

Abstract
When concentrated in mildly acidic solutions, bovine pancreatic ribonuclease (RNase A) forms long-lived oligomers including two types of dimer, two types of trimer, and higher oligomers. In previous crystallographic work, we found that the major dimeric component forms by a swapping of the C-terminal β-strands between the monomers, and that the minor dimeric component forms by swapping the N-terminal α-helices of the monomers. On the basis of these structures, we proposed that a linear RNase A trimer can form from a central molecule that simultaneously swaps its N-terminal helix with a second RNase A molecule and its C-terminal strand with a third molecule. Studies by dissociation are consistent with this model for the major trimeric component: the major trimer dissociates into both the major and the minor dimers, as well as monomers. In contrast, the minor trimer component dissociates into the monomer and the major dimer. This suggests that the minor trimer is cyclic, formed from three monomers that swap their C-terminal β-strands into identical molecules. These conclusions are supported by cross-linking of lysyl residues, showing that the major trimer swaps its N-terminal helix, and the minor trimer does not. We verified by X-ray crystallography the proposed cyclic structure for the minor trimer, with swapping of the C-terminal β-strands. This study thus expands the variety of domain-swapped oligomers by revealing the first example of a protein that can form both a linear and a cyclic domain-swapped oligomer. These structures permit interpretation of the enzymatic activities of the RNase A oligomers on double-stranded RNA.
Keywords: 3D domain swapping, bovine pancreatic ribonuclease, protein oligomerization, enzyme activity on double stranded RNA, cross-linking
3D domain swapping is a mechanism for protein oligomerization, in which two or more molecules of the same protein form an oligomer by exchanging identical domains. In a 3D domain-swapped dimer, one subunit lends a domain to replace the identical domain of the other subunit, and vice versa. The interface that exists between the swapped domain and the rest of the protein in both the monomer and the domain-swapped oligomer is termed the closed interface. The interface that exists only in the domain-swapped oligomer is termed the open interface. The loop presenting different conformations in the monomer and the domain-swapped oligomer is termed the hinge loop. Since 3D domain swapping was found in the dimeric structure of diphtheria toxin (Bennett et al. 1994), more than 30 proteins have been reported to be domain swapped. 3D domain swapping is a possible mechanism for the formation of protein aggregates including amyloids (Klafki et al. 1993; Bennett et al. 1995; Schlunegger et al. 1997; Cohen and Prusiner 1998; Liu et al. 1998, 2001). 3D domain swapping also endows proteins with additional properties, for example, metal binding in the case of CksHs2 (Parge et al. 1993), and allostery and antitumor and immunosuppression activity in the case of bovine seminal ribonuclease (Vescia and Tramontano 1980; Piccoli et al. 1988; Cafaro et al. 1995). To explore the variety, mechanism, and physiological significance of 3D domain swapping, we have investigated structures of RNase A oligomers. We determined previously the structures of the two domain-swapped RNase A dimers. Here, we report the structural characterization of the two RNase A trimers.
RNase A forms oligomers after lyophilization in acetic acid. Among these oligomers are the two types of trimers, as well as the two types of dimers. The formation of these oligomers was first observed by Crestfield et al. (1962). Further biochemical studies on these oligomers were carried out by Libonati et al. (1996) and Gotte et al. (1999). One trimer (the major trimer) predominates over the other trimer (the minor trimer), similar to the two dimers (Gotte et al. 1999). The two trimers show different biophysical properties on gel-filtration chromatography, ion-exchange chromatography, and native gel electrophoresis, suggesting that they have different quaternary structures. Both trimers have higher enzyme activity on double-stranded (ds) RNA than do the two dimers and the monomer (Gotte et al. 1999). RNase A has also been found as a domain-swapped dimer under conditions similar to physiological conditions (Park and Raines 2000).
The structures of the RNase A dimers offer hints about the structures of the trimers. In our studies of the structures of the RNase A dimers, we found the major dimer forms by swapping its C-terminal β-strand (Liu et al. 2001), whereas the minor dimer forms by swapping its N-terminal α-helix (Liu et al. 1998). On the basis of these structures, we constructed a model of an RNase A trimer, with both types of swapping taking place in the same molecule (Liu et al. 2001). By this mechanism, monomers can continuously be added to either end of the domain-swapped complex to form longer linear oligomers with no exposed closed interface (no dangling domains). However, until the structures of the two trimers are known, this model remains to be verified. In addition, it is unknown whether the model, if correct, represents the structure of the major trimer or the minor trimer. Furthermore, what is the structure of the other trimer? To verify our model, to answer these questions, and to continue to explore the repertoire of 3D domain swapping, we conducted dissociation and cross-linking studies of the RNase A trimers and determined the crystal structure of the minor trimer.
Results
Dissociation of the two trimers
We observed that the two trimers dissociate over weeks into dimers and monomers when they are stored at a high concentration at 4°C. Thermal treatment accelerates the dissociation. Because there are two types of dimers, studying the components of the dissociation products may help understand the structures of the trimers. On the basis of our previous trimer model (Fig. 1A ▶), the dissociation product should comprise the monomer (Fig. 1B ▶), the major dimer (Fig. 1C ▶), and the minor dimer (Fig. 1D ▶). These components can be distinguished by cation exchange chromatography. To test the trimer model, we carried out a thermal dissociation experiment. Figure 2 ▶ shows the elution patterns of the dissociation products from the major and the minor trimers on a cation exchange column. From the elution patterns, it is clear that the major trimer dissociates into the major dimer, the minor dimer, and the monomer, whereas the minor trimer mainly dissociates into the major dimer and the monomer. The dissociation products of the major trimer are consistent with what is predicted from the previous trimer model, suggesting that the model represents the quaternary structure of the major trimer.
Fig. 1.

Speculative dissociation pathways of the model of RNase A linear trimer. The proposed model of a trimer (A) can dissociate in two ways. (Top pathway) The blue subunit dissociates from the trimer and refolds to form the monomer (B). The remaining two subunits (red and green) refold to form the major dimer (C). (Bottom pathway) The red subunit dissociates from the trimer and refolds to form the monomer (B). The remaining two subunits (green and blue) refold to form the minor dimer (D). The figure was created using Raster 3D (Merritt and Bacon 1997).
Fig. 2.

Cation exchange chromatograms of the dissociation of the two RNase A trimers. The two trimers were heated at 55°C for 5 min, loaded onto a cation exchange Vydac S column, and eluted as described in Materials and Methods. The major trimer (broken line) dissociates into the minor dimer, the major dimer, and the monomer, whereas the minor trimer (solid line) dissociates into the major dimer and the monomer. There is also a trace amount of the minor dimer in the dissociation product of the minor trimer. The pattern of dissociation of the major trimer is consistent with the linear trimer model of Fig. 1A ▶.
Simultaneously with this dissociation experiment at UCLA, Nenci et al. (2001) found in Verona that the major trimer dissociates into both major and minor dimers. Their work by the alternative method of gel electrophoresis reaches the same conclusion as the present work by gel filtration, and is described independently.
There is also a trace amount of the minor dimer in the dissociation product of the minor trimer (Fig. 2 ▶). This could be either a contamination from the minor dimer or an interconversion between the minor and the major dimers. To test the possibility of the interconversion, the RNase A major and minor dimers were individually incubated at 55°C for 5 min, and then subjected to cation exchange chromatography. The elution patterns show there is no interconversion between the two dimers. The possibility of contamination is tested by the cross-linking experiment below.
Cross-linking of the trimers
DFDNB cross-links Lys 7 and Lys 41 of RNase A (Lin et al. 1984). In the minor dimer, Lys 7 is located on the swapped helix and Lys 41 is located on the main domain. The cross-linking, therefore, occurs intermolecularly in the minor dimer and results in a covalent dimer that can survive the thermal treatment. However, the major dimer forms by swapping the C-terminal β-strand, on which neither Lys 7 nor Lys 41 is located (Liu et al. 2001). The cross-linking of Lys 7 and Lys 41, therefore, occurs intramolecularly in the major dimer and does not prevent the major dimer from dissociation during thermal treatment. Consequently, the cross-linked major dimer dissociates into cross-linked monomer upon thermal treatment. To test the possibility of contamination and to further confirm the thermal dissociation results, the two trimers were further purified on a cation exchange column. The purified trimers were individually cross-linked with DFDNB, heated, and loaded onto a Vydac S column. RNase A monomer and the two dimers were cross-linked as standards.
The elution patterns of the cross-linked dimers and trimers are shown in Figure 3 ▶. In the elution pattern of the cross-linked minor dimer, there are peaks of the cross-linked monomer, the cross-linked minor dimer, and the intact monomer. In the elution pattern of the cross-linked major dimer, there are peaks of the cross-linked monomer, the intact monomer, and unidentified components. These results are consistent with what is predicted, on the basis of the structures of the two dimers of RNase A. After cross-linking and thermal treatment, the major trimer yields the cross-linked monomer, the cross-linked minor dimer, and the intact monomer (Fig. 3A ▶). The minor trimer, however, yields the cross-linked monomer and the intact monomer, but no cross-linked minor dimer (Fig. 3B ▶). Therefore, domain swapping of the N-terminal helix occurs in the major trimer, but not in the minor trimer. These results further support the data from the thermal dissociation experiment, and indicate that the trace amount of the minor dimer in the dissociation products of the minor trimer in Figure 2 ▶ is from contamination (i.e., incomplete separation by ion exchange chromatography).
Fig. 3.

Cation exchange chromatograms of the cross-linked RNase A major trimer (A) and the cross-linked minor trimer (B). The major dimer (broken line) and the minor dimer (dotted line) were also cross-linked and used as standards. All the major and minor trimers dissociate during the cross-linking reaction. Cross-linking of the major trimer (A) yields the cross-linked monomer, the intact monomer, and the cross-linked minor dimer, whereas cross-linking of the minor trimer (B) yields the cross-linked monomer and the intact monomer.
Models for the two RNase A trimers
The dissociation of the major trimer into the major dimer, the minor dimer, and the monomer is consistent with our earlier trimer model (Liu et al. 2001; Fig. 1 ▶). These results indicate that the quaternary structure of our previous trimer model, with two types of domain swapping in the same molecule, is correct and that the model corresponds to the major trimer. The fact that the minor trimer dissociates into the major dimer and the monomer indicates that only one type of domain swapping (the swapping of the C-terminal β-strand) takes place in the minor trimer. For a protein to form an oligomer with one type of domain swapping and without an exposed closed interface, the oligomer must be in cyclic form, as proposed before (Bennett et al. 1995). Therefore, we suggest that the minor trimer is a cyclic, domain-swapped molecule. On the basis of these results, a model was constructed for the minor trimer with a threefold axis overlapping the twofold axis of the major dimer (Fig. 4 ▶). The hinge loop (residues 112–115) of the minor trimer was modeled with the program CODA (Deane and Blundell 2000). The model was then subjected to energy minimization by use of the program CNS (Brunger et al. 1998). On the basis of this model, the minor trimer dissociates into the major dimer and the monomer (Fig. 4 ▶), which is consistent with the results of the thermal dissociation experiment.
Fig. 4.

Speculative dissociation mechanism of the proposed RNase A minor trimer. A model for the RNase A minor trimer was constructed, with its threefold axis replacing the twofold axis in the major dimer. During dissociation, one subunit (blue) departs the trimer molecule and refolds into the monomer, whereas the other two subunits (red and green) form the major dimer. Because the proposed model of the minor trimer is symmetric, the dissociation of the green or red subunit will result in the same products. The figure was created using Raster 3D (Merritt and Bacon 1997).
Crystallization of the trimers
To verify the models of the two trimers proposed above, we carried out crystallization of the two trimers. Crystals of the minor trimer were first obtained from conditions 30 and 31 of Crystal Screen I (Hampton Research). Larger crystals were obtained from 13% PEG 10,000 (Sigma), 0.2 M ammonium sulfate. The addition of 4% 2-propanol yielded a cubic crystal form, as well as the needle-shape crystals. However, both types of crystals diffracted X-rays poorly, only to 3.5 Å on Rigaku RU-200. Polyethylene glycol (PEG) undergoes oxidation in solution, producing free radicals, which could damage the crystals and contribute to their poor diffraction (Jurnak 1986). Considering this possibility, a fresh PEG 10,000 solution was made for more crystallization trials. However, the fresh PEG 10,000 solution failed to yield any crystals. It appears that the aged and the freshly prepared PEG 10,000 solutions have different pH values, with pH 2.5 for the aged PEG 10,000 and pH 3.7 for the fresh one. Further investigation showed that the pH of the droplets yielding crystals, that is, the mixture of the protein solution and the mother liquor prepared from the aged PEG 10,000, is ∼3.5, but the pH of the droplets from the fresh PEG 10,000 is ∼4.7. At this low pH, the RNase A trimers are unstable. To test whether the crystals were the minor trimer, the crystals were filtered by centrifuge filtration, washed with the mother liquor, and dissolved in 20 mM phosphate buffer (pH 6.0), 0.1 M Na2SO4. The sample was subjected to gel filtration chromatography. The retention time of the redissolved crystals on the column corresponds to that of the minor trimer, indicating that the crystals are the minor trimer. It is unexpected that the minor trimer can be stable at low pH for such a long period. However, macromolecular crowding can affect the association of protein molecules (Minton 2001). It was reported that PEG enhances self-association of spectrin (Cole and Ralston 1994) and polymerization of actin (Lindner and Ralston 1997). Therefore, PEG 10,000 in the crystallization conditions may enhance the stability of the minor trimer under acidic conditions through a macromolecular crowding effect.
Overall structure of the RNase A minor trimer
A more intense and focused X-ray beam significantly improved the diffraction of the crystals. The needle-shape crystals diffracted to 2.3 Å with a Rigaku FRD generator, and to 2.1 Å on a synchrotron X-ray beam. The crystals belong to the space group P21212, with three monomers per asymmetric unit. The structure of the minor trimer of RNase A was determined by molecular replacement by use of the RNase A monomer as a probe, and was refined to 2.2 Å resolution (Table 1).
Table 1.
X-ray diffraction data collection and atomic refinement of the RNase A minor trimer
| Data collection | ||
| Crystal | RNase A minor trimer | RNase A minor trimer |
| X-ray source | Rigaku FRD, UCLA | NSLS Beam Line X8C |
| Temperature (°C) | −170 | −170 |
| Cryo protectant | 25% glycerol | 25% glycerol |
| Wavelength (Å) | 1.5418 | 1.072 |
| Resolution range (Å) | 40–2.3 | 40–2.1 |
| Total reflections | 152,811 | 327,545 |
| Unique reflections | 17,206 | 22,989 |
| Completeness (%) | 96.9 | 99.7 |
| Rmergea(%) | 7.6 | 9.6 |
| Space group | P2l2l2 | P2l2l2 |
| Unit cell dimensions | a = 114.48 Å, b = 122.05 Å, c = 26.82 Å | a = 114.59 Å, b = 122.17 Å, c = 26.84 Å |
| Refinement | |||
| a Rmerge = ∑hkl ∑i | I(hkl)i − <I(hkl)> | /∑hkl ∑i <I(hkl)i>. | |||
| b R = ∑hkl | F(hkl)o − <F(hkl)c> | /∑hkl F(hkl)o. (rmsd) Root-mean-square deviation. | |||
| Resolution range (Å) | 10–2.2 | No. of reflections | 19,343 |
| Rb (%) | 18.4 | No. of protein atoms | 2,853 |
| Free Rb (%) | 25.7 | No. of water molecules | 270 |
| rmsd bond length (Å) | 0.014 | No. of sulfate ions | 4 |
| rmsd bond angle | 1.906° | Average B factor (Å2) | 21.91 |
The X-ray structure reveals that the minor trimer is cyclic and 3D domain-swapped at the C-terminal β-strand (residues 116–124, Fig. 5 ▶), as proposed from the dissociation results. However, the orientation of the subunits in the structure of the minor trimer is different from that in our model, which was constructed on the basis of the structure of the major dimer. This different orientation makes the structure (Fig. 5 ▶) more compact than the model (Fig. 4 ▶). The shape of the minor trimer is like a propeller, with the blades (subunits) related by a threefold axis. The active site of RNase A contains catalytic residues His 12, Lys 41, and His 119. In the minor trimer, the active sites are composite, consisting of His 12 and Lys 41 from one subunit and His 119 from another subunit. Domain swapping does not disrupt the active sites of the minor trimer, which is consistent with the observation that the minor trimer retains enzyme activity (Gotte et al. 1999).
Fig. 5.

Ribbon diagrams of the structures of the RNase A monomer (A), the minor dimer (B), the major dimer (C), the major trimer model (D), and the minor trimer (E). The N- and C-termini are labeled. The N-terminal helix and the C-terminal strand that are swapped in the oligomers are colored in blue and red, respectively, in the monomer (A). The minor dimer (B) swaps the N-terminal helix, whereas the major dimer (C) swaps the C-terminal strand. Both types of swapping take place in the major trimer model (D): The green subunit swaps the C-terminal strand with the red subunit and swaps the N-terminal helix with the blue subunit. The minor trimer (E) is 3D domain-swapped at the C-terminal strands. The three subunits of the molecule are related by a threefold axis, giving the molecule the shape of a propeller. The figure was created using Raster 3D (Merritt and Bacon 1997).
Open interface of the minor trimer
The closed interface of the minor trimer is similar to that of the major dimer, which also swaps its C-terminal strands, whereas the open interface of the minor trimer is different from the major dimer. The area of the closed interface of the minor trimer is 1780 Å2 per subunit, similar to that of the major dimer (1720 Å2 per subunit). The open interface area of the minor trimer is 355 Å2 per subunit, larger than that of the major dimer (200 Å2 per subunit, Liu et al. 2001). In the major dimer of RNase A, the interactions at the open interface involve only the hinge loop. There is no interaction between the two functional units (FU). The functional unit is composed of the swapped domain of one subunit and the main domain of another subunit (Liu et al. 1998). It is similar to the monomer except that the monomer consists of one polypeptide chain, whereas an FU is composed of portions of two polypeptide chains. In the minor trimer of RNase A, the interactions at the open interface not only involve the hinge loop, but also the functional units. These interactions are mainly hydrogen bonds: Gln 69 OE1 (FU1)–Tyr 73 OH (FU2), Thr 70 OG1 (FU1)–Thr 70 N (FU2), Pro 114 O (FU1)–Asn 113 N (FU2), and Thr 70 OG1 (FU1)–Thr 70 OG1 (FU2). These hydrogen bonds are also present between FU2 and FU3, as well as FU3 and FU1. The open interface interactions of the minor trimer are also mediated by a sulfate ion and a few waters through an intricate hydrogen bond network, which is described below.
Sulfate ion-binding site at the open interface
During refinement, we observed strong electron density peaks, not modeled by the protein. The shape of these peaks suggests they are either sulfate ions or phosphate ions. The crystallization conditions contain 0.18 M phosphate buffer and 0.2 M ammonium sulfate. Because the concentrations of the sulfate and phosphate ions are similar to each other, it is difficult to distinguish which ion is bound to the minor trimer. Because the concentration of the sulfate ion is slightly higher than that of the phosphate ion, we assigned sulfate ions to the unoccupied electron densities. There are four sulfate ions bound to the minor trimer, three of which are bound to the active site from each subunit, similar to what was reported in other RNase A monomer and dimer structures (Fedorov et al. 1996; Liu et al. 1998, 2001).
The fourth sulfate ion is bound at the open interface, located on the threefold axis of the minor trimer. Closer examination of this binding site reveals that the open interface of the minor trimer forms a trap for the sulfate ion (Fig. 6 ▶). There is an intricate hydrogen bond network in the trap. Thr 70 residues from the three subunits form the base of the trap. The water molecules in the trap show a layer structure. OG1 of Thr 70 from each subunit hydrogen bonds individually with one water molecule in the first layer. These three waters, in turn, form hydrogen bonds with another three waters in the second layer, which then form hydrogen bonds with three waters in the third layer. The waters in the third layer form hydrogen bonds with the three oxygen atoms of the sulfate ion. These three oxygen atoms are related by the threefold axis of the minor trimer molecule. Three tyrosyl residues (Tyr 115 from each subunit) surround the sulfate ion with their aromatic rings aligned parallel to the threefold axis of the trimer, creating a relatively hydrophobic environment in the trap, which apparently orientates the sulfate ion. The fourth oxygen atom of the sulfate ion is liganded by the backbone nitrogen of Gly 112 from each subunit. As one oxygen atom can accept at most two hydrogen bonds, this oxygen may form hydrogen bonds alternatively with these three backbone nitrogens. Cα atoms of the three Gly 112 residues form the bottleneck of the trap. Above the neck is one water forming hydrogen bonds with the carboxyl oxygen of Pro 114 from subunits 1 and 3 (cyan and yellow in Fig. 6 ▶). The trap is sealed by the carboxyl oxygen of Asn 113 from each subunit, forming hydrogen bonds with one water in the center. Again, these hydrogen bonds may form alternatively because one water molecule can be the donor for two hydrogen bonds and there are three carboxyl oxygen atoms surrounding this water molecule.
Fig. 6.
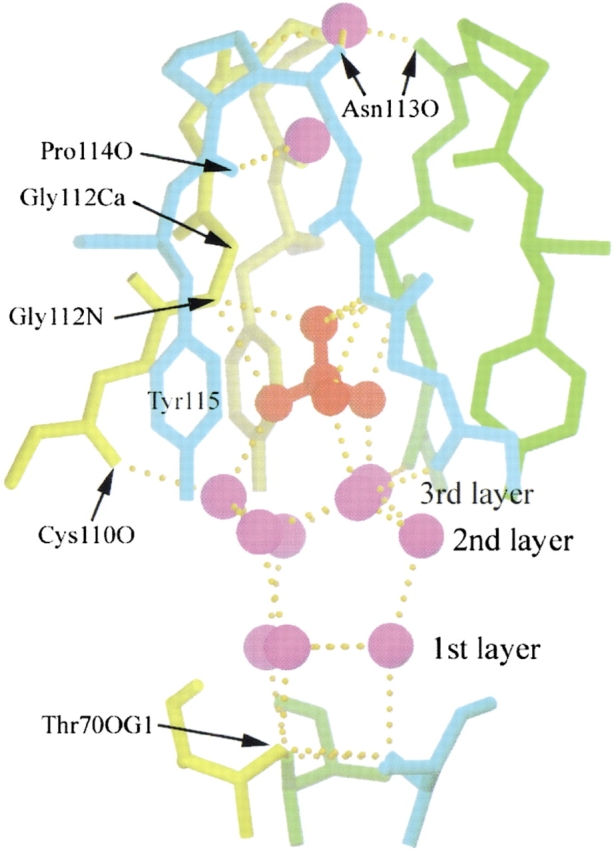
The trap for a sulfate ion at the open interface of the RNase A minor trimer. The sulfate ion and the water molecules are in red and purple, respectively. The protein chains from the three subunits of the minor trimer are in cyan, green, and yellow, respectively. The protein atoms and residues that hydrogen bond with the water molecules and sulfate ion are indicated. There is an intricate hydrogen bond network in the trap. The waters in the trap are aligned in three layers as labeled. The structure is viewed perpendicular to the threefold axis of the minor trimer. For clarity, the sidechains of Cys 110, Glu 111, and Asn 113 are omitted. The figure was created using SETOR (Evans 1993).
Discussion
Additional function through 3D domain swapping
The sulfate ion bound at the open interface shows that 3D domain swapping can create a new binding site for ions. New ion binding sites created through 3D domain swapping have been reported previously. In the structure of a domain-swapped trimer of barnase, there is also a sulfate ion bound at the open interface (Zegers et al. 1999). The open interface of the trimeric barnase forms a concave cavity. The sulfate binds to this cavity by hydrogen bonding to OG of Ser 38 from each subunit. Therefore, this sulfate ion is solvent accessible. In the minor trimer of RNase A, the sulfate ion is buried in the open interface.
Another example of ion binding at the open interface is shown in the structure of a domain-swapped dimer of CksHs2, in which a divalent metal ion is bound to Glu 63 from each subunit at the open interface. In this case, 3D domain swapping creates a metal binding site and the metal ion in turn stabilizes the dimer by neutralizing the charge repulsion at the open interface (Parge et al. 1993).
All of these three structures indicate that 3D domain swapping can create a binding site for ions, whereas the monomer alone cannot form such a binding site. The RNase A monomer was reported to bind to sulfate ions outside of the active site (Fedorov et al. 1996). However, this sulfate is bound at the interface between two monomers through crystal packing. This suggests that the sulfate ion is not bound to the monomer in solution, further suggesting that the monomer alone cannot form a sulfate-binding site. On the contrary, the intricate hydrogen-bonding network between the sulfate ion and the open interface of the RNase A minor trimer implies that the sulfate ion will be bound to the minor trimer in solution, and is not an artifact from crystal packing. Therefore, the sulfate ion bound at the open interface of the minor trimer of RNase A supports the suggestion that 3D domain swapping can endow a protein with additional functions (Bennett et al. 1995).
Different enzymatic activities on dsRNA
Both RNase A dimers and trimers have higher activities on dsRNA than does the monomer. In addition, bovine seminal ribonuclease (BS-RNase), a natural domain-swapped dimeric ribonuclease, also shows enzymatic activity on dsRNA (Libonati and Floridi 1969). Among these three dimers, BS-RNase has the highest activity, whereas the minor dimer has the lowest activity on dsRNA polyA–polyU (Opitz et al. 1998; Gotte et al. 1999). Libonati and his coworkers suggested that destabilization of dsRNA is required for the digestion (Sorrentino and Libonati 1994; Gotte et al. 1999). This suggestion is supported by the crystal structure of RNase A complexed with d(pA)4, in which the polynucleotide chain adopts a conformation different from a double strand helix (McPherson et al. 1986). This is also supported by the observation that DNA is destabilized upon its binding to RNase A (Jensen and von Hippel 1976). Crystal structures of RNase A complexed with polynucleotides suggest there are multiple binding subsites on RNase A involved in the enzyme activity (Pares et al. 1991).
To study the mechanism of the enzymatic activity of the dimers on dsRNA, we firstly compared the distributions of the subsites on these dimers. The residues interacting with the substrate are colored in red, and the basic residues outside of the binding subsites are labeled in blue (Fig. 7 ▶). In the figure, the two patches formed by the multiple binding subsites from each subunit are close to each other in all of the dimers. Therefore, these two binding patches from each subunit may form a binding site for dsRNA, and thus increase the binding affinity to dsRNA. In addition, Arg 33 and Lys 34 from BS-RNase, Arg 85 and Lys 98 from the RNase A minor dimer are located between the two binding patches. These residues may provide additional binding subsites for phosphate groups on dsRNA. Secondly, we examined the relationship between the enzymatic activity and the distance between the active sites. We obtained the distance between the two active sites in each dimer by measuring the distance of the phosphate/sulfate anions at the active sites. The distance is 36.8Å in BS-RNase, 38.7Å in the major dimer, and 39.6 Å in the minor dimer, respectively. These distances between active sites on the dimers are correlated with their enzymatic activity on dsRNA: The enzymatic activity on dsRNA decreases as the distance between the active sites increases. Because there are positive charges around the active site of RNase A, decreasing the distance of the active sites in RNase A oligomers increases the density of positive charges around the active sites. Therefore, the correlation of the enzymatic activity and the distance of the active sites is consistent with the previous proposal that the number of positive charges close to the binding site is correlated with the activity on dsRNA (Sorrentino and Libonati 1994).
Fig. 7.

Proposed binding sites for RNA on BS-RNase (A), RNase A major dimer (B), RNase A minor dimer (C), RNase A major trimer model (D), and RNase A minor trimer (E). The subunits are in gray with different shades. The residues interacting with RNA are in red. The active site residues are in magenta and the basic residues outside of the binding sites are in blue. Basic residues between the two binding sites in BS-RNase and RNase A minor dimer are indicated on the structure. The proposed binding sites on BS-RNase, RNase A major dimer, and RNase A minor trimer are twisted around the molecules, whereas the binding sites on RNase A minor dimer are located on one side of the molecule. The figure is plotted with SETOR (Evans 1993).
Thirdly, we analyzed the relative orientation of the binding patches in each dimer. The two binding patches in BS-RNase and in the RNase A major dimer are oriented in such a way that they are twisted around the dimer molecules, whereas in the minor dimer, these two patches are located on the same side of the dimer molecule (Fig. 7 ▶). The twisted orientation of the two binding patches may help destabilize dsRNA, and, therefore, increase the enzymatic activity on dsRNA. Therefore, we suggest that the relative orientation of the two binding patches is also related to the enzymatic activity on dsRNA of the dimeric ribonucleases. In summary, we propose that three factors can affect the enzymatic activity of the dimeric ribonucleases on dsRNA: The activity on dsRNA increases as there are more positive charges around the active sites; the activity on dsRNA increases as the distance of the active sites in the dimer decreases; the activity on dsRNA increases as the relative orientation of the two binding patches is more twisted around the molecules.
This proposal can be extended to the RNase A trimers. The activities on dsRNA and the distances between the active sites of each oligomer are listed in Table 2. The results fit the proposal above well, especially for the activities on polyA–polyU. The minor trimer has the shortest distance between the active sites and possesses the highest enzyme activities on dsRNA. The major trimer is a combination of the major dimer and the minor dimer, and its enzymatic activity on dsRNA is similar to that of the major dimer. In addition, the binding patches in the minor trimer are twisted to a greater extent than the major trimer and the dimers (Fig. 7 ▶), consistent with its highest enzymatic activity among the dimers and trimers of RNase A, supporting the proposal above that the relative orientation of the binding patches affects the enzymatic activity on dsRNA by destabilizing dsRNA. The enzymatic activities on viral dsRNA also fit the proposal over all, but not as perfectly as do the activities on polyA–polyU. This could be due to an effect from the tertiary structure of viral dsRNA.
Table 2.
Structure-activity correlation of various RNase A molecules for hydrolysis of double stranded RNA (dsRNA)
| RNase A species | Distance between active sites (Å) | Specific activity on poly(A)-poly(U) (activity/mg) | Specific activity on viral dsRNA (activity/nmol) |
| Monomer | N/A | 2.7 ± 0.5 | 0.44 ± 0.02 |
| Minor dimer | 39.6 | 4.5 ± 0.8 | 1.43 ± 0.19 |
| Major dimer | 38.7 | 30 ± 3 | 1.37 ± 0.19 |
| Major trimer | 39.6 and 38.7 | 32 ± 6 | 5.00 ± 0.24 |
| Minor trimer | 28.8 | 61 ± 5 | 5.52 ± 0.27 |
Notice that the activity increases as the distance between the active sites decreases. Activity data on viral dsRNA are from Gotte et al (1999).
Conclusions
We characterized the two trimers of RNase A using dissociation and cross-linking experiments. We showed that the major trimer has the properties of our previous linear trimer model. On the basis of the fact that the minor trimer dissociates into the major dimer and the monomer only, we constructed a cyclic model for the minor trimer, with the C-terminal strands swapped. We then determined the structure of the minor trimer, showing that the proposed structure for the minor trimer is correct.
From these studies, we have confirmed that two types of domain swapping can take place in the same molecule by the swapping of two different domains. We have also shown that RNase A can form both linear and cyclic oligomers. The structures of the RNase A oligomers suggests the possibility that other proteins may form oligomers in the similar way, which will significantly expand the database of domain-swapped proteins. Both the major dimer and the minor trimer of RNase A are formed through the swapping of the C-terminal β-strands. They have the same closed interface and the same hinge loop. However, their open interfaces are different from each other due to the different conformations of the hinge loop in the two oligomers, indicating the structural flexibility of the hinge loop. In addition, our studies reveal the first example of a protein that can form both a linear and a cyclic domain-swapped oligomer and bring our knowledge of 3D domain swapping to a new level. RNase A also forms tetramers and pentamers. Structural studies of these oligomers will illuminate the role of 3D domain swapping in protein oligomerization. A major unanswered question remains: can domain swapping lead to high oligomers, including amyloid-like fibers?
Materials and methods
Materials
All chemicals were from Fisher unless specifically mentioned.
Preparation of the trimers
RNase A trimers were prepared according to the reported method (Gotte et al. 1999). In brief, RNase A was lyophilized in 40% acetic acid. The trimers were separated from the monomer and other oliogmers by gel filtration chromatography on a Superdex 75 HL 26/60 column. The major and the minor trimers were then separated from each other by a cation exchange Pharmacia Source 15S HR 10/10 column. The column was eluted with a 0.085–0.180 M of phosphate (pH 6.7) gradient.
Dissociation of the trimers
RNase A trimers slowly dissociate into dimers and monomer. Mild thermal treatment accelerates this dissociation. The purified trimers (0.5 mg/mL in 20 mM phosphate buffer at pH 6.0) were kept at 55°C for 5 min, and then placed on ice. The products of the dissociation were analyzed on a cation exchange Vydac S column.
Cross-linking of the RNase A monomer and oligomers
Lys 7 and Lys 41 of RNase A are close to each other in three-dimensional space. 1,5-difluoro-2,4-dinitro-benzene (DFDNB) has been used to cross-link these two residues (Lin et al. 1984). Following this protocol, the RNase A monomer, the two dimers, and the two trimers were individually dissolved in 2 mL of 50 mM borate buffer (pH 8.5) at a concentration of 0.5 mg/mL. An aliquot of 20 μL of DFDNB [Sigma, 0.5 mg/mL in 2% (v/v) methanol solution] was added every 30 min over a period of 3 h, while the solutions were stirred in the dark at room temperature. The solutions were stirred for another 20 h before the reactions were quenched by adding glacial acetic acid to adjust the solutions to pH 3.0. The reaction mixtures were concentrated, dialyzed against 20 mM phosphate buffer (pH 6.0), and subjected to chromatographic analyses.
Gel filtration chromatography
A TosoHaas analytical size exclusion column TSK-GEL G3000SWXL was equilibrated and eluted with 20 mM phosphate buffer (pH 6.0), 0.1 M Na2SO4 at a flow rate of 0.4 mL/min. The elution pattern was monitored at 214 nm.
Cation exchange chromatography
The RNase A samples were loaded onto a Vydac S cation exchange column equilibrated with 20 mM phosphate buffer (pH 6.0) at a flow rate of 0.5 mL/min. The column was washed for 10 min, and then eluted with a 20–80-mM phosphate (pH 6.0) gradient over a period of 50 min. The intact monomer, dimers, and trimers were used as standards.
Enzymatic activity assay
The assay for the enzymatic activities on dsRNA of different RNase A oligomers was essentially the same as described before (Sorrentino and Libonati 1994) with slight modifications. The reactions were carried out at 25°C in 0.4 mL of standard saline citrate (0.15 M NaCl and 0.015 M sodium citrate at pH 7.0) containing 2.5 mM sodium phosphate buffer (pH 6.7). The substrate is poly(A)–poly(U) (Sigma, 40μg/mL). The concentration for each RNase A oligomer is as follows: monomer, 25 μg/mL; the minor dimer, 5 μg/mL; the major dimer, 10 μg/mL; the major trimer, 5 μg/mL; the minor trimer, 5 μg/mL.
Crystallization, X-ray diffraction, and structure determination
Crystallization trials of the RNase A trimers were carried out by use of the hanging-drop vapor diffusion method at 4°C. Each trimer (10 mg/mL in 0.18 M phosphate buffer at pH 6.7) was mixed with an equal volume of mother liquors from Crystal Screen I (Hampton Research). Crystals were soaked in mother liquor containing 25% glycerol and fast frozen. X-ray diffraction analysis was first carried out on a Rigaku R-AXIS IV imaging plate detector at −170°C. The X-ray source was a Rigaku RU-200 generator operating at 50 kV, 100 mA with focusing mirrors. Diffraction data were collected on a Rigaku R-AXIS IV++ imaging plate detector at −170°C, with Rigaku FRD as X-ray source to 2.3 Å resolution. Then, diffraction data to 2.1 Å were collected at −170°C at synchrotron beam line X8C at Brookhaven National Laboratory. The data were indexed and processed with the programs DENZO and SCALEPACK (Otwinowski and Minor 1996). The structure of the minor trimer was solved from the FRD data by molecular replacement using the program EPMR (Kissinger et al. 1999). The RNase A monomer (PDB code 1RTB, Birdsall and McPherson 1992) was used as a search model. The model from the FRD data was then refined against the synchrotron data to 2.2 Å resolution. Alternating cycles of model building with the program O (Jones et al. 1991) and refinement with the program CNS (Brunger et al. 1998) were used to determine the final structure. Coordinates and structure factors for the Rnase A minor trimer have been deposited with the PDB codes 1JS0 and r1js0sf.
Acknowledgments
We thank D. Cascio, D.H. Anderson, and M. Sawaya for technical advice. This work was supported by NSF, NIH, DOE, and Italian MURST-Prin 1999 and 2000.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BS-RNase, bovine seminal ribonuclease
DFDNB, 1,5-difluoro-2,4-dinitro-benzene
dsRNA, double stranded RNA
d(pA)4, deoxy-adenosine-tetranucleotide
FU, functional unit
PEG, polyethylene glycol
RNase A, bovine pancreatic ribonuclease
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.36602
References
- Bennett, M.J., Choe, S., and Eisenberg, D. 1994. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. 91 3127–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.J., Schlunegger, M.P., and Eisenberg, D. 1995. 3D domain swapping: A mechanism for oligomer assembly. Protein Sci. 4 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsall, D.L. and McPherson, A. 1992. Crystal structure disposition of thymidylic acid tetramer in complex with ribonuclease A. J. Biol. Chem. 267 22230–22236. [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta. Crystallogr. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Cafaro, V., De Lorenzo, C., Piccoli, R., Bracale, A., Mastronicola, M.R., Di Donato, A., and D'Alessio, G. 1995. The antitumor action of seminal ribonuclease and its quaternary conformations. FEBS Lett. 359 31–34. [DOI] [PubMed] [Google Scholar]
- Cohen, F.E. and Prusiner, S.B. 1998. Pathologic conformations of prion proteins. Annu. Rev. Biochem. 67 793–819. [DOI] [PubMed] [Google Scholar]
- Cole, N. and Ralston, G. 1994. Enhancement of self-association of human spectrin by polyethylene glycol. Int. J. Biochem. 26 799–804. [DOI] [PubMed] [Google Scholar]
- Crestfield, A.M., Stein, W.H., and Moore, S. 1962. On the aggregation of bovine pancreatic ribonuclease. Arch. Biochem. Biophys. Suppl. 1 217–222. [PubMed] [Google Scholar]
- Deane, C.M. and Blundell, T.L. 2000. CODA: A combined algorithm for predicting the structurally variable regions of protein models. Proteins 40 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, S.V. 1993. SETOR: Hardware-lighted three-dimensional solid representations of macromolecules. J. Mol. Graphics 11, 134–138. [DOI] [PubMed] [Google Scholar]
- Fedorov, A.A., Joseph-McCarthy, D., Fedorov, E., Sirakova, D., Graf, I., and Almo, S.C. 1996. Ionic interactions in crystalline bovine pancreatic ribonuclease A. Biochemistry 35 15962–15979. [DOI] [PubMed] [Google Scholar]
- Gotte, G., Bertoldi, M., and Libonati, M. 1999. Structural versatility of bovine ribonuclease A. Distinct conformers of trimeric and tetrameric aggregates of the enzyme. Eur. J. Biochem. 265 680–687. [DOI] [PubMed] [Google Scholar]
- Jensen, D.E. and von Hippel, P.H. 1976. DNA "melting" proteins. I. Effects of bovine pancreatic ribonuclease binding on the conformation and stability of DNA. J. Biol. Chem. 251 7198–7214. [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Jurnak, F. 1986. Effect of chemical impurities in polyethylene glycol on macromolecular crystallization. J. Crystal Growth 76 577–582. [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D55 484–491. [DOI] [PubMed] [Google Scholar]
- Klafki, H.W., Pick, A.I., Pardowitz, I., Cole, T., Awni, L.A., Barnikol, H.U., Mayer, F., Kratzin, H.D., and Hilschmann, N. 1993. Reduction of disulfide bonds in an amyloidogenic Bence Jones protein leads to formation of "amyloid-like" fibrils in vitro. Biol. Chem. Hoppe-Seyler 374 1117–1122. [DOI] [PubMed] [Google Scholar]
- Libonati, M. and Floridi, A. 1969. Breakdown of double-stranded RNA by bull semen ribonuclease. Eur. J. Biochem. 8 81–87. [DOI] [PubMed] [Google Scholar]
- Libonati, M., Bertoldi, M., and Sorrentino, S. 1996. The activity on double-stranded RNA of aggregates of ribonuclease A higher than dimers increases as a function of the size of the aggregates. Biochem. J. 318 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.H., Konishi, Y., Dentor, M.E., and Scheraga, H.A. 1984. Influence of an extrinsic cross-link on the folding pathway of ribonuclease A. Conformational and thermodynamic analysis of cross-linked (lysine7-lysine41)-ribonuclease a. Biochemistry 265 5504–5512. [DOI] [PubMed] [Google Scholar]
- Lindner, R. and Ralston, G. 1997. Macromolecular crowding: Effects on actin polymerization. Biophys. Chem. 66 57–66. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Hart, P.J., Schlunegger, M.P., and Eisenberg, D. 1998. The crystal structure of a 3D domain-swapped dimer of RNase A at a 2.1-A resolution. Proc. Natl. Acad. Sci. 95 3437–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., Gotte, G., Libonati, M., and Eisenberg, D. 2001. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Structu. Biol. 8 211–214. [DOI] [PubMed] [Google Scholar]
- McPherson, A., Brayer, G., and Morrison, R. 1986. Crystal structure of RNase A complexed with d(pA)4. Biophys. J. 49 209–219. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Meth. Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Minton, A.P. 2001. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 276 10577–10580. [DOI] [PubMed] [Google Scholar]
- Nenci, A., Gotte, G., Bertoldi, M., and Libonati, M. 2001. Structural properties of trimers and tetramers of ribonuclease A. Protein Sci. 10 2017–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz, J.G., Ciglic, M.I., Haugg, M., Trautwein-Fritz, K., Raillard, S.A., Jermann, T.M., and Benner, S.A. 1998. Origin of the catalytic activity of bovine seminal ribonuclease against double-stranded RNA. Biochemistry 37 4023–4033. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1996. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pares, X., Nogues, M.V., de Llorens, R., and Cuchillo, C.M. 1991. Structure and function of ribonuclease A binding subsites. Essays Biochem. 26 89–103. [PubMed] [Google Scholar]
- Parge, H.E., Arvai, A.S., Murtari, D.J., Reed, S.I., and Tainer, J.A. 1993. Human CksHs2 atomic structure: A role for its hexameric assembly in cell cycle control. Science 262 387–394. [DOI] [PubMed] [Google Scholar]
- Park, C. and Raines, R.T. 2000. Dimer formation by a "monomeric" protein, Protein Sci. 9 2026–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli, R., Di Donato, A., and D'Alessio, G. 1988. Co-operativity in seminal ribonuclease function. Kinetic studies. Biochem. J. 253 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlunegger, M., Bennett, M., and Eisenberg, D. 1997. In Advances in protein chemistry, (eds. F.M. Richards et al.), Vol. 50, pp. 61–122. Academic Press, New York. [DOI] [PubMed]
- Sorrentino, S. and Libonati, M. 1994. Human pancreatic-type and nonpancreatic-type ribonucleases: A direct side-by-side comparison of their catalytic properties. Arch. Biochem. Biophys. 312 340–384. [DOI] [PubMed] [Google Scholar]
- Vescia, S. and Tramontano, D. 1980. Antitumoral action of bovine seminal ribonuclease. Mol. Cell. Biol. 36 125–128. [DOI] [PubMed] [Google Scholar]
- Zegers, I, Deswarte, J., and Wyns, L. 1999. Trimeric domain-swapped barnase. Prot. Natl. Acad. Sci. 96 818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]