3). In some ganglion cells (9% of all cells recorded) (Fig. 3A) glial-cell stimulation increases cell spiking. In many more ganglion cells, however (47%) (Fig. 3B), glial-cell stimulation decreases cell spiking. Glial modulation of ganglion cell activity is blocked when glial cell Ca2+ increases are reduced with thapsigargin, suggesting that glia to neuron signaling depends on glial Ca2+ increases.
Fig. 3. Glial modulation of light-evoked spiking in rat ganglion cells.
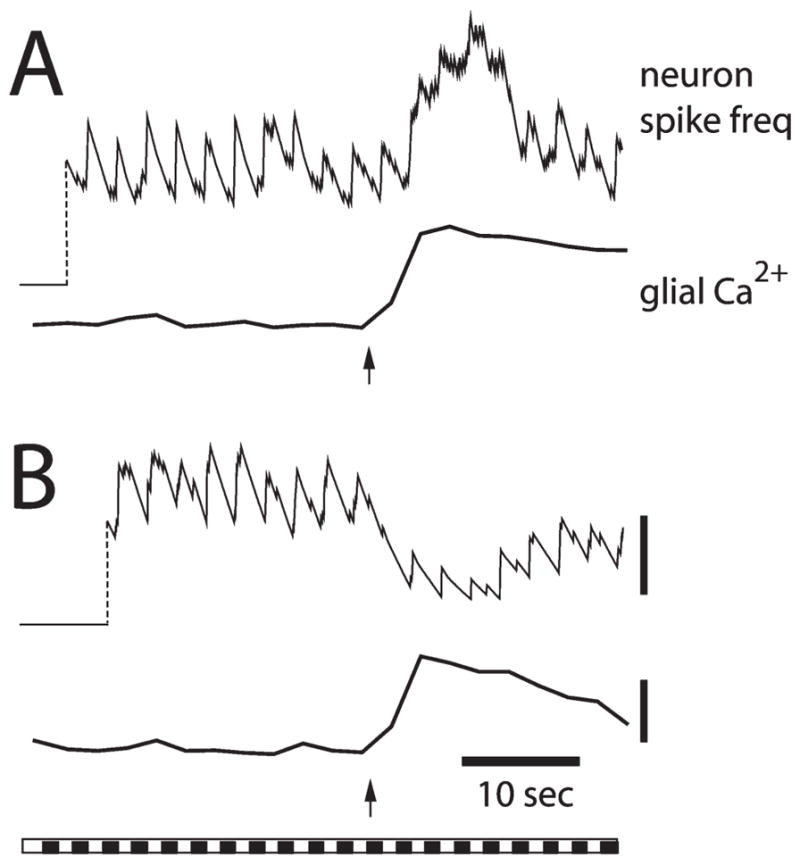
Facilitation of neuron spiking (A) and depression of spiking (B) are illustrated in recordings from two ganglion cells. A frequency plot of spike activity (top trace) and Ca2+ levels in glial cells adjacent to the neuron (bottom trace) are shown for each trial. Arrows indicate initiation of the glial Ca2+ wave. The bar at the bottom shows the repetitive light stimulus that evokes neuronal spiking. Calibration bars; 3 spikes second−1; 20% ΔF/F. Modified from Newman and Zahs (1998).
Mechanisms of glia to neuron signaling
The modulation of neuronal spiking by glial cells described above could be mediated by several mechanisms. Stimulated glial cells might release a transmitter that activates receptors on presynaptic terminals. For example, glial cells release glutamate (Parpura et al., 1994; Jeftinija et al., 1996; Bezzi et al., 1998; Innocenti et al., 2000) and ATP (Cotrina et al., 1998; Newman, 2001b). These gliotransmitters might activate pre-synaptic glutamate, ATP and adenosine metabotropic receptors, either facilitating or depressing synaptic transmission.
Gliotransmitter release could also modulate postsynaptic receptors on ganglion cells. The release of D-serine will facilitate NMDA synaptic transmission by binding to the NMDA co-agonist site (Wolosker et al., 1999; Miller, 2004). Release of glutamate, in contrast, might desensitize glutamatergic receptors, thus, reducing glutamate synaptic transmission.
Gliotransmitters might also directly modulate the excitability of ganglion cells. Release of ATP from glial cells could activate P2X receptors on ganglion cells (Taschenberger et al., 1999), depolarizing the cells. ATP, if converted to adenosine by ectoenzymes, might also activate A1 adenosine receptors (Braas et al., 1987) and hyperpolarize ganglion cells.
Our results indicate that retinal glial cells modulate ganglion cell activity by several of these mechanisms, as shown in the following experiments.
Presynaptic modulation of ganglion-cell activity
Previously unpublished experiments from our laboratory indicate that glial cells modulate synaptic transmission onto ganglion cells by presynaptic mechanisms. We stimulated the retina with brief light flashes repeated at 5-second intervals. These stimuli evoke excitatory post-synaptic potentials (EPSCs) in ganglion cells. ATPγS ejections, which elicit glial Ca2+ increases, result in a rise in EPSC amplitude in some ganglion cells (Fig. 4A, middle). It is possible that the ATPγS ejection used to stimulate glial cells facilitates synaptic transmission, not by stimulating glial cells, but by acting directly on presynaptic terminals. This seems unlikely, however, because activation of glial cells by mechanical stimulation produces a similar facilitation of synaptic transmission (Fig. 4A, bottom).
Fig. 4. Glial facilitation and depression of synaptic transmission.

Light flashes repeated at 5-second intervals evoke EPSCs in ganglion cells. (A) Facilitation of synaptic transmission. Sample ganglion cell EPSCs are shown at the top. EPSC amplitudes are plotted as a function of time in the middle and bottom. Stimulation of glial cells with either ATPγS (middle) or a mechanical stimulus (bottom) results in a transient increase in EPSC amplitude. The two trials are from the same ganglion cell. (B) Depression of synaptic transmission. Sample ganglion cell synaptic currents (an IPSC followed by an EPSC) are shown in the middle. The amplitudes of IPSCs (top) and EPSCs (bottom) are shown. Stimulation of glial cells with ATPγS results in a transient decrease in EPSC amplitude but little change in IPSC amplitude. Unpublished results.
In some ganglion cells, glial cell stimulation results in a decrease in the amplitude of light-evoked EPSCs. In the example illustrated in Fig. 4B, light flashes evoke a complex synaptic current composed of an initial inhibitory post-synaptic current (IPSC) followed by an EPSC. Glial stimulation reduces the EPSC but has little effect on the IPSC, indicating that glial modulation of synaptic transmission can be selective. Additional experiments to monitor changes in paired pulse facilitation indicate that glial modulation of synaptic transmission reflects a presynaptic mechanism.
Recordings from many ganglion cells reveal that glial modulation of synaptic transmission similar to that illustrated in Fig. 4 occurs relatively rarely. Only a small percentage of ganglion cells show either facilitation or depression of synaptic transmission. We believe that glial modulation of synaptic transmission by presynaptic mechanisms, although present, might play a relatively minor role in glial modulation of ganglion-cell activity.
Postsynaptic modulation of ganglion cell activity
Additional experiments demonstrate that robust glial modulation of ganglion-cell activity occurs by direct hyperpolarization of ganglion cells (Newman, 2003a). When glial Ca2+ increases are evoked by ATPγS ejection, a hyperpolarization, generated by a slow outward current, is elicited in neighboring ganglion cells (Fig. 5A). Of the ganglion cells monitored, 52% show moderate hyperpolarizations (<5 mV), 12% show large hyperpolarizations (>5 mV) and 36% show little or no response.
Fig. 5. Stimulation of glial cells inhibits ganglion cells.

(A) Ejection of ATPγS onto the retinal surface evokes a Ca2+ increase in glial cells and a hyperpolarization (current-clamp recording) and an outward current (voltage-clamp recording) in a neighboring ganglion cell (Newman, 2003a). (B) ATPγS stimulation of glial cells hyperpolarizes a ganglion cell and blocks spontaneous action potentials in the cell. Unpublished results.
The hyperpolarizing response is mediated by the stimulated glial cells rather than by a direct neuronal response to the ATPγS ejection (Newman, 2003a). Diverse stimuli, including ejection of ATP, dopamine, thrombin, lysophosphatidic acid and direct mechanical stimulation, evoke glial Ca2+ increases. Each stimulus also elicits ganglion cell hyperpolarization.
Glial hyperpolarization of ganglion cells can produce substantial changes in neuronal activity. In the experiment illustrated in Fig. 5B, for example, a high rate of spontaneous spiking present in a ganglion cell is blocked completely when neighboring glial cells are stimulated with ATPγS.
Ganglion-cell hyperpolarization is mediated by the release of ATP from glial cells and the activation of adenosine A1 receptors on ganglion cells, as demonstrated by several types of experiments (Newman, 2003a). Glial-mediated hyperpolarization of ganglion cells is abolished by DPCPX, an A1 receptor antagonist (Fig. 6A). Ejection of adenosine onto ganglion cells mimics the response produced by glial-cell stimulation (Fig. 6B). Inhibition of ecto-ATPases and ectonucleotidases, both of which are needed to convert ATP to adenosine following ATP release from glial cells, substantially reduce the glial-mediated ganglion-cell response (Fig. 6C).
Fig. 6. Glial inhibition of ganglion cells is mediated by glial release of ATP and activation of neuronal adenosine receptors.

(A) Stimulation of glial cells with ATPγS evokes an inhibitory outward current in a ganglion cell (control), which is abolished by addition of DPCPX, an A1 adenosine receptor antagonist. (B) Adenosine ejection evokes a larger, shorter-latency current in a ganglion cell than ATPγS ejection at the same retinal location. (C) Addition of AOPCP, an ectonucleotidase inhibitor that blocks the conversion of AMP to adenosine, reduces and slows the time-course of outward ganglion cell current evoked by ATPγS ejection. The effect is largely reversible (Newman, 2003a).
We have used the luciferin–luciferase chemiluminescence assay to image ATP release following glial-cell stimulation. In the presence of Cd2+, which blocks transmitter release from neurons, mechanical stimulation of glial cells evokes an ATP release into the inner plexiform (synaptic) layer. This release is most likely from Müller cells. Indeed, selective stimulation of Müller cells and astrocytes demonstrates that stimulation of Müller cells, but not astrocytes, is both necessary and sufficient to elicit ganglion cell hyperpolarization (Newman, 2003a).
Activation of ganglion cell A1 receptors evokes cell hyperpolarization by opening Ba2+-sensitive K+ channels (Newman, 2003a). The reversal potential of the slow outward current evoked by glial cell stimulation is near the K+ equilibrium potential. The outward current is blocked by Ba2+, indicating that inwardly rectifying K+ channels mediate the response. Recent experiments in our laboratory demonstrate that the adenosine-evoked current is reduced substantially by tertiapin, which blocks G-protein-coupled inwardly rectifying K+ channels (GIRKs).
Together, these experiments demonstrate that retinal glial cells can inhibit ganglion cells by releasing ATP. The ATP is converted to adenosine by ectoenzymes, which stimulates ganglion cell A1 receptors. It is interesting to speculate that released ATP might also directly excite ganglion cells. Ganglion cells express P2X receptors (Taschenberger et al., 1999) and activation of these receptors would depolarize ganglion cells. Thus, retinal glial cells, by releasing ATP, might be able to either excite or inhibit ganglion cells, depending on the type of receptors expressed by the neurons.
D-serine release from Müller cells
D-serine released from glial cells is believed to potentiate NMDA receptor responses in the brain (Miller, 2004). D-serine binds to the co-agonist (glycine)-binding site on NMDA receptors (Wolosker et al., 1999) and might be the endogenous co-agonist in the CNS (Mothet et al., 2000; Schell et al., 1995).
Release of D-serine from Müller cells might also potentiate NMDA receptor transmission in the retina (Stevens et al., 2003). Both D-serine and serine racemase, the enzyme that synthesizes D-serine, are localized in Müller cells in the retina. In addition, D-serine potentiates NMDA responses in the retina. D-serine augments the NMDA component of light-evoked ganglion-cell responses and potentiates ganglion-cell responses to NMDA ejection (Fig. 7A). Addition of D-amino acid oxidase, which degrades D-serine, reduces both the NMDA component of light-evoked responses and ganglion-cell responses to NMDA ejection (Fig. 7B). The results demonstrate that endogenous D-serine, which is likely to be released from Müller cells, modulates NMDA synaptic transmission in the retina.
Fig. 7. Modulation of NMDA receptor transmission by D-serine in the retina.

(A) Addition of D-serine, an NMDA receptor co-agonist, potentiates the inward current recorded from a ganglion cell in response to NMDA ejection. (B) Addition of D-amino-acid-oxidase, an enzyme that degrades D-serine, reduces the inward current evoked by NMDA ejection (Stevens et al., 2003).
Glia to glia signaling
The findings outlined above demonstrate bidirectional signaling between neurons and glial cells in the retina. Neuronal activity evokes Ca2+ increases in Müller cells. Stimulated Müller cells, in turn, release transmitters that modulate synaptic transmission and neuronal excitability. These interactions are likely to take place within a confined area, perhaps within a few microns of the synapse. Release of transmitter from the presynaptic terminal will evoke a Ca2+ increase within glial processes that contact the synapse. The stimulated processes will then release ATP which would feed back onto either the pre-synaptic or post-synaptic elements of the synapse.
This neuron-glia dialogue need not be restricted to a single synapse, however. Stimulation of a glial process might result in Ca2+ increases in other regions of the glial cell or in the initiation of a Ca2+ wave that propagates from the stimulated cell to other glial cells. In this manner, glial cells could influence the excitability of neurons distant from the initial site of glial cell stimulation.
Glial Ca2+ waves that propagate both within and between cells do occur in brain tissue (Peters et al., 2003) and the retina (Newman and Zahs, 1997). In retinal astrocytes and Müller cells, stimulation of one cell region results in a Ca2+ increase that propagates into other cells regions (Newman, 2001b). Focal stimulation also elicits intercellular Ca2+ waves (Fig. 8) (Newman and Zahs, 1997; Newman, 2001b). These waves travel at a velocity of ~23 μm s−1 and propagate up to ~200 μm from the stimulation site before they die out.
Fig. 8. Intercellular Ca2+ wave in retinal glial cells.
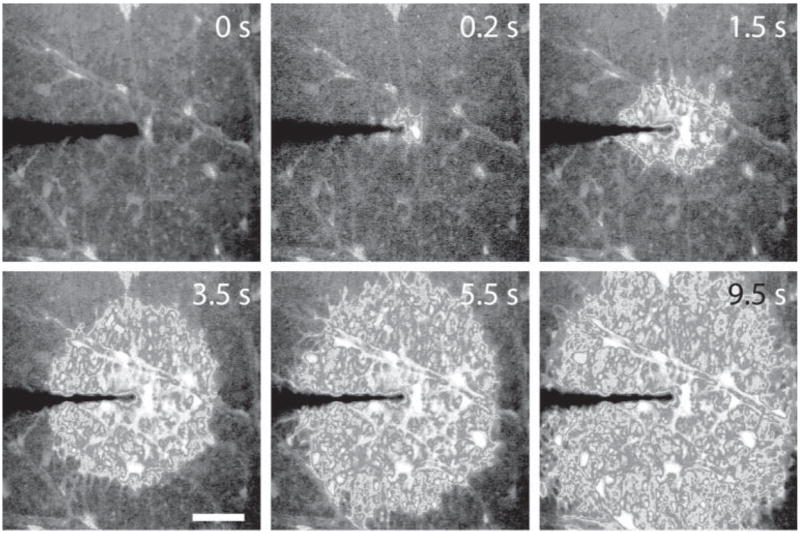
Mechanical stimulation of an astrocyte evokes an intercellular Ca2+ wave that propagates through both astrocytes and Müller cells. Calcium fluorescence images at the vitreal surface of the retina are shown. Numbers indicate elapsed time following stimulation. Scale bar, 50 μM. Unpublished results.
Ca2+ waves propagate between retinal glial cells by two mechanisms (Newman, 2001b). Astrocyte to astrocyte propagation is mediated principally by diffusion of an intracellular messenger between gap-junction-coupled glial cells (Robinson et al., 1993; Zahs and Newman, 1997). In contrast, propagation from astrocytes to Müller cells and propagation between Müller cells is mediated by glial release of ATP, which functions as an extracellular messenger. ATP release also contributes to wave propagation between retinal astrocytes.
Glial Ca2+ waves, previously characterized in the retina and brain slices, have been initiated by artificial stimuli such as agonist ejection, mechanical stimulation and electrical stimulation. It remains an open question whether neuronal activity can also initiate intercellular Ca2+ waves.
Recently, we have obtained evidence that intercellular Ca2+ waves can be evoked by neuronal activity in the retina. As described above, light stimuli evoke large, transient, Müller cell Ca2+ increases in the presence of adenosine. In some trials, the initial light-evoked Ca2+ increase is followed by a delayed Ca2+ rise in a few Müller cells. This secondary Ca2+ increase then propagates into neighboring Müller cells as a Ca2+ wave. Although these Ca2+ waves propagate into a limited number of glial cells and have, to date, only been observed in the presence of adenosine, their existence indicates that intercellular Ca2+ waves might occur in vivo.
CONCLUSION
Bidirectional communication between neurons and glial cells has been described in several brain-slice preparations and is believed to be responsible for glial modulation of synaptic transmission in the brain. We have observed similar bidirectional signaling between neurons and Müller cells in the mammalian retina. Light-evoked neuronal activity leads to increases in the generation of Ca2+ transients in Müller cells. These light-evoked glial responses are potentiated greatly by adenosine. These experiments represent the first demonstration that a natural stimulus can evoke Ca2+ increases in CNS glia.
Signaling in the reverse direction, from glia to neurons, occurs by a number of mechanisms in the retina. Glial stimulation can either facilitate or depress synaptic transmission onto ganglion cells by presynaptic mechanisms. Glia might also modulate synaptic transmission by releasing D-serine and potentiating postsynaptic NMDA receptors. In addition, Müller cells can inhibit ganglion cells directly by releasing ATP.
Although bidirectional neuron–glia signaling occurs in both the brain and the retina, the principal transmitters that mediate signaling appear to be different in these two CNS regions. In the brain, glutamate is the primary neurotransmitter that is responsible for eliciting Ca2+ increases in astrocytes. Similarly, glial modulation of neurons in the brain is mediated primarily by astrocytic release of glutamate. By contrast, in the retina ATP appears to be the principal transmitter and is responsible for both neuron to glia signaling and for glia to neuron signaling. It is unclear what accounts for this difference in signaling between the brain and the retina. One possible reason is that glutamate is released tonically from retinal neurons (Miller, 2001) and, thus, might not be well suited to function as the transmitter mediating neuron–glia communication in the retina.
As in the brain, the existence of signaling between neurons and glia in the retina indicates that retinal glial cells might play a role in information processing. Given the relatively slow time course of signaling observed, it is likely that glial cells play a modulatory role in visual processing rather than contributing directly to the generation of rapid, light-evoked signals. Our current understanding of neuron–glia interactions does not permit us to speculate on the precise role of retinal glial cells in the processing of visual information.
Acknowledgments
I thank J.I. Gepner and M. Metea for helpful discussions. This research is supported by NIH grant EY004077.
References
- Araque A, Martin ED, Perea G, Arellano JI, Buno W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. Journal of Neuroscience. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. European Journal of Neuroscience. 1998a;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. Journal of Neuroscience. 1998b;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Lodi, et al. Prostaglandins stimulate calcium-dependent glutamate release in a strocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. ATP Excites Interneurons and Astrocytes to Increase Synaptic Inhibition in Neuronal Networks. Journal of Neuroscience. 2004;24:8606–8620. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braas KM, Zarbin MA, Snyder SH. Endogenous adenosine and adenosine receptors localized to ganglion cells of the retina. Proceedings of the National Academy of Sciences of the USA. 1987;84:3906–3910. doi: 10.1073/pnas.84.11.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JHC, Alves-Rodriques A, Liu S, Li J, Azmi-Ghadimi H, et al. Connexins regulate calcium signaling by controlling ATP release. Proceedings of the National Academy of Sciences of the USA. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- Innocenti B, Parpura V, Haydon PG. Imaging extracellular waves of glutamate during calcium signaling in cultured astrocytes. Journal of Neuroscience. 2000;20:1800–1808. doi: 10.1523/JNEUROSCI.20-05-01800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeftinija SD, Jeftinija KV, Stefanovic G, Liu F. Neuroligand-evoked calcium-dependent release of excitatory amino acids from cultured astrocytes. Journal of Neurochemistry. 1996;66:676–684. doi: 10.1046/j.1471-4159.1996.66020676.x. [DOI] [PubMed] [Google Scholar]
- Kang J, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nature Neuroscience. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Matyash V, Filippov V, Mohrhagen K, Kettenmann H. Nitric oxide signals parallel fiber activity to Bergmann glial cells in the mouse cerebellar slice. Molecular and Cell Neuroscience. 2001;18:664–670. doi: 10.1006/mcne.2001.1047. [DOI] [PubMed] [Google Scholar]
- Miller RF. The physiology and morphology of the vertebrate retina. In: Ryan SJ, editor. Retina. Mosby; 2001. pp. 138–170. [Google Scholar]
- Miller RF. D-Serine as a glial modulator of nerve cells. Glia. 2004;47:275–283. doi: 10.1002/glia.20073. [DOI] [PubMed] [Google Scholar]
- Mothet J-P, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, et al. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proceedings of the National Academy of Sciences of the USA. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glia of the retina. In: Ryan SJ, editor. Retina. Mosby; 2001a. pp. 89–103. [Google Scholar]
- Newman EA. Propagation of intercellular calcium waves in retinal astrocytes and Müller cells. Journal of Neuroscience. 2001b;21:2215–2223. doi: 10.1523/JNEUROSCI.21-07-02215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial cell inhibition of neurons by release of ATP. Journal of Neuroscience. 2003a;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. New Roles for astrocytes: Regulation of synaptic transmission. Trends in Neurosciences. 2003b;26:536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
- Newman EA. Glial modulation of synaptic transmission in the retina. Glia. 2004;47:268–274. doi: 10.1002/glia.20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glia and synaptic transmission. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford University Press; 2005. [Google Scholar]
- Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847. doi: 10.1126/science.275.5301.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. Journal of Neuroscience. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. Journal of Neuroscience. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters O, Schipke CG, Hashimoto Y, Kettenmann H. Different mechanisms promote astrocyte Ca2+ waves and spreading depression in the mouse neocortex. Journal of Neuroscience. 2003;23:9888–9896. doi: 10.1523/JNEUROSCI.23-30-09888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. Journal of Neuroscience. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SR, Hampson ECGM, Munro MN, Vaney DI. Unidirectional coupling of gap junctions between neuroglia. Science. 1993;262:1072–1074. doi: 10.1126/science.8093125. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: Localization to astrocytes and glutamate-stimulated release. Proceedings of the National Academy of Sciences of the USA. 1995;92:3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipke CG, Kettenmann H. Astrocyte responses to neuronal activity. Glia. 2004;47:226–232. doi: 10.1002/glia.20029. [DOI] [PubMed] [Google Scholar]
- Stevens ER, Esquerra M, Kim P, Newman EA, Snyder SH, Zahs KR, et al. D-serine and serine racemase are present in the vertebrate retina and contribute to the functional expression of NMDA receptors. Proceedings of the National Academy of Sciences of the USA. 2003;100:6789–6794. doi: 10.1073/pnas.1237052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Juttner R, Grantyn R. Ca2+-permeable P2X receptor channels in cultured rat retinal ganglion cells. Journal of Neuroscience. 1999;19:3353–3366. doi: 10.1523/JNEUROSCI.19-09-03353.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Magistretti PJ, Haydon PG. Glia in synaptic transmission. Oxford University Press; 2002. The Tripartite Synapse. [Google Scholar]
- Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proceedings of the National Academy of Sciences of the USA. 1999;96:13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahs KR, Newman EA. Asymetric gap junctional coupling between glial cells in the rat retina. Glia. 1997;20:10–22. [PubMed] [Google Scholar]