

Abstract
During vertebrate limb development, growth plate chondrocytes undergo temporally and spatially coordinated differentiation that is necessary for proper morphogenesis. Parathyroid hormone-related peptide (PTHrP), its receptor, the PTH/PTHrP receptor, and Indian hedgehog are implicated in the regulation of chondrocyte differentiation, but the specific cellular targets of these molecules and specific cellular interactions involved have not been defined. Here we generated chimeric mice containing both wild-type and PTH/PTHrP receptor (−/−) cells, and analyzed cell–cell interactions in the growth plate in vivo. Abnormal differentiation of mutant cells shows that PTHrP directly signals to the PTH/PTHrP receptor on proliferating chondrocytes to slow their differentiation. The presence of ectopically differentiated mutant chondrocytes activates the Indian hedgehog/PTHrP axis and slows differentiation of wild-type chondrocytes. Moreover, abnormal chondrocyte differentiation affects mineralization of cartilaginous matrix in a non-cell autonomous fashion; matrix mineralization requires a critical mass of adjacent ectopic hypertrophic chondrocytes. Further, ectopic hypertrophic chondrocytes are associated with ectopic bone collars in adjacent perichondrium. Thus, the PTH/PTHrP receptor directly controls the pace and synchrony of chondrocyte differentiation and thereby coordinates development of the growth plate and adjacent bone.
The bones of the developing limb form through the process of endochondral bone formation (1, 2). First, mesenchymal cells condense; cells in the core differentiate into chondrocytes, and the cells at the periphery differentiate into the perichondrium. Then, chondrocytes in the center stop proliferating, differentiate into prehypertrophic and then hypertrophic chondrocytes, induce mineralization of surrounding matrix, and undergo apoptosis. Concomitantly, the perichondrium abutting prehypertrophic and hypertrophic chondrocytes becomes bone collar, and blood vessels invade the mineralized cartilage. In association with vascular invasion, osteoblasts and hematopoietic cells replace the mineralized cartilage with bone and bone marrow. Thus, in this process, differentiation and proliferation of various types of cells must be properly coordinated.
Parathyroid hormone-related peptide (PTHrP), the PTH/PTHrP receptor, and Indian hedgehog (Ihh) are involved in the regulation of chondrocyte differentiation, and their interrelationships have been explored (3–13). Overexpression of PTHrP or expression of a constitutively active PTH/PTHrP receptor delays chondrocyte differentiation (3, 4). Mice missing PTHrP or the PTH/PTHrP receptor show accelerated differentiation of chondrocytes (5–9). Overexpression of Ihh or administration of its surrogate, Sonic hedgehog, also delays chondrocyte differentiation in association with increased PTHrP mRNA expression, but Sonic hedgehog has no effect on limbs from either PTHrP (−/−) or PTH/PTHrP receptor (−/−) mice (8, 9). On the basis of these data, a model was proposed that PTHrP, the PTH/PTHrP receptor, and Ihh form a negative feedback loop that regulates the rate of chondrocyte differentiation (9). However, the model suggested by these studies lacks critical pieces of information necessary for understanding how bone formation is regulated. First, the specific target cells on which PTHrP and Ihh act in a stage-specific manner have not been defined. Proliferating chondrocytes most obviously affected by PTHrP or PTH/PTHrP receptor knockout normally express little PTH/PTHrP receptor mRNA, while more differentiated prehypertrophic chondrocytes express large amounts of this mRNA (14, 15), and these prehypertrophic chondrocytes have been thought to be the target of PTHrP signaling (9). Moreover, many other tissues that could influence bone development, such as the kidney, also express the PTH/PTHrP receptor (16). The specific target cells of Ihh signaling are also incompletely defined. Second, the aforementioned feedback model for regulation of chondrocyte differentiation was based on experiments that involved the use of supraphysiological amounts of PTHrP and Ihh, and, therefore, the model needs to be tested in a more physiological setting. Third, the functional relationship of chondrocyte differentiation to other critical processes in endochondral bone development, such as mineralization of cartilaginous matrix and bone collar formation, is poorly understood. To address these questions, we have generated chimeric mice containing both wild-type and PTH/PTHrP receptor (−/−) cells, and have analyzed cell–cell interactions in the growth plate in vivo.
MATERIALS AND METHODS
Generation of Embryonic Stem (ES) Cell Lines.
To introduce a β-galactosidase transgene as a marker, mice heterozygous for the PTH/PTHrP receptor-null mutation in a C57BL/6–129/SvJ background (8) were mated with C57BL/6 mice carrying a β-galactosidase transgene that was engineered to be expressed ubiquitously (17). By further mating, mice were made heterozygous for the PTH/PTHrP receptor-null mutation and homozygous for the β-galactosidase transgene. They were then mated with mice heterozygous for the PTH/PTHrP receptor-null mutation to produce blastocysts. ES cell lines were established as previously described (18) and were genotyped by Southern blot analysis. The animal care was in accordance with the policies of the Massachusetts General Hospital.
Southern Blot Analysis.
Southern blot analysis was performed as previously described (5). To detect disrupted alleles of the PTH/PTHrP receptor gene, 10 μl (≈10 μg) of genomic DNA was digested with SacI, Southern blotted, and probed with a 1.0-kb SacI-KpnI fragment containing a part of the first intron. The sizes of genomic DNA fragments expected from the normal and disrupted alleles are 5.6 and 4.9 kb, respectively. To detect β-galactosidase transgene alleles, 10 μl (≈10 μg) of genomic DNA were digested with StuI, Southern blotted, and probed with a 140-bp fragment containing a 5′-flanking region of the transgene (17). The sizes of DNA fragments expected from the normal and the mutant alleles are ≈7 and ≈12 kb, respectively.
Generation of Chimeric Mice.
Chimeras were generated by blastocyst injection as previously described (19). ES cell lines homozygous for the PTH/PTHrP receptor-null mutation were injected into wild-type BALB/cJ or C57BL/6 blastocysts. As a control, wild-type ES cells established at the same time were injected into wild-type blastocysts. For production of chimeras with various degrees of ES cell contribution, the number of ES cells injected into the blastocele cavity was varied from 3 to 15. Progeny of three independent homozygous clones yielded the identical phenotype. All the ES cell lines gave germ line transmission. All observations in the results were verified in at least five different chimeric mice. The fraction of PTH/PTHrP receptor (−/−) cells was estimated by β-galactosidase staining of the skin and the periarticular growth plate.
Histological Analysis.
Chimeras were sacrificed at various ages, dissected, and fixed in 4% paraformaldehyde/PBS at 4°C for 4 hr. Tissues were stained with X-Gal (5-bromo-4-chloro-3-indolyl β-d-galactoside) as described previously (20). Subsequently, they were processed, embedded in paraffin, and cut. Sections were stained with H&E or nuclear fast red (Vector Laboratories) for morphological study. For detection of mineralization, sections were stained with 1% alizarin red S; mineral is stained bright red.
In Situ Hybridization.
Tissues were fixed in 4% paraformaldehyde/PBS overnight at 4°C, processed, embedded in paraffin, and cut. In situ hybridization was performed as described previously (15) by using complimentary 35S-labeled riboprobes. The probes for mouse type X collagen, mouse Patched (Ptc), mouse Ihh, and rat PTHrP were obtained from Dr. Bjorn Olsen (Harvard Medical School, Boston, MA), Dr. Ron Johnson (Stanford University, Stanford, CA), Dr. Benoit Saint-Jacques (Harvard Medical School, Boston MA) and Dr. Andrew C. Karaplis (McGill University, Montreal, Canada), respectively.
RESULTS
Generation of Chimeric Mice and Ectopic Differentiation of Mutant Chondrocytes.
ES cell lines homozygous for the PTH/PTHrP receptor-null mutation were generated de novo from inner cell masses. Four homozygous mutant, four heterozygous mutant, and five wild-type clones were isolated. Each carried one copy of a β-galactosidase transgene widely expressed in mouse tissues (17). All cells in the growth plate from the mice carrying the β-galactosidase transgene alone exhibited β-galactosidase activity and normal morphology (data not shown). In mice generated by injecting wild-type cells expressing the β-galactosidase transgene (stained blue) into wild-type blastocysts, cells descended from the ES cells and from the host blastocysts behaved indistinguishably, as expected (Fig. 1, A and B). In contrast, in the growth plates of chimeric mice generated by injecting PTH/PTHrP receptor (−/−) ES cells (stained blue) into wild-type blastocysts, mutant cells prematurely differentiated into cells resembling hypertrophic chondrocytes while surrounded by wild-type proliferating chondrocytes (Fig. 1 C and D). All mutant cells above a line approximately corresponding to the level at which the growth plate narrowed (arrowhead) resembled proliferating chondrocytes; those below the line appeared hypertrophic. These ectopic hypertrophic-like cells expressed type X collagen and Ihh mRNAs (Fig. 2), markers for hypertrophic and prehypertrophic/hypertrophic chondrocytes, respectively (8, 9). They also underwent apoptosis, detected by terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling and stopped expressing H4 mRNA, a marker for cells in S phase (data not shown). A comparison of the fraction of blue mutant cells in the periarticular and distal portions of the growth plate (Fig. 1C) shows that these ectopic hypertrophic cells became gradually outnumbered by surrounding wild-type cells, which continued to proliferate. Thus, the mutant cells ectopically differentiated into hypertrophic chondrocytes independently of any signals from surrounding wild-type proliferating chondrocytes. We conclude that the specific target of PTHrP signaling is the proliferating chondrocyte. PTHrP acts directly on the low level of PTH/PTHrP receptors on proliferating chondrocytes to slow and synchronize their differentiation, instead of relying on a cascade involving more distally situated, prehypertrophic cells.
Figure 1.
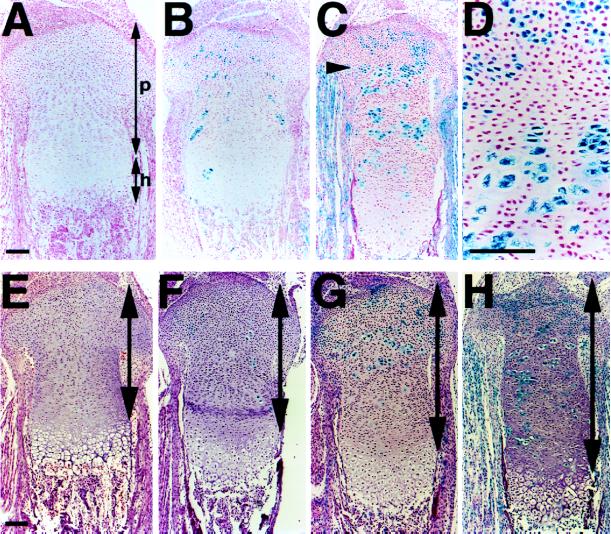
Ectopic differentiation of PTH/PTHrP receptor (−/−) chondrocytes and elongation of chimeric growth plates. (A and B) Sections of the tibiae from d17.5 wild-type embryos (A) and d17.5 wild-type embryos containing cells with one β-galactosidase transgene (stained blue) and cells with no transgene (B) were stained for β-galactosidase activity and counterstained with nuclear fast red. Cells with and without the transgene behave indistinguishably. p, proliferating layer; h, hypertrophic layer including prehypertrophic chondrocytes. (C and D) Sections of the tibiae from d17.5 chimeric embryos containing both wild-type and PTH/PTHrP receptor (−/−) cells were stained for β-galactosidase activity and counterstained with nuclear fast red. PTH/PTHrP receptor (−/−) cells (stained blue) ectopically differentiate into hypertrophic-like cells in the middle of wild-type proliferating chondrocytes. All mutant cells below a line approximately corresponding to the level at which the growth plate narrows (arrowhead) appear hypertrophic. (E–H) Sections of the tibiae from wild-type (E) and chimeric embryos with various contributions of PTH/PTHrP receptor (−/−) cells (F–H) were stained for β-galactosidase activity and counterstained with H&E. Contributions of mutant cells are estimated ≈10% (F), 30% (G), and 60% (H). In proportion to the contribution of mutant cells, columns of wild-type proliferating chondrocytes elongate (two-headed arrows). (Bar = 100 μm.)
Figure 2.
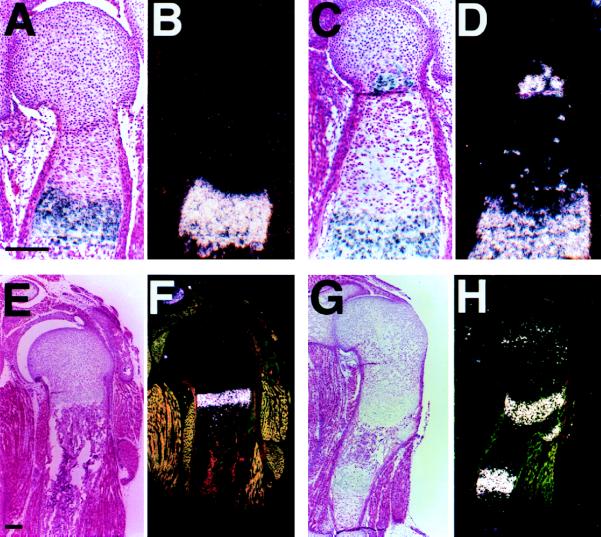
Expression of type X collagen and Ihh by ectopically differentiated PTH/PTHrP receptor (−/−) chondrocytes. (A–D) In situ hybridization of sections of the humeri from d17.5 embryos with a mouse type X collagen antisense probe. In the wild-type (A, bright field; B, dark field), type X collagen is expressed in hypertrophic chondrocytes. In the chimera containing both wild-type and PTH/PTHrP receptor (−/−) cells (C, bright field; D, dark field), ectopically differentiated mutant cells also express type X collagen in the proliferating layer. (E–H) In situ hybridization of sections of the humeri from d17.5 embryos with a mouse Ihh antisense probe. In the wild-type (E, bright field; F, dark field), Ihh is expressed in prehypertrophic and hypertrophic chondrocytes. In the chimera (G, bright field; H, dark field), ectopically differentiated mutant cells also express Ihh in the proliferating layer. (Bar = 200 μm.)
Up-Regulation of the Ihh/PTHrP Axis and Elongation of Chimeric Growth Plates.
The independent patterns of differentiation of wild-type and PTH/PTHrP receptor (−/−) chondrocytes within the chimeric growth plate allowed us to assess effects of subtle perturbation of the Ihh/PTHrP axis in an in vivo setting. A key prediction of the Ihh/PTHrP negative feedback model is that the periarticular perichondrium directly or indirectly senses the production of Ihh by prehypertrophic and hypertrophic chondrocytes and responds by slowing chondrocyte differentiation through the actions of PTHrP. Because mutant chondrocytes ectopically differentiate and produce Ihh much closer to the articular surface than do wild-type hypertrophic chondrocytes (Fig. 2 G and H), this ectopic production of Ihh might lead to an increase in PTHrP expression in the periarticular perichondrium and consequent slowing of differentiation of wild-type proliferating chondrocytes in chimeras. In the growth plates of chimeras, the length of the columns of wild-type proliferating chondrocytes increased as the contribution of PTH/PTHrP receptor (−/−) chondrocytes increased (Fig. 1 E–H). Thus, wild-type chondrocytes slowed their differentiation in the presence of ectopic hypertrophic chondrocytes. In wild-type mice, expression of Patched (Ptc), a marker of Ihh action (21–23), was noted in proliferating chondrocytes, most strongly in cells adjacent to Ihh-producing prehypertrophic and hypertrophic chondrocytes, with expression decreasing toward the end of the bone (Fig. 3 A and B). In contrast, in chimeras, proliferating chondrocytes surrounding ectopic hypertrophic chondrocytes overexpressed Ptc mRNA, evidence of ectopic action of Ihh (Fig. 3 C and D). Further, in chimeras, PTHrP mRNA expression was increased in the periarticular growth plate including not only perichondrium but also proliferating chondrocytes (Fig. 3 E–H). Thus, the presence of ectopic hypertrophic chondrocytes in the growth plate led to the activation of the Ihh/PTHrP axis, as well.
Figure 3.
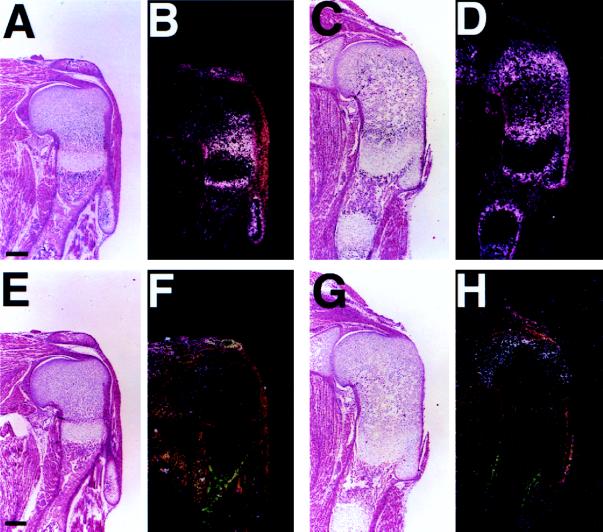
Up-regulation of Ptc and PTHrP expression in chimeric growth plates. (A–D) In situ hybridization of sections of the humeri from d17.5 embryos with a mouse Ptc antisense probe. In the wild-type (A, bright field; B, dark field), Ptc is expressed in proliferating chondrocytes, most strongly adjacent to Ihh-producing prehypertrophic chondrocytes, with expression decreasing toward the articular surface. In the chimera containing both wild-type and PTH/PTHrP receptor (−/−) cells (C, bright field; D, dark field), proliferating chondrocytes surrounding ectopic hypertrophic chondrocytes overexpress Ptc mRNA. (E–H) In situ hybridization of sections of the humeri from d17.5 embryos with a rat PTHrP antisense probe. In the wild-type (E, bright field; F, dark field), PTHrP is weakly expressed in the periarticular region. In the chimera (G, bright field; H, dark field), PTHrP expression is strongly up-regulated in the same region. (Bar = 200 μm.)
Ectopic Mineralization of Cartilaginous Matrix in Chimeric Bones.
During the final steps of chondrocyte differentiation, hypertrophic chondrocytes produce a mineralized matrix (1, 2) (Fig. 4A, arrows). This mineralized cartilaginous matrix provides a rigid scaffold on which osteoblasts deposit and mineralize bone matrix. The heterogeneous pattern of chondrocyte differentiation in the chimeric growth plate allowed us to consider the relative cell autonomy of this process. The pattern of matrix mineralization was examined in a series of chimeras with varying contributions of PTH/PTHrP receptor (−/−) cells. In chimeras with up to ≈60% contribution of mutant cells (Fig. 4B), matrix adjacent to ectopic hypertrophic chondrocytes did not mineralize in the proliferating layer. However, in chimeras with a very high (>90%) contribution, matrix adjacent to ectopic hypertrophic chondrocytes did mineralize when the cells were present in large clusters (Fig. 4 C and D, arrows). These data demonstrate that chondrocyte differentiation affects mineralization of cartilaginous matrix in a non-cell autonomous fashion and that matrix mineralization requires a critical mass of adjacent hypertrophic chondrocytes.
Figure 4.
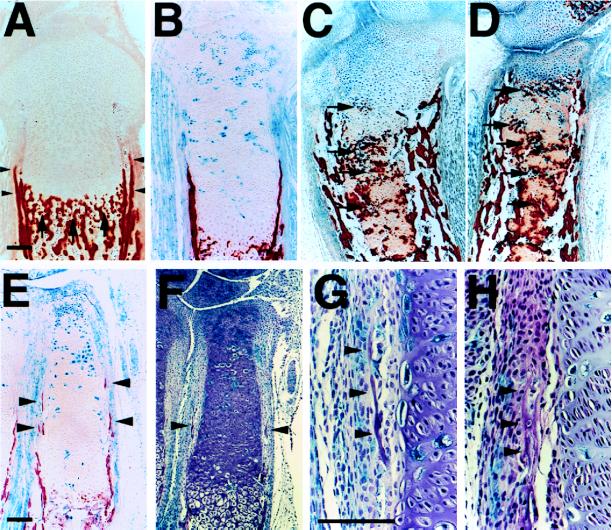
Ectopic mineralization of cartilaginous matrix and ectopic bone collar formation in chimeric bones. (A–D) Sections of the tibiae from d17.5 embryos were stained for β-galactosidase activity and also with alizarin red S for detection of mineralization. In the wild-type (A), mineralization of cartilaginous matrix occurs adjacent to late hypertrophic chondrocytes (arrows). No mineralization is observed adjacent to ectopic hypertrophic chondrocytes in the tibiae from chimeras with ≈60% contribution of PTH/PTHrP receptor (−/−) cells (B), whereas in chimeras with >90% contribution, matrix adjacent to ectopic hypertrophic chondrocytes in large clusters mineralize (C and D, arrows). (E–H) Sections of the radii from d17.5 chimeric embryos with ≈60% contribution of mutant cells were stained for β-galactosidase activity and also stained with alizarin red S (E) or H&E (F–H). In the wild-type (A), bone collars are formed in the perichondrium abutting prehypertrophic and hypertrophic chondrocytes (arrowheads). No ectopic bone collar formation is observed in the tibiae of chimeras with ≈60% contribution (B). However, in smaller bones such as the radii of the same chimeras, ectopic bone collars are formed (E and F, arrowheads). At higher magnification, ectopic bone collars are seen near clusters of ectopic hypertrophic chondrocytes; ectopic bone collars include both wild-type and PTH/PTHrP receptor (−/−) osteoblasts (stained blue) (G and H, arrowheads). In the tibiae of chimeras with >90% contribution, there is intensive ectopic bone collar formation (C and D). (Bar = 100 μm.)
Ectopic Bone Collar Formation in Chimeric Bones.
Normally, bone collars are formed in the perichondrium abutting prehypertrophic and hypertrophic chondrocytes (Fig. 4A, arrowheads) (1, 2). The mechanisms that assure coordinated formation of the bone collar at the site of differentiation of hypertrophic chondrocytes are unknown. Interestingly, bone collars in PTHrP (−/−) and PTH/PTHrP receptor (−/−) mice are also formed in the perichondrium abutting hypertrophic chondrocytes, despite much shorter layers of proliferating chondrocytes (refs. 5 and 8; data not shown). Two kinds of hypothesis can explain these observations. Bone collar formation and differentiation of growth plate chondrocytes might be independent processes, each regulated by PTHrP. Alternatively, PTHrP might only regulate chondrocyte differentiation, with the differentiated chondrocytes then regulating bone collar formation. Ectopic differentiation of PTH/PTHrP receptor (−/−) cells into hypertrophic chondrocytes in chimeras allowed us to distinguish these alternatives. If the first hypothesis is correct, then mutant perichondrial cells will form bone collars much closer to the articular surface than will wild-type cells. If the second hypothesis is correct, then ectopic bone collars will be formed by both wild-type and mutant cells in the perichondrium close to ectopic hypertrophic chondrocytes. No ectopic bone collar formation was observed in the tibiae of chimeras with up to a ≈60% of contribution of mutant cells (Fig. 4B). However, in smaller bones such as the radii and ulnae of the same chimeras, ectopic bone collars were observed (Fig. 4 E and F, arrowheads). At higher magnification, ectopic bone collars were seen near clusters of ectopic hypertrophic chondrocytes; both wild-type and mutant osteoblasts were found with no preference in the ectopic bone collar (Fig. 4 G and H, arrowheads). In chimeras with >90% contribution, ectopic bone collars were found in the tibiae, too (Fig. 4 C and D). In total, in 12 chimeras with >60% contribution, 23 bones had 36 ectopic bone collars. All ectopic bone collars were formed near (<100 μm away from) clusters of ectopic hypertrophic chondrocytes and contained mixtures of wild-type and mutant osteoblasts. Thus, these data suggest that bone collars are induced in the perichondrium by hypertrophic chondrocytes and that a critical mass of hypertrophic chondrocytes is required for this induction.
DISCUSSION
Our results demonstrate that the specific target of PTHrP signaling during limb development is the proliferating chondrocyte. Although proliferating chondrocytes express only low levels of the PTH/PTHrP receptor, the pattern of differentiation of PTH/PTHrP receptor (−/−) chondrocytes in chimeric growth plates shows that the receptors on proliferating chondrocytes are needed to slow their differentiation at a normal rate. In chimeras with up to ≈60% contribution of mutant cells, all mice evaluated were normocalcemic (data not shown), unlike PTHrP (−/−) or PTH/PTHrP receptor (−/−) mice (24), so we could exclude an effect of hypocalcemia on the observed bone phenotype.
Elongation of columns of wild-type proliferating chondrocytes in chimeric growth plates, in association with the activation of the Ihh/PTHrP axis, suggests that the growth plate senses the presence of ectopic hypertrophic chondrocytes and slows chondrocyte differentiation by a local feedback loop involving Ihh and PTHrP. This loop controls both the rate and the synchrony of chondrocyte differentiation. The importance of the feedback loop in controlling the synchrony of chondrocyte differentiation explains the admixture of proliferating and hypertrophic chondrocytes seen in PTHrP (−/−) and PTH/PTHrP receptor (−/−) mice (9, data not shown).
The increase in PTHrP expression in the periarticular growth plate, which includes proliferating chondrocytes as well as perichondrium, suggests that this region is a target of Ihh signaling during limb development, but whether Ihh directly acts on the periarticular growth plate remains to be studied.
The patterns of chondrocyte mineralization of chimeric growth plates illustrate the functional importance of coordinated differentiation of adjacent chondrocytes. Presumably some combination of secreted stimulators or inhibitors of mineralization requires a critical mass of hypertrophic chondrocytes to provide an appropriate milieu for mineralization. The precise mechanism involved will require further analysis. The induction of bone collars adjacent to ectopic hypertrophic chondrocytes further stresses the importance of coordinated chondrocyte differentiation. The identity of the relevant molecular signals for bone collar induction cannot be determined from our studies, but the involvement of Ihh seems plausible. Ihh is expressed by ectopic hypertrophic chondrocytes. Ihh stimulated osteogenic differentiation of mesenchymal cell lines in vitro, and Sonic hedgehog induced ectopic bone formation in vivo (25, 26). When chicken limbs were infected with Ihh-producing retrovirus, bone collar formation was accelerated despite suppression of formation of hypertrophic chondrocytes (9). Further, in our chimeric mice, expression of Ptc was up-regulated in osteoblasts surrounding ectopic bone collars (data not shown). These data suggest that Ihh may have a role in bone collar formation, but the precise mechanism remains to be established.
Acknowledgments
We thank Drs. H. Jüppner, A. McMahon, and C. Tabin for critical reading of the manuscript. This work was supported by the National Institutes of Health Grant DK47038.
ABBREVIATIONS
- PTH
parathyroid hormone
- PTHrP
parathyroid hormone-related peptide
- Ihh
Indian hedgehog
- Ptc
Patched
- ES
embryonic stem
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1. Erlebacher A, Filvaroff E H, Gitelman S E, Derynck R. Cell. 1995;80:371–378. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- 2.Marks S C, Hermey D C. In: Principles of Bone Biology. Bilezikian J P, Raisz L G, Rodan G A, editors. San Diego: Academic; 1996. pp. 3–14. [Google Scholar]
- 3.Weir E C, Philbrick W M, Amling M, Neff L A, Baron R, Broadus A E. Proc Natl Acad Sci USA. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs C S, Lee K, Pirro A, Kronenberg H M, Jüppner H. Proc Natl Acad Sci USA. 1997;94:13689–13694. doi: 10.1073/pnas.94.25.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karaplis A C, Luz A, Glowacki J, Bronson R T, Tybulewicz V L J, Kronenberg H M, Mulligan R C. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 6.Amizuka N, Henderson J E, Warshawsky H, Goltzman D, Karaplis A C. J Cell Biol. 1994;126:1611–1623. doi: 10.1083/jcb.126.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee K, Lanske B, Karaplis A C, Deeds J D, Kohno H, Nissenson R A, Kronenberg H M, Segre G V. Endocrinology. 1996;137:5109–5118. doi: 10.1210/endo.137.11.8895385. [DOI] [PubMed] [Google Scholar]
- 8.Lanske B, Karaplis A C, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize L H K, Mulligan R C, Abou-Samra A-B, et al. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 9.Vortkamp A, Lee K, Lanske B, Segre G V, Kronenberg H M, Tabin C J. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 10.Riddle R D, Johnson R L, Laufer E, Tabin C. Cell. 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 11.Echelard Y, Epstein D J, St-Jacques B, Shen L, Mohler J, McMahon J A, McMahon A P. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 12.Krauss S, Concordet J-P, Ingham P W. Cell. 1993;75:1431–1444. doi: 10.1016/0092-8674(93)90628-4. [DOI] [PubMed] [Google Scholar]
- 13.Bitgood M J, McMahon A P. Dev Biol. 1995;172:126–138. doi: 10.1006/dbio.1995.0010. [DOI] [PubMed] [Google Scholar]
- 14.Lee K, Deeds J D, Chiba S, Un-no M, Bond A T, Segre G V. Endocrinology. 1994;134:441–450. doi: 10.1210/endo.134.1.8275957. [DOI] [PubMed] [Google Scholar]
- 15.Lee K, Deeds J D, Segre G V. Endocrinology. 1995;136:453–463. doi: 10.1210/endo.136.2.7835276. [DOI] [PubMed] [Google Scholar]
- 16.Jüppner H, Abou-Samra A-B, Freeman M, Kong X F, Schipani E, Richards J, Kolakowski L F, Jr, Hock J, Potts J T, Kronenberg H M, et al. Science. 1991;254:1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- 17.Zambrowicz B P, Imamoto A, Fiering S, Herzenberg L A, Kerr W G, Soriano P. Proc Natl Acad Sci USA. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson E J. In: Teratocarcinomas and Embryonic Stem Cells. Robertson E J, editor. Oxford: IRL; 1987. pp. 71–112. [Google Scholar]
- 19.Bradley A. In: Teratocarcinomas and Embryonic Stem Cells. Robertson E J, editor. Oxford: IRL; 1987. pp. 113–152. [Google Scholar]
- 20.Rossert J, Eberspaecher H, de Crombrugghe B. J Cell Biol. 1995;129:1421–1432. doi: 10.1083/jcb.129.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stone D M, Hynes M, Armanini M, Swanson T A, Gu Q, Johnson R A, Scott M P, Pennica D, Goddard A, Phillips H, et al. Nature (London) 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 22.Marigo V, Davey R A, Zuo Y, Cunningham J M, Tabin C J. Nature (London) 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- 23.Goodrich L V, Johnson R L, Milenkovic L, McMahon J A, Scott M P. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- 24.Kovacs C S, Lanske B, Hunzelman J L, Guo J, Karaplis A C, Kronenberg H M. Proc Natl Acad Sci USA. 1996;93:15233–15238. doi: 10.1073/pnas.93.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinto N, Iwamoto M, Enomoto-Iwamoto M, Noji S, Ohuchi H, Yoshioka H, Kataoka H, Wada Y, Yuhao G, Takahashi H E, et al. FEBS Lett. 1997;404:319–323. doi: 10.1016/s0014-5793(97)00014-8. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura T, Aikawa T, Iwamoto-Enomoto M, Iwamoto M, Higuchi Y, Maurizio P, Kinto N, Yamaguchi A, Noji S, Kurisu K, et al. Biochem Biophys Res Commun. 1997;237:465–469. doi: 10.1006/bbrc.1997.7156. [DOI] [PubMed] [Google Scholar]