

Abstract
Purpose
To evaluate the safety, toxicity, in vivo immunologic activation, and maximum-tolerated dose (MTD) of EMD 273063 (hu14.18-IL-2) in patients with metastatic melanoma.
Patients and Methods
Thirty-three patients were treated with EMD 273063, a humanized anti-GD2 monoclonal antibody (mAb) linked to interleukin-2 (IL-2). EMD 273063 was given as a 4-hour intravenous infusion on days 1, 2, and 3 of week 1. Patients with stabilization or regression of disease could receive a second course of treatment at week 5. Dose levels evaluated were 0.8, 1.6, 3.2, 4.8, 6.0, and 7.5 mg/m2/d.
Results
Nineteen of 33 patients completed course 1 with stable disease and went on to receive course 2. Eight patients had stable disease on completion of course 2. Grade 3 adverse events included hypophosphatemia (11 patients), hyperglycemia (three patients), hypotension (two patients), thrombocytopenia (one patient), hypoxia (three patients), elevated hepatic transaminases (two patients), and hyperbilirubinemia (one patient). Opioids were required for treatment-associated arthralgias and/or myalgias during 17 of 52 treatment courses. No grade 4 adverse events were observed. Dose-limiting toxicities at the MTD included hypoxia, hypotension, and elevations in AST/ALT. Grade 3 toxicities were anticipated based on prior studies of IL-2 or anti-GD2 mAbs, and all resolved. Immune activation was induced, as measured by lymphocytosis, increased peripheral-blood natural killer activity, and cell numbers, and increased serum levels of the soluble alpha chain of the IL-2 receptor complex.
Conclusion
Treatment with the immunocytokine EMD 273063 induced immune activation and was associated with reversible clinical toxicities at the MTD of 7.5 mg/m2/d in melanoma patients.
INTRODUCTION
Interleukin-2 (IL-2) has been used alone and in combination with other therapies in the treatment of patients with metastatic melanoma and has well-documented antitumor effects in a minority of patients.1 Murine models suggest two distinct mechanisms by which IL-2 treatment can mediate antitumor effects.2 IL-2 treatment augments the activation of pre-existing antigen-specific T cells to enhance their recognition and destruction of neoplastic tissue and activates a separate population of lymphocytes known as natural killer (NK) cells. NK cells lack T-cell markers and antigen-specific receptors and have demonstrated the ability to destroy tumor targets in vitro and in vivo.3,4 Novel approaches that selectively induce immune attack at sites of tumor while minimizing the known side effects of IL-2 are needed. A more selective induction of tumor-specific T cells, or localization of activated NK cells to sites of tumor, may provide better tumor specificity and more effective tumor destruction.
One way to potentially improve the antitumor efficacy of activated NK cells is to direct their lytic activity more selectively toward tumor cells.5,6 This can be demonstrated in vitro with the addition of tumor-reactive monoclonal antibody (mAb) to facilitate antibody-dependent cell-mediated cytotoxicity (ADCC). Fc receptor–bearing effector cells, including NK cells, primarily mediate ADCC. In vitro studies have demonstrated that ADCC of tumor cells is increased when effector cells are initially activated with IL-2. This IL-2 activation can be performed in vitro or in vivo. Mice receiving IL-2 plus tumor-specific mAb demonstrate improved antitumor effects compared with animals treated with either agent alone.7–9 Only intact mAb, not fragment antigen-binding regions, were able to induce ADCC in vitro or provide the antitumor effects in vivo.8 These results suggest that direct effector cell–tumor cell contact is involved in the antitumor mechanism and that it may be possible, with in vivo activation of effector cells by IL-2, to improve clinical antitumor effects of mAb treatment.
Preclinical studies in selected murine models bearing syngeneic tumors have tested the antitumor activity and immunologic effects of tumor-reactive mAbs genetically linked to IL-2, designated immunocytokines (IC). These studies document that IC can induce potent antitumor effects in vivo that exceed the antitumor activity of comparable amounts of mAb and IL-2 given in combination as separate molecules. Furthermore, depending on the model system evaluated, the antitumor activity is mediated primarily by T cells10 or by NK cells.11 It appears that the IC molecule localizes to the tumor cell surface because of the recognition by the mAb component of the IC molecule. This would allow NK cells to be activated by the IL-2 component of the IC molecule and then mediate ADCC using the Fc component of the IC molecule.
The EMD 273063 molecule evaluated in this clinical trial consists of the humanized 14.18 antibody genetically linked to a molecule of human IL-2 at the carboxy terminus of each immunoglobulin heavy chain. The hu14.18 mAb recognizes the GD2 disialoganglioside, expressed on tumors of neuroectodermal origin including melanoma, neuroblastoma, and certain sarcomas.12,13 The primary objectives of this study were to assess the safety, toxicity, and in vivo immunologic effects and to determine the maximum-tolerated dose (MTD) of EMD 273063 when administered as three daily 4-hour intravenous (IV) infusions in patients with melanoma. Furthermore, the pharmacokinetics of EMD 273063 at different dose levels was investigated.
PATIENTS AND METHODS
Patients
Potentially eligible patients required histologically confirmed melanoma (or other GD2+ malignancy confirmed by immunohistochemistry) that was considered surgically and medically incurable. These patients could have either measurable or nonmeasurable but evaluable metastatic disease, or they could have no evidence of disease after surgical resection of either distant metastases or regionally recurrent disease. Patients with multiple (two or more) local or regional recurrences were included only if they had prior evidence of lymph node involvement and if each recurrence was separated in time by at least 2 months. All patients needed to have adequate bone marrow function (defined by total WBC > 3,500/μL or total granulocytes > 2,000/μL, platelets > 100,000/μL, and hemoglobin > 10.0 g/dL), adequate liver function (defined by an AST less than three times the normal level and a total bilirubin less than 2.0 mg/dL), and adequate renal function (defined by a serum creatinine < 2.0 mg/dL or a creatinine clearance of > 60 mL/min). All patients had an Eastern Cooperative Oncology Group performance status of 0 or 1 and a life expectancy of at least 12 weeks. Patients who had previously received chemotherapy, radiation therapy, or other immunosuppressive therapy within 4 weeks before study were excluded. Patients could have prior CNS metastases if treated and stable for at least 4 weeks before starting the study. Written informed consent that was approved by the University of Wisconsin Health Sciences institutional review board was obtained from all patients.
Hu14.18-IL-2 Immunocytokine
The hu14.18-IL-2 IC (EMD 273063) was provided by EMD Pharmaceuticals Inc. (Durham, NC). Preclinical evaluation has shown that 1 mg of the fusion protein contains approximately 3 × 106 U of IL-2 (based on a proliferative assay with IL-2 responsive Tf-1β cells) and approximately 0.8 mg of the hu14.18 mAb14 (J.A. Hank, unpublished data; Tf-1β cells are described in the cell lines subsection of Immunologic Monitoring).
Study Design
This phase I trial was designed as an open-label, nonrandomized dose-escalation study. Patient accrual followed a two-stage design as described by Storer.15 In the first stage, escalation to the next dose level was permitted after a single patient had been evaluated at the current dose level without the occurrence of any toxicity (other than fever) one grade or worse above baseline. Once that level of toxicity was observed, three to six patients were to be entered into that dose level as well as into subsequent treatment cohorts. The MTD was defined as that dose that was estimated to cause dose-limiting toxicity (DLT) during course 1 in 33% of patients. EMD 273063 was administered on an inpatient basis as a 4-hour IV infusion over 3 consecutive days during the first week of each course. Patients were discharged from the hospital, if stable, approximately 24 hours after the completion of the third infusion. Adverse events and toxicities were graded as per the National Cancer Institute Common Toxicity Criteria (version 2.0) and the University of Wisconsin Comprehensive Cancer Center Toxicity Grading Scale for IL-2 (performance status, weight gain, and temperature).16 DLT was defined as the occurrence of grade 3 or 4 toxicity other than grade 3 lymphopenia, hyperbilirubinemia, or hypophosphatemia. A protocol amendment excluded patients with a known history of diabetes, as this therapy may alter blood glucose levels. Patients with grade 3 treatment-related toxicities were required to recover to at least grade 1 before they could resume treatment at a 50% dose reduction for course 2. Disease assessment was performed between 20 to 26 days after initiation of each treatment course. Patients with ≥ 25% disease progression were removed from the study. Patients with stable disease were eligible for course 2, starting on day 29. Patients were required to have an objective partial or complete clinical response to be eligible for any additional courses of therapy past course 2.
Clinical Laboratory Monitoring
Clinical hematology, chemistry, and C-reactive protein (CRP) evaluations were performed by standard methodology at the University of Wisconsin Hospital Clinical Laboratory. Blood samples, unless otherwise indicated, were taken in the morning on the indicated protocol day, before administration of EMD 273063. Thus day 1 samples from course 1 reflect blood samples obtained before any EMD 273063 treatment.
Immunologic Monitoring
Cell lines
The Daudi, K562, LA-N-5 (GD-2–positive neuroblastoma) and M21 (GD-2–positive melanoma) cell lines were maintained as previously described.14,17 The IL-2 responsive Tf-1β cell line was created by transfecting the gene for the IL-2 receptor beta chain into the myeloid leukemia Tf-1 cell line, already expressing the IL-2 receptor gamma chain.18
Flow cytometry
Fluorescent cell-bound antibody was detected by standard indirect immunofluorescence methods (Becton Dickinson, San Diego, CA). To detect intact hu14.18-IL-2 IC binding to GD2 positive cells lines, phycoerythrin-conjugated monoclonal rat antihuman IL-2 (PharMingen, San Diego, CA) was used.10
ADCC and NK assays
These 51Cr release assays were performed in RPMI-HS, as previously described.10 For ADCC assays, IC, serum, or antibody as indicated was added to the effectors. Target cells at 5 × 103 were added to the effector cells. Fresh peripheral-blood mononuclear cells (PBMCs) from the patients were used as effector cells, and PBMCs from a healthy control donor were tested in parallel, to insure the validity of the assays.
In vitro proliferative response to IL-2
The IL-2 responsive Tf-1β cell line was cultured at a concentration of 1 × 105 cells/well. Patient serum was added to each microwell to determine the IL-2–inducing capacity of the IC present in the serum obtained after an infusion of IC. Cultures were incubated at 37°C for 48 hours, pulsed with 1 μCi of [3H]thymidine for 18 hours, collected with a Filtermate 196 (Packard-Canberra, Meriden, CT) harvester, and [3H]thymidine incorporation was quantified with a Matrix 9600 direct beta counter with a 5-minute counting period (Packard-Canberra).
Enzyme-linked immunosorbent assay (ELISA) methods
Soluble IL-2 receptor alpha (sIL-2Rα) was measured by a commercial (Immunotech, Marseilles, France) double-mAb ELISA kit, according to the manufacturer’s specifications. Measurement of EMD 273063 levels in patients’ sera by ELISA were performed as previously described.19,20 Plates coated with 1A7, a mouse anti-idiotype antibody to 14.18, were used. Diluted serum specimens (in duplicates) were incubated overnight at 4°C. They were then serially washed and incubated with biotinylated goat antihuman IL-2 antibody (R&D Systems, Minneapolis, MN) followed by ExtrAvidin-AP (Sigma, St Louis, MO). A standard curve was based on dilutions of lot no. 31,916 of hu 14.18-IL-2 and ranged from 0 to 20 ng/mL. The detection limit for this assay was determined as 0.16 ng/mL. The intra-assay and inter-assay precision was estimated as 4.9% and 3.2%, respectively.
Statistical Methods
Pharmacokinetic parameters and immunologic end points are described using descriptive statistics. The area under the curve from time zero to 24 (AUC0–24) hours was calculated with the Lagrange method. Blood samples for determining pharmacokinetic parameters were collected on day 1, course 1, at 0, 0.5, 4, 4.5, 5, 6, 8, 16, and 24 hours from the start of the 4-hour infusion. The terminal half-life (T1/2) was determined from the terminal slope on a log-linear plot of concentration versus time (after 4 hours).
Jonckheere-Terpstra trend test was performed to determine the significance of the association between increasing dose level and each of the pharmacokinetic parameters AUC0–24 and peak concentration. A Spearman rank correlation analysis was also performed to determine the relationship between actual dose administered and the pharmacokinetic parameters.
Linear regression methods for repeated measures were used to evaluate the relationships between dose and immunologic end points.21 Wilcoxon signed-rank test was used to determine changes from baseline in CRP levels. All statistical analyses were performed with SAS software (version 8.2, SAS Institute Inc, Cary, NC). All P values are two-sided and were not adjusted for the number of parameters evaluated. As such, they should only be interpreted as exploratory.
RESULTS
Patient Characteristics
Pretreatment characteristics are outlined in Table 1. The median age of the 33 patients was 49 years (range, 27 to 69 years), and all had Eastern Cooperative Oncology Group performance status 0 or 1. Prior therapies included surgery (29 patients), biologic therapy (20 patients), radiotherapy (10 patients), and chemotherapy (nine patients). Sites of metastatic disease included lung (19 patients), distant nodal, skin, or subcutaneous disease (26 patients), other visceral sites (13 patients), and bone (three patients). Two patients had previously treated brain metastases. Five patients (15%) had no evidence of disease at the time of treatment.
Table 1.
Patient Demographics
| No. of Patients* | |
|---|---|
| Total No. of patients | 33 |
| Sex | |
| Male | 22 |
| Female | 11 |
| Prior therapy | |
| Surgery | 29 |
| Biologic | 20 |
| Radiation | 10 |
| Chemotherapy | 9 |
| Sites of disease | |
| Distant node, skin, subcutaneous | 26 |
| Lung | 19 |
| Other visceral metastases | 13 |
| CNS† | 2 |
| Bone | 3 |
| No evidence of disease‡ | 5 |
| Disease status | |
| M1a§ | 5 |
| M1b|| | 10 |
| M1c¶ | 13 |
| Performance status# | |
| 0 | 23 |
| 1 | 10 |
Median age, 49 years; range, 27 to 69 years.
Previously treated CNS metastases.
Four patients with resection of distant metastases and one patient with prior resection and radiation for regionally recurrent disease.
Metastatic disease to distant nodes, skin or subcutaneous lesions.
Metastatic disease to lung.
Metastatic disease to other visceral sites or M1a, M1b disease with elevated lactate dehydrogenase.
Eastern Cooperative Oncology Group performance status.
The dose-escalation schedule is shown in Table 2. The first patient had toxicity requiring expansion of the treatment group to three patients (grade 2 elevation of AST and grade 2 headache), and subsequent dose escalation occurred in treatment cohorts of three to six patients at one of the following dose levels: 0.8, 1.6, 3.2, 4.8, 6.0, or 7.5 mg/m2/d.
Table 2.
Dose-Escalation Schedule
| Dose Level | mg/m2/d | No. of Patients in Course 1 |
|---|---|---|
| 1 | 0.8 | 3 |
| 2 | 1.6 | 6 |
| 3 | 3.2 | 6 |
| 4 | 4.8 | 6 |
| 5 | 6.0 | 6 |
| 6 | 7.5 | 6 |
Pharmacokinetics
EMD 273063 serum levels were evaluated in serial samples from all 33 patients immediately after the first 4-hour infusion (day 1, course 1). These values were used to calculate the peak serum level of EMD 273063, as well as its clearance, half-life, and area under the curve (Table 3). The overall mean half-life was found to be 3.7 hours (± standard deviation of 0.9 hours), which is intermediate between the half-lives of its two components (approximately 45 minutes for IL-2 and 3 days for the chimeric 14.18 mAb) and comparable to that which was observed for the half-life of chimeric 14.18-IL-2 in mice.22 After the clearance of EMD 273063 from the sera of these patients, neither the IL-2 nor hu14.18 mAb components could be detected (data not shown), which contrasts with data from mice.22 The peak serum levels of EMD 273063 and AUC during course 1 showed a significant dose-dependent increase (P < .001), whereas clearance showed a dose-dependent decrease (P < .0001). This inverse relationship between dose and clearance is not linear (P < .001), suggesting that the clearance may be influenced by several factors, as expected for a molecule that binds to GD2, IL-2 receptors, and Fc receptors. The clearance values for the five patients who had no measurable disease at the time of study entry did not show any difference in their clearance values from those 28 patients with measurable disease (P = .37; data not shown), suggesting that the presence or absence of macroscopic tumor does not influence the clearance of EMD 273063.
Table 3.
Pharmacokinetic Parameters of EMD 273063
| Peak Concentration (ng/mL)*† |
Clearance (L/h)
|
Half-Life (h)
|
AUC (ng/mL*h)*‡ |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Dose (mg/m2) | No. of Patients | Mean | SD | Mean | SD | Mean | SD | Mean | SD |
| 0.8 | 3 | 185 | 31 | 1.39 | 0.45 | 2.7 | 0.21 | 1,138 | 248 |
| 1.6 | 6 | 696 | 390 | 0.67 | 0.16 | 3.5 | 1.10 | 5,106 | 2,540 |
| 3.2 | 6 | 1,490 | 693 | 0.66 | 0.22 | 3.8 | 0.45 | 11,448 | 4,970 |
| 4.8 | 6 | 3,965 | 2,001 | 0.39 | 0.11 | 4.2 | 1.25 | 25,529 | 6,794 |
| 6.0 | 6 | 6,339 | 1,519 | 0.33 | 0.09 | 3.8 | 0.86 | 38,944 | 12,294 |
| 7.5 | 6 | 5,514 | 2,808 | 0.38 | 0.11 | 3.9 | 0.66 | 40,870 | 18,958 |
Abbreviations: AUC, area under the curve; SD, standard deviation.
Peak concentration and AUC were obtained at course 1, day 1. Values shown are mean and SD for all patients at each dose level.
The association between maximal concentration and dose level was highly significant (P < .001).
The association between AUC and dose level was highly significant (P < .001).
Toxicity
Clinical toxicities are listed in Table 4. All patients had transient fevers that were successfully treated with acetaminophen and/or indomethacin. A majority of patients experienced grade 1 or 2 rigors, and 16 patients (48%) required meperidine for grade 2 rigors. The rigors were transient and resolved on completion of the infusion. Thirteen patients (39%) required opioids for treatment-related neuropathic pain. This pain was predominantly localized to the lower back, pelvis, or lower extremities, usually occurred toward the end of the 4-hour infusion, and resolved within 2 to 4 hours. No grade 2 or worse functional neuropathies (motor or sensory disturbances) were observed at any dose level. Twenty patients (61%) experienced pruritus requiring oral medications for symptom control, but no severe allergic reactions were observed.
Table 4.
Adverse Events* Observed With EMD 273063
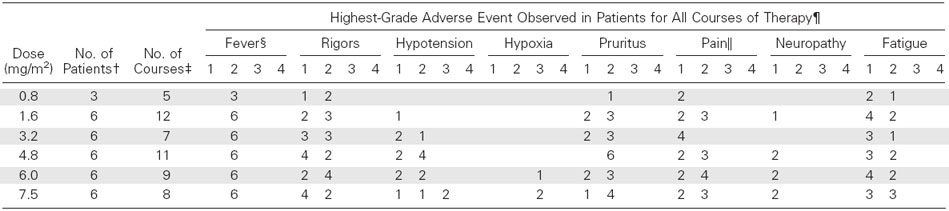
Adverse events graded 1 to 4 as per National Cancer Institute Common Toxicity Criteria, version 2.0, except as indicated.
Total number of patients treated at each dose level.
Total number of courses administered for all patients at the indicated dose level.
Fever and pain graded 1 to 4 as per the University of Wisconsin Comprehensive Cancer Center Toxicity Criteria.
For each category of toxicity, each patient was scored based on their highest grade of toxicity for either course of treatment. The number of patients showing each grade of toxicity as their highest grade is shown for each dose level of EMD 273063.
The clinical toxicities at the MTD that were considered dose-limiting were hypoxia and hypotension. Two patients (6%) experienced grade 3 hypoxia during course 1 and received supplemental oxygen. One patient at dose level 5 (6.0 mg/m2/d) experienced mild hypoxia (pulse oximetry recorded at 89%) during an episode of rigors. The hypoxia resolved immediately after administration of low-flow oxygen by nasal cannula, and no further supplemental oxygen was required after the rigors resolved. A patient at dose level 6 (7.5 mg/m2/d) experienced grade 3 hypoxia requiring supplemental oxygen, as well as grade 3 hypotension requiring vasopressors, intensive care unit monitoring, and discontinuation of treatment. Both grade 3 toxicities for this patient resolved within 12 hours, and no respiratory support, beyond supplemental oxygen, was required. An additional patient at dose level 6 (7.5 mg/m2/d) experienced transient grade 3 hypoxia during course 2 and required supplemental oxygen, as well as grade 3 hypotension that resolved with IV fluids and did not require vasopressors. Eight patients (24%) at dose levels 3 to 6 required supplemental IV fluids for transient grade 2 hypotension that resolved after completion of the hu14.18-IL-2 infusion. No patient had substantial weight gain or significant peripheral edema, and no grade 4 toxicities were observed.
Significant laboratory changes observed for each dose level for two courses of treatment are listed in Table 5. Four patients (12%) had grade 2 thrombocytopenia and one patient, at dose level 4, had grade 3 thrombocytopenia. One patient at dose level 4 experienced a grade 3 elevation of bilirubin on day 2 of course 1. Treatment was held on day 3 with resolution of the hyperbilirubinemia. The patient did not receive course 2 secondary to progression of disease. Eight patients (24%) had a grade 2 elevation of AST and one patient at dose level 6 had a grade 3 elevation of AST and ALT.
Table 5.
Laboratory Changes* Observed With EMD 273063
Adverse events graded 1 to 4 as per National Cancer Institute Common Toxicity Criteria, version 2.0, except as indicated.
Total number of patients treated at each dose level.
Total number of courses administered for all patients at the indicated dose level.
For each category of laboratory toxicity, each patient was scored based on their highest grade of toxicity for either course of treatment.
The number of patients showing each grade of toxicity as their highest grade is shown for each dose level of EMD 273063.
Grade 1 hyperglycemia above baseline values was observed in 18 patients (55%). Five additional patients (15%) had grade 2 hyperglycemia. These events occurred on the days of EMD 273063 administration, and all episodes of hyperglycemia resolved on completion of therapy. Three patients (9%) had grade 3 hyperglycemia. Two of these were patients with known diabetes mellitus requiring oral hypoglycemic therapy. These patients responded well to insulin administration to control their hyperglycemia. Because the transient grade 3 hyperglycemia was readily managed and was without significant adverse consequences, a protocol amendment was approved to exclude patients with a known history of diabetes mellitus from this study, as this therapy may alter blood glucose levels. Hypophosphatemia was also frequently observed. Thirteen patients (39%) had grade 2 hypophosphatemia, and 11 patients (33%) had grade 3 hypophosphatemia. Patients with hypophosphatemia were monitored closely with daily serum levels of phosphate, creatinine, sodium, and potassium, in addition to venous blood pH and 24-hour urine phosphate. These patients did not have significant increases in creatinine, and no additional electrolyte abnormalities were detected. There was no substantial change in venous blood pH. Twenty-four hour urine phosphate measurements in patients with grade 3 hypophosphatemia demonstrated no renal wasting of phosphate. A typical course of hypophosphatemia is listed in Table 6 for a patient at dose level 5. This treatment-associated hypophosphatemia resolved after completion of therapy without phosphate supplementation. A protocol amendment was approved to exclude grade 3 hypophosphatemia as a DLT for the purpose of defining MTD.
Table 6.
Characteristic Course of Hypophosphatemia*
| Day
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 22 | 26 | |
| Serum PO4, mg/dL | 3.2 | 2.9 | 2.1 | 1.3 | 1.5 | 2.4 | 2.6 | 3.1 | 3.4 | |
| Serum creatinine, mg/dL | 1.2 | 1.3 | 1.3 | 1.1 | 1.2 | 1.2 | ||||
| Serum Na+, mg/dL | 142 | 139 | 137 | 143 | 138 | 141 | 140 | 140 | ||
| Serum K+, mg/dL | 4.4 | 4.0 | 4.0 | 4.5 | 3.7 | 4.3 | 4.5 | 4.4 | ||
| Venous blood pH | 7.39 | 7.38 | 7.43 | 7.40 | ||||||
| Urine PO4, g/24 h | 0.2 | 0.0 | 0.1 | 0.7 | 0.6 | |||||
A typical course of hypophosphatemia (grade 3) is demonstrated in a patient receiving EMD 273063 (6 mg/m2/d). Data shown are from course 1: hu14.18-IL2 was given on days 1, 2, and 3.
Table 7 shows the DLTs that occurred during course 1 for all dose levels. All of the observed toxicities were reversible and resolved on completion of protocol therapy. The MTD was determined to be 7.5 mg/m2/d, as two of six patients had dose-limiting toxicities (one with hypoxia, hypotension, and hyperglycemia and one with elevated ALT/AST) during course 1 at this dose level.
Table 7.
Dose-Limiting Toxicities Observed During Course 1 in Patients Receiving EMD 273063
| Dose (mg/m2) | Total Patients | Dose-Limiting Toxicity* and No. of Patients |
|---|---|---|
| 0.8 | 3 | None |
| 1.6 | 6 | AST, 1† |
| 3.2 | 6 | None |
| 4.8 | 6 | Thrombocytopenia, 1 |
| 6.0 | 6 | Hypoxia, 1 |
| 7.5§ | 6 | Hypoxia, 1; AST/ALT, 1‡; hypotension, 1; hyperglycemia, 1 |
Protocol amendments excluded the following grade 3 toxicities as dose-limiting toxicities; hyperglycemia in patients with known diabetes mellitus (patients with a known history of diabetes mellitus were excluded), hypophosphatemia, and hyperbilirubinemia.
Grade 3 elevation of AST.
Grade 3 elevation of AST and ALT.
The dose of 7.5 mg/m2/d was found to be the maximum-tolerated dose, as two to six patients during course 1 showed reversible dose-limiting toxicity at this dose level (one patient with hypoxia, hypotension, and hyperglycemia and one patient with elevated AST/ALT).
Clinical Outcome
Thirty-three patients were treated on this study. Two patients (6%) completed only the first 2 of 3 days for course 1. One of these patients (dose level 4) had a grade 3 hyperbilirubinemia on day 2 of treatment, and the other patient (dose level 6) had grade 3 hypoxia and hypotension requiring treatment to be held. Both of these patients had progression of disease and did not receive a second course of therapy. Nineteen patients (58%) had stable disease after the first course of therapy and received a second course of therapy. Five patients (15% of all patients) required a 50% dose reduction for course 2 secondary to adverse events in course 1. Seventeen patients (52% of all patients) completed course 2. One patient (dose level 4) declined to receive the final infusion during course 2, and one patient (dose level 6) had the final infusion during course 2 held because of hypotension.
Although this phase I study was not designed to determine overall tumor response rate to treatment, all patients were followed for antitumor activity. None showed improvement of measurable disease to qualify as a complete or partial response. Eight patients (24% of all patients) had stable disease after the second course of treatment. Four of these eight patients continue with no evidence of progressive disease (one with stable disease treated at 4.8 mg/m2/d and three with no evidence of disease treated at 0.8, 3.2, and 6.0 mg/m2/d) for 26 to 60 months since completing protocol therapy. Five of the 33 patients entered the study with no measurable disease after surgical resection of recurrences or metastases. Two of these five patients have experienced disease progression, whereas the remaining three patients continue with no evidence of disease. One additional patient had an objective decrease in a lung nodule after two courses of therapy, but the overall disease response was scored as disease progression because of growth in a distant node. The node was resected after hu14.18-IL-2 therapy, and the patient remained free from disease progression for 3 years, after which disease progression was noted and alternate therapy begun.
Immunologic Monitoring
A peripheral-blood lymphopenia occurred on days 2 to 4 (P < .0001), and this was followed by a rebound lymphocytosis on days 5 to 22 (P < .0001) in course 1 (Fig 1A). Both of these changes were dose-dependent (P < .01 and P < .05, respectively). Lymphocyte counts for the 12 patients who received course 2 of EMD 273063 without a dose reduction are shown in Figure 1B. The baseline lymphocyte count for course 2 (day 29 of course 1) was increased over the baseline lymphocyte count for course 1, indicating that effects of the first course of treatment are still present on day 29 (Fig 1B). In addition, the lymphocyte counts during course 2 on days5, 8. and 15 are greater than the corresponding values for days 5, 8, and 15 during course 1 for these 12 patients.
Fig 1.
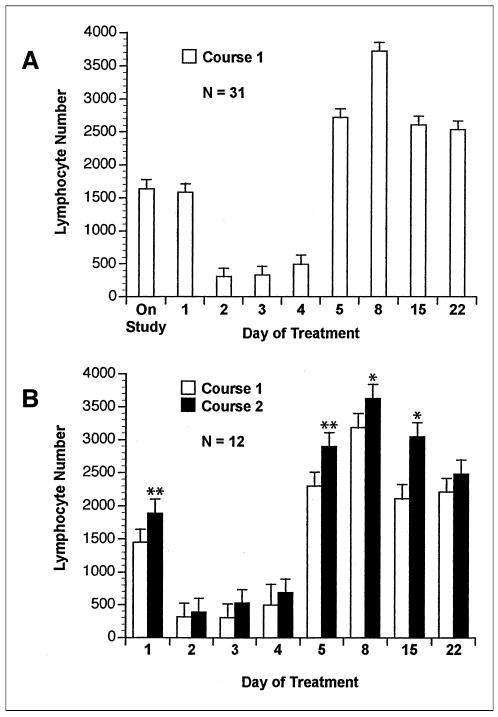
Mean peripheral-blood lymphocyte count (cell number per mcL ± SE) for (A) the 31 patients completing course 1 and (B) the 12 patients completing both courses at the same dose level. In A, values after d1 are different from pretreatment (P < .0001). Significant differences in B are shown (course 2 v course 1). (*), P < .05; (**), P < .01.
Lymphocyte cell surface phenotype showed an expansion of CD16+ and CD56+ lymphocytes (NK cell markers) after the first week of EMD 273063 therapy (Fig 2A). This effect was still present on day 29 of course 1 (day 1, course 2; Fig 2B). For patients 19 through 33 (receiving 4.8 to 7.5 mg/m2/d), lymphocyte cell surface phenotype was determined on days 15 and 22 in addition to days 1 and 8. This analysis demonstrated that the augmentation of CD56 and CD56/CD16 coexpressing cells remains significantly elevated (P < .01) on days 8, 15, and 22 (data not shown).
Fig 2.
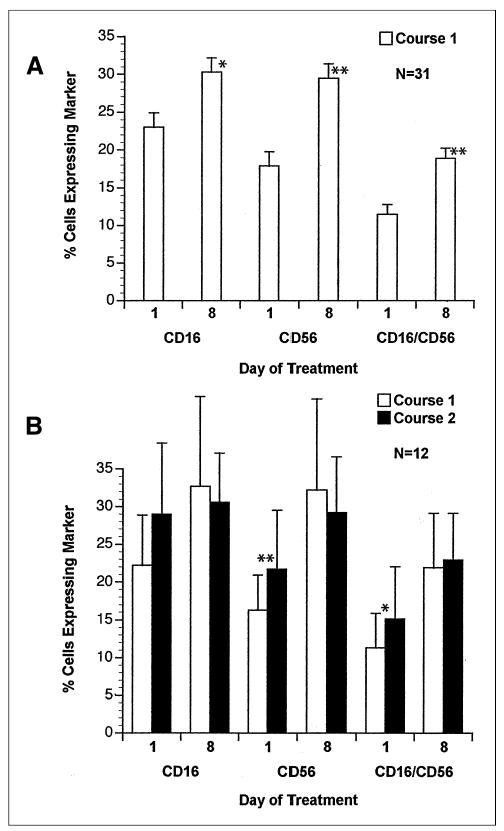
Mean (+ SE) percentage of peripheral-blood mononuclear cell–expressing marker before treatment (day 1) and on day 8 for (A) patients completing course 1 and (B) patients completing courses 1 and 2 at the same dose level. Significant differences are shown for (A) day 1 versus day 8 and (B) for course 2 versus course 1. (*), P < .05; (**), P < .01.
As a measure of immune activation, CRP levels for patients 13 through 33 and sIL-2R∞ levels for all patients were obtained. As shown in Table 8, a significant increase in mean CRP level was detected on days 3 to 5 of treatment in both course 1 and course 2 compared with baseline for each course. This increase in CRP returned to baseline levels by the third week of each treatment course. The sIL-2R∞ level was significantly increased over baseline starting 24 hours after the EMD 273063 infusion during both course 1 (Fig 3A) and course 2 (Fig 3B), which peaked at day 4 to 5 and then declined. The increase in sIL-2R∞ was found to be dose dependent (P = .014). sIL-2R∞ values for course 2 were increased compared with corresponding values in course 1 for days 1 to 5 for patients receiving the same dose in both courses (P < .05).
Table 8.
CRP Monitoring*
| Course No. | Time† | No. of Patients‡ | Mean CRP§ | SD|| | P¶ |
|---|---|---|---|---|---|
| 1 | Pretreatment | 20 | 2.4 | 4.87 | |
| After 2 doses | 16 | 14.9 | 4.95 | .0005 | |
| After 3 doses | 19 | 9.9 | 3.86 | .0003 | |
| After 1 week | 17 | 3.3 | 4.27 | .16 | |
| After 3 weeks | 16 | 2.6 | 6.22 | .46 | |
| 2 | Pretreatment | 9 | 1.9 | 4.57 | |
| After 2 doses | 10 | 13.3 | 6.18 | .009 | |
| After 3 doses | 8 | 9.9 | 3.68 | .02 | |
| After 1 week | 8 | 2.9 | 3.27 | .41 | |
| After 3 weeks | 6 | 0.7 | 0.52 | 1.00 |
Abbreviations: CRP, C-reactive protein; SD, standard deviation.
CRP levels were monitored for patients 13 through 33 (dose levels 3.2 to 7.5 mg/m2/d).
Samples were obtained pretreatment (day 0 or 1), after two doses (day 3), after three doses (day 4 or 5), after 1 week (day 8 or 9) and after 3 weeks (day 22 or 23).
Number of patients for whom C-reactive protein data are available at each time point.
Mean CRP (mg/dL).
Standard deviation.
P value when compared to day 0, 1.
Fig 3.
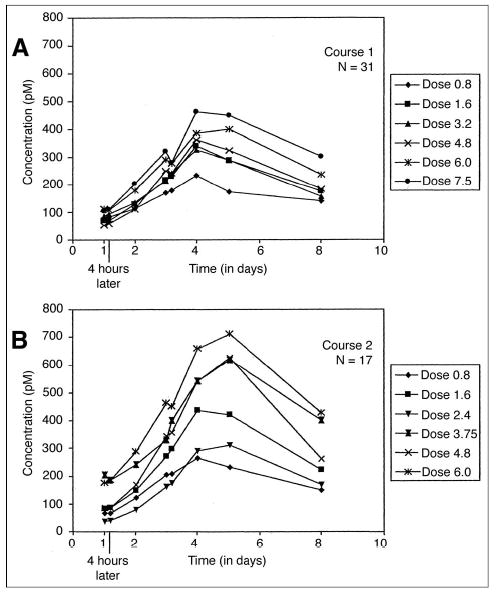
Mean soluble interleukin-2 receptor levels in serum by dose level pre-infusion and 4 hours after starting infusion (days 1 and 3) and on days 2, 4, 5, and 8 for (A) patients completing course 1 and (B) for patients completing course 2.
NK function, IL-2–activated NK function, and ADCC mediated by PBMC from 31 patients completing course 1 are shown in Figure 4. There was a significant increase in killing mediated by lymphocytes collected on day 8 when compared with day 1 in the IL-2 activated NK function and ADCC assays. This increase is seen when IL-2 or EMD 273063 is added to the medium during the assay. There was no significant dose response effect for this difference, potentially reflecting the patient-to-patient and day-to-day variability in these assays. The 12 patients who received course 2 at the same dose as in course 1 showed ADCC results that were very similar to those obtained during course 1. The only parameter that was found to be different for course 2 from course 1 was increased killing in the presence of IL-2 on day 1 (P ≤ .05), indicating that augmented killing measured by this assay remained elevated on day 29 (day 1, course 2; data not shown.)
Fig 4.
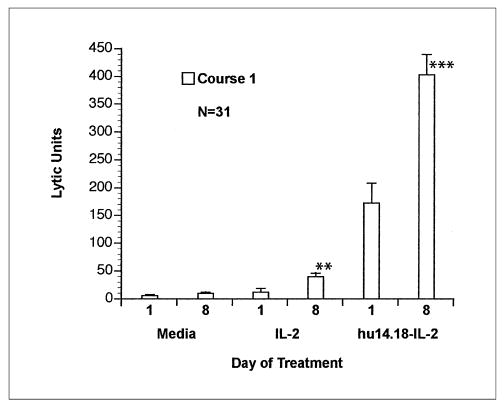
Antibody-dependent cell-mediated cytotoxicity using the LA-N-5 target with peripheral-blood mononuclear cells obtained from patients on day 1 (before treatment) and day 8. Mean lytic units (+ SE) for cultures containing medium alone, interleukin-2 (IL-2; 100 U/mL), or EMD 273063 (0.25 μg/mL). Significant differences are shown for day 8 versus day 1. (**), P < .01; (***), P < .001.
Because the LA-N-5 target is relatively resistant to fresh NK cells, it is useful for measuring IL-2 augmented killing and ADCC. However, the weak killing of LA-N-5 mediated by fresh PBMC in medium (without supplemental IL-2 in vitro) was not significantly greater on day 8 than on day 1. For patients 19 through 33, standard NK assays were performed using the NK susceptible K562 target cell. A significant increase in percentage cytotoxicity of K562 target cells, when tested either in medium (P < .05) or in the presence of IL-2 (P < .01), was observed on day 8 when compared with day 1 at an effector to target ratio of 50:1 (data not shown).
Serum samples from selected patients were also evaluated to determine functional IL-2 activity of circulating IC and anti-GD2 binding ability of the antibody component of IC. As shown in Figure 5A, the IL-2 responsive Tf-1β cell line demonstrated IL-2–induced proliferation with patient serum obtained after infusion of EMD 273063. An increase in proliferation was seen during the first 4 hours after the start of the 4-hour infusion. Values returned to baseline by 16 hours after this infusion. Serum samples collected at these time points were also examined by flow cytometry for the presence of intact EMD 273063 IC that retains its IL-2 component and its anti-GD2 antibody activity. As shown in Figure 5B, patient serum samples obtained after the EMD273063 infusion contained EMD 273063 capable of binding to the M21 (GD2-positive) cell line as detected by flow cytometry using a PE-labeled anti–IL-2 detection antibody. The amount of IC able to bind to M21 increased during the first 4 hours after the start of the 4-hour infusion. Values returned to baseline by 8 to 16 hours after this infusion.
Fig 5.
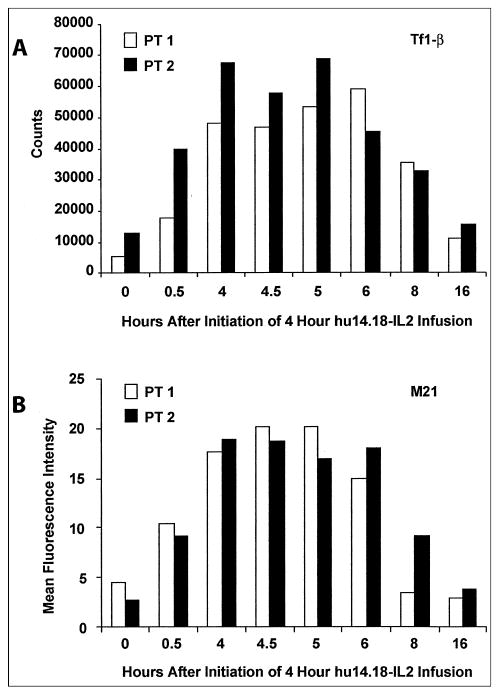
Serum samples were obtained at the indicated times from patients (PT) 1 and 2 (0.8 mg/m2/d) for assays (see Immunologic Monitoring section of Patients and Methods). (A) Proliferation of interleukin-2 responsive Tf-1β cells (cpm); (B) binding of EMD 273063 to M21 (mean fluorescence intensity units). IL-2, interleukin-2.
Finally, in vitro assays were performed with specimens from patients to determine whether administration of EMD 273063 results in conditions in vivo consistent with those needed to achieve ADCC. As shown previously in Figure 4, PBMCs from day 8 show augmented ADCC on GD2+ target cells when EMD 273063 is added to the cytotoxic assay. This same ADCC assay was performed with PBMC from day 8. However, instead of adding EMD 273063 to the assay, serum obtained from the patient before or after EMD 273063 administration was added. As shown in Figure 6, PBMC obtained from patients on day 8 of course 2 were able to mediate augmented killing of the LA-N-5 cell line in the presence of serum obtained after EMD 273063 administration compared with that observed with serum obtained before infusion. This increase was due to a combination of the effectors obtained on day 8 and the serum obtained after treatment with EMD-273063, as the day 8 effectors with pretreatment serum (P < .05), or the postreatment serum without effectors (P < .05) mediated significantly less cytotoxicity. Thus the EMD 273063 circulating in patients after IV administration is able to facilitate ADCC with PBMCs activated in vivo by EMD 273063 from that same patient.
Fig 6.
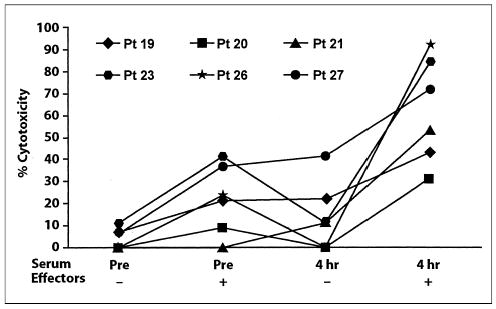
Antibody-dependent cell-mediated cytotoxicity using the LA-N-5 target (% cytotoxicity; effector: target ratio of 50:1) with and without effectors (day 8 of course 2) in medium containing autologous serum obtained pretreatment (pre) or after the initial 4-hour infusion, for six consecutive assays on patients at 4.8 to 6.0 mg/m2/d. The killing by effectors in 4 hours’ serum was six-fold greater than with effectors in preserum when calculated using lytic units (not shown).
DISCUSSION
Antibody-based therapies aimed at gangliosides on melanoma have demonstrated in vivo targeting to melanoma and potential as treatment approaches for metastatic melanoma patients.23 We report the results of the initial clinical trial of EMD 273063, an IC molecule composed of the humanized form of the 14.18 mAb linked to IL-2. The EMD 273063 IC molecule binds well to human melanoma and neuroblastoma cells in vitro and mediates potent ADCC against these cells in vitro using human NK cells as effectors.14,24 It activates potent in vivo destruction of human melanoma tumors grown in immunodeficient mice when combined with IL-2–activated human effector cells.25 It also causes more antitumor effects than comparable amounts of 14.18 mAb combined with IL-2 when administered daily for 3 to 5 days in immunocompetent mice bearing localized or metastatic B78 murine melanoma or NXS2 murine neuroblastoma.25,26
This phase I dose escalation study enrolled 33 adults with melanoma. The MTD15 was determined to be 7.5 mg/m2/d, as two of six patients showed reversible DLT at that dose level during course 1, using the specific DLT criteria defined for this study. The 90% CI for this value is 1.2 to 13.8 mg/m2/d, reflecting the low statistical power of a study with three to six patients per dose level. In a previous clinical trial in which we combined the chimeric form of this mAb (not the IC) with separate constant infusions of IL-2, the MTD of the ch14.18 mAb was found to be 7.5 mg/m2/d.16 DLTs in that study were pain requiring morphine, allergic reactions, weakness, and decreased performance status. As the hu14.18-IL-2 is composed of 16.7% IL-2 and 83.3% hu14.18, the amount of hu14.18 mAb in 7.5 mg of hu14.18-IL-2 is 6.25 mg. Thus the 7.5 mg/m2/d MTD of the chimeric mAb when combined with IL-2 is similar to the amount of hu14.18 mAb (6.25 mg/m2/d) present in the MTD of the hu14.18-IL-2 molecule determined here. Dose-limiting side effects of EMD 273063 at the MTD were IL-2–related and included transient, reversible hypoxia and hypotension. Most of the clinical toxicities seen in this study were anticipated and were similar to those previously reported for IL-2 and anti-GD2 mAb treatments.16,27,28 Most patients in the three highest dose levels had pelvic, abdominal, chest, or extremity pain requiring IV morphine. This pain was similar to what we previously reported for patients treated with ch14.18.16,29 Although this pain could be moderate to severe and require use of intermittent or constant IV infusion of morphine, it could be adequately controlled to the satisfaction of most patients and was not dose-limiting. There was no evidence of motor neuropathy associated with this treatment. The remaining toxicities were similar to those frequently seen with IL-2 given alone. These toxicities were reversible and resolved after therapy.
Two common toxicities with this therapy, as noted by laboratory values, were hypophosphatemia and hyperglycemia. A majority of patients had grade 2 or 3 hypophosphatemia that was noted during routine laboratory monitoring. This hypophosphatemia was not associated with any clinical symptoms and resolved spontaneously in all patients without supplementation. This laboratory finding is thought to represent the effects of IL-2–related intracellular phosphate migration rather than renal wasting of phosphate.30,31 The lack of renal wasting was demonstrated here in patients with grade 3 hypophosphatemia. Treatment-associated hyperglycemia was also present in most patients. The majority of this hyperglycemia was grade 1 (18 patients), but five patients had grade 2 and three patients had grade 3 hyperglycemia. Two patients with known diabetes previously requiring an oral agent required temporary use of insulin for management of this treatment-associated hyperglycemia. Although the etiology of this hyperglycemia is not known, treatment with IL-2 has previously been associated with exacerbation of diabetes mellitus.32 It is important to note that, in all cases, the hyperglycemia was managed without difficulty and without additional clinical sequelae, and this toxicity resolved after therapy was completed.
There were immunologic changes associated with this EMD 273063 therapy, including an increase in lymphocyte count, an increase in the percentage of CD16+ and CD56+ PBMCs, an increase in NK lysis, and an increase in ADCC. Additional evidence for immune activation included an increase in serum levels of CRP and of sIL-2R∞. All of these changes can be seen in patients receiving treatment with IL-2; thus these responses provide evidence that the EMD 273063 molecule has an IL-2 component that functions in vivo and induces similar systemic immune activation as is seen with IL-2. Laboratory analyses of sera and PBMCs showed that the EMD 273063 molecule circulating in patient sera after IV administration retained its ability to activate IL-2–responsive cells through the IL-2 receptor and retained its ability to bind to GD-2–positive tumor cells and deliver IL-2 to their surface, as detected by flow cytometry. All patients achieved conditions necessary for immune activation. Our previous trial combining the ch14.18 mAb with IL-2 infusion in patients with melanoma reported the development of anti-idiotypic antibody against the ch14.18 mAb in treated patients.16 Our preliminary analysis of sera from these 33 patients reported here shows that 16 developed an anti-idiotypic antibody after treatment with hu14.18-IL-2 using a similar assay to that used in our prior report (Hank et al, manuscript in preparation).
In conclusion, a treatment dose and schedule for the IC EMD 273063 that can induce immune activation with acceptable toxicity was identified in patients with melanoma. The finding of four high-risk patients who continue without evidence of disease progression for 26 to 60 months after only two cycles (six doses) of treatment suggests possible clinical benefit from this immunotherapeutic intervention. The MTD of 7.5 mg/m2/d is suggested for phase II testing but will require clear guidelines for cessation of treatment and dose adjustment for DLT. Phase II testing is now planned to determine antitumor activity of hu14.18-IL-2 in patients with metastatic melanoma.
Acknowledgments
We thank our colleagues, Drs Alexander Rakhmilevich, Zane Neal, and Ilia Buhtoiarov of University of Wisconsin–Madison for helpful discussions; Drs Tom Dahl and Mike Super of EMD–Lexigen in Billerica, MA; and Drs Oscar Kashala, Lothar Finke, Knut Sturmhoefel, Raj Malik, Denise Wayne, and Tom Wong of EMD Pharmaceuticals–Durham, NC, for provision of hu14.18-IL-2 and involvement in the conduct of this study; and Kathy Neish for assistance with manuscript preparation.
Footnotes
Work previously presented in part at the 35th Annual Meeting of the American Society of Clinical Oncology, Atlanta, GA, May 15–18, 1999; the 36th Annual Meeting of the American Society of Clinical Oncology, New Orleans, LA, May 20–23, 2000; the 38th Annual Meeting of the American Society of Clinical Oncology, Orlando, FL, May 18–21, 2002; American Association for Cancer Research Annual Meeting, Washington DC, July 11–15, 2003; and Society for Biologic Therapy Annual Meetings, Bethesda, MD, November 8–10, 2001, and San Diego, CA, November 7–9, 2002.
Authors’ disclosures of potential conflicts of interest are found at the end of this article.
Authors’ Disclosures of Potential Conflicts of Interest
The following authors or their immediate family members have indicated a financial interest. No conflict exists for drugs or devices used in a study if they are not being evaluated as part of the investigation. Acted as a consultant within the last 2 years: Ralph Reisfeld, EMD. Served as an officer or member of the Board of a company: Stephen D. Gillies, EMD. Received more than $2,000 per year from a company for either of the last 2 years: Stephen D. Gillies, EMD.
Supported by grant Nos. CA32685, CA14520, CA87025, CA81403, and RR03186 from the National Institutes of Health and a grant from the Midwest Athletes for Childhood Cancer Fund. Partial personnel support was provided by EMD for data management required by EMD for this study, which was beyond the clinical research and data monitoring required for this National Cancer Institute–supported study.
References
- 1.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 2.Mule JJ, Yang JC, Lafeniere R, et al. Identification of cellular mechanisms operation in vivo during the regression of established pulmonary metastases by the systemic administration of high dose recombinant interleukin-2. J Immunol. 1987;139:285–295. [PubMed] [Google Scholar]
- 3.Albertini MR, Sondel PM: Tumor immunology and immunotherapy, in Abeloff MD, Armitage JO, Lichter AS, et al (eds): Clinical Oncology (ed 2). Philadelphia, PA, Churchill Livingstone, 2000, pp 214–241
- 4.Colucci F, Caligiuri MA, Di Santo JP. What does it take to make a natural killer? Nat Rev Immunol. 2003;3:413–425. doi: 10.1038/nri1088. [DOI] [PubMed] [Google Scholar]
- 5.Harel W, Shau H, Hadley CG, et al. Increased lysis of melanoma by in vivo-elicited human lymphokine-activated killer cells after addition of antiganglioside antibodies in vitro. Cancer Res. 1990;50:6311–6315. [PubMed] [Google Scholar]
- 6.Hank JA, Robinson RR, Surfus J, et al. Augmentation of antibody dependent cell mediated cytotoxicity following in vivo therapy with recombinant interleukin-2. Cancer Res. 1990;50:5234–5239. [PubMed] [Google Scholar]
- 7.Shiloni E, Euisenthal A, Sachs D, et al. Antibody-dependent cellular cytotoxicity mediated by murine lymphocytes activated in recombinant interleukin-2. J Immunol. 1987;138:1992–1998. [PubMed] [Google Scholar]
- 8.Bernstein N, Stames C, Levy R. Specific enhancement of the therapeutic effect of anti-idiotype antibodies on a murine B cell lymphoma by IL-2. J Immunol. 1988;140:2839–2845. [PubMed] [Google Scholar]
- 9.Schultz KR, Peace DJ, Badger CC, et al. Monoclonal antibody therapy of murine lymphoma: Enhanced efficacy by concurrent administration of interleukin-2 or lymphokine-activated-killer cells. Cancer Res. 1990;50:5421–5425. [PubMed] [Google Scholar]
- 10.Becker JC, Pancook JD, Gillies SD, et al. T cell-mediated eradication of murine metastatic melanoma induced by targeted interleukin 2 therapy. J Exp Med. 1996;183:2361. doi: 10.1084/jem.183.5.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lode HN, Xiang R, Dreier T, et al. Natural killer cell-mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin-2 therapy. Blood. 1998;91:1706. [PubMed] [Google Scholar]
- 12.Mujoo K, Cheresh D, Yang HM, et al. Disialoganglioside GD2 on human neuroblastoma: Target antigen for monoclonal antibody mediated cytolysis and suppression of tumor growth. Cancer Res. 1987;47:1098–1104. [PubMed] [Google Scholar]
- 13.Cheung NK, Saarinen UM, Neely JE, et al. Monoclonal antibodies to a glycolipid antigen on human neuroblastoma cells. Cancer Res. 1985;45:2642–2649. [PubMed] [Google Scholar]
- 14.Hank JA, Surfus JE, Gan J, et al. Activation of human effector cells by a tumor reactive recombinant anti-ganglioside- GD2/interleukin-2 immunocytokine (ch14.18-IL-2) Clin Cancer Res. 1996;2:1951–1959. [PubMed] [Google Scholar]
- 15.Storer B. Design and analysis of phase I clinical trials. Biometrics. 1989;45:925–937. [PubMed] [Google Scholar]
- 16.Albertini MR, Hank JA, Schiller JH, et al. Phase IB trial of chimeric anti-GD2 antibody plus interleukin-2 for melanoma patients. Clin Cancer Res. 1997;3:1277–1288. [PubMed] [Google Scholar]
- 17.Hank JA, Weil-Hillman G, Surfus JE, et al. Addition of interleukin-2 in vitro augments detection of lymphokine-activated killer activity generated in vivo. Cancer Immunol Immunother. 1990;31:53–59. doi: 10.1007/BF01742496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farner NL, Gan J, de Jong JLO, et al. Alteration of the CD343 Tf-1beta cell line profile in response to long-term exposure to IL-15. Cytokine. 1997;9:316–327. doi: 10.1006/cyto.1996.0171. [DOI] [PubMed] [Google Scholar]
- 19.Gan J, Kendra K, Ricci M, et al. Specific ELISA systems for quantitation of antibody-cytokine fusion proteins. Clin Diagn Lab Immunol. 1999;6:236–242. doi: 10.1128/cdli.6.2.236-242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hank JA, Surfus JE, Gan J, et al: Determination of peak serum levels and immune response to the humanized anti-ganglioside antibody: Interleukin-2 immunocytokine, in Buolamwini JK, Adjei AA (eds): Methods in Molecular Medicine (vol 85). Totowa, NJ, Humana Press, 2003, pp 123–131 [DOI] [PubMed]
- 21.Lindsey JK: Models for Repeated Measurements. Oxford, United Kingdom, Oxford University Press, 1993
- 22.Kendra K, Gan J, Ricci M, et al. Pharmacokinetics and stability of the 14.18-IL-2 fusion protein in mice. Cancer Immunol Immunother. 1999;48:219–229. doi: 10.1007/s002620050569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott AM, Lee F-T, Hopkins W, et al. Specific targeting, biodistribution, and lack of immunogenicity of chimeric anti-GD3 monoclonal antibody KM871 in patients with metastatic melanoma: Results of a phase I trial. J Clin Oncol. 2001;19:3976–3987. doi: 10.1200/JCO.2001.19.19.3976. [DOI] [PubMed] [Google Scholar]
- 24.Sondel PM, Hank JA, Gan J, et al. Preclinical and clinical development of immunocytokines. Curr Opin Investig Drugs. 2003;4:696–700. [PubMed] [Google Scholar]
- 25.Becker JC, Pancook JD, Gillies SD, et al. Eradication of human hepatic and pulmonary melanoma metastases in SCID mice by antibody-interleukin-2 fusion proteins. Proc Natl Acad Sci U S A. 1996;93:2702–2707. doi: 10.1073/pnas.93.7.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lode HN, Xiang R, Varki NM, et al. Targeted interleukin-2 therapy of spontaneous neuroblastoma to bone marrow. J Natl Cancer Inst. 1997;89:1586–1591. doi: 10.1093/jnci/89.21.1586. [DOI] [PubMed] [Google Scholar]
- 27.Frost JD, Ettinger LJ, Hank JA, et al. Phase I/IB trial of murine monoclonal anti-GD2 antibody 14. G2a plus IL-2 in children with refractory neuroblastoma: A report of the Children’s Cancer Group. Cancer. 1997;80:317–333. doi: 10.1002/(sici)1097-0142(19970715)80:2<317::aid-cncr21>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 28.Yu AL, Uttenreuther-Fischer MM, Huang CS, et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol. 1998;16:2169–2180. doi: 10.1200/JCO.1998.16.6.2169. [DOI] [PubMed] [Google Scholar]
- 29.Ozkaynak F, Sondel P, Krailo M, et al. A phase I study of chimeric human/murine anti-ganglioside GD2 monoclonal antibody (ch14.18) with GM-CSF in children with neuroblastoma immediately post hematopoietic stem cell transplantation: A Children’s Cancer Group Study. J Clin Oncol. 2000;18:4077–4085. doi: 10.1200/JCO.2000.18.24.4077. [DOI] [PubMed] [Google Scholar]
- 30.Webb DE, Austen HA, Belldegrun A, et al. Metabolic and renal effects of interleukin-2 immunotherapy for metastatic cancer. Clin Nephrol. 1988;30:141–145. [PubMed] [Google Scholar]
- 31.Kozeny GA, Nicolas JD, Creekmore S, et al. Effects of interleukin-2 immunotherapy on renal function. J Clin Oncol. 1988;6:1170–1176. doi: 10.1200/JCO.1988.6.7.1170. [DOI] [PubMed] [Google Scholar]
- 32.Soni N, Meropol NJ, Porter M, et al. Diabetes mellitus induced by low-dose interleukin-2. Cancer Immunol Immunother. 1996;43:59–62. doi: 10.1007/s002620050304. [DOI] [PubMed] [Google Scholar]