

Abstract
Many protein pairs that share the same fold do not have any detectable sequence similarity, providing a valuable source of information for studying sequence-structure relationship. In this study, we use a stringent data set of structurally similar, sequence-dissimilar protein pairs to characterize residues that may play a role in the determination of protein structure and/or function. For each protein in the database, we identify amino-acid positions that show residue conservation within both close and distant family members. These positions are termed "persistently conserved". We then proceed to determine the "mutually" persistently conserved (MPC) positions: those structurally aligned positions in a protein pair that are persistently conserved in both pair mates. Because of their intra- and interfamily conservation, these positions are good candidates for determining protein fold and function. We find that 45% of the persistently conserved positions are mutually conserved. A significant fraction of them are located in critical positions for secondary structure determination, they are mostly buried, and many of them form spatial clusters within their protein structures. A substitution matrix based on the subset of MPC positions shows two distinct characteristics: (i) it is different from other available matrices, even those that are derived from structural alignments; (ii) its relative entropy is high, emphasizing the special residue restrictions imposed on these positions. Such a substitution matrix should be valuable for protein design experiments.
Keywords: Molecular evolution, sequence conservation, protein structure, protein folding, bioinformatics
The protein structure space is considerably smaller than the sequence space (Brenner and Levitt 2000; Koppensteiner et al. 2000). This means that many protein sequences, including highly dissimilar ones, assume similar folding patterns (Orengo et al. 1994; Brenner and Levitt 2000; Koppensteiner et al. 2000). It is not uncommon for structurally similar protein pairs to have only 10% sequence identity, suggesting that many positions have no critical role in structure determination, and that the folding determinants are restricted to a limited number of sequence residues. This was demonstrated both experimentally and computationally. Experimentally, it was shown for a number of proteins that many of the mutations introduced along the sequence have had no effect on the protein's activity (Rennell et al. 1991; Markiewicz et al. 1994; Suckow et al. 1996), and on its stability (Milla et al. 1994). Most of the mutations that have had an effect were located either at the core of the fold or at the protein's functional sites. The core residues, where conservation seemed to be important, were mostly hydrophobic, but their identity was not crucial and different hydrophobic residues could replace one another without affecting the structure (Lim and Sauer 1989; Bowie et al. 1990).
Recent computational studies have reached similar conclusions. Mirny and colleagues (Mirny et al. 1998; Mirny and Shakhnovich 1999) and Ptitsyn and Ting (1999) compared the sequences within large protein superfamilies of solved structures to search for positions that conserve a certain type of residues (hydrophobic, charged, etc.). The comparison was done within each family that is contained in the superfamily, and conserved positions were examined across the families within a superfamily. Thus, a position can be conserved across the superfamily but contain different types of residues in the different families. Both studies have identified only a small number of such positions that were also spatially close, and suggested that they form folding nuclei. Stabilization centers that are formed by long-range interactions also were suggested by Dosztányi et al. (1997), who demonstrated their conservation within the corresponding protein families. Similar findings also were described in computational design protocols, where compatible sequences have been sought for a given structural template (Koehl and Levitt 1999), and in lattice models (Mirny et al. 1998). The designed sequences showed high conservation in a relatively small number of specific positions. The potential role of key residues in structure determination was also demonstrated by the Gaussian network model, by which residues involved in the highest frequency fluctuations near the native state coordinates were identified (Demirel et al. 1998). Recently, Reddy et al. (2001) have analyzed common substructures that recur in the protein data bank (PDB), and identified in them conserved key amino acids positions (CKAAPs). All these observations show that a very small number of residues play a key role in fold determination, enabling different sequences that contain these key residues at appropriate positions to assume similar folds. These can function either as folding nuclei (Mirny and Shakhnovich 1999, 2001), establish the molecule's active site (e.g., Reddy et al. 2001), or play key roles in critical positions of secondary structure elements (Reddy et al. 2001). In this study, we use a novel automated technique on a global, stringently constructed structural database to identify and characterize such significant positions. A global analysis of proteins from many different folds enables us to draw general conclusions regarding soluble proteins whose structures have been solved. Stringency is applied at the level of database construction and at the level of positional conservation analysis, enabling us to separate those positions that are conserved as a result of nondivergence from a common ancestor, from those that are conserved for structural/functional reasons.
The database that is used in the current analysis is of pairs of proteins that share a common fold but are dissimilar in sequence (12% identical residues on the average). By aligning the two structures of paired proteins, structurally aligned positions are determined. These positions may possess identical residues in the two proteins or different residues. One way to distinguish structurally aligned positions that may be critical for the structure and/or function of the proteins in a pair is to follow their pattern of conservation in their respective protein sequence families. Positions that contain residues that are conserved in both protein families are denoted mutually conserved. Because the two families of the structurally similar, sequence-dissimilar (SSSD) aligned proteins are so remote, mutually conserved positions, by virtue of their intra- and interfamily conservation, may be important for structure or function. More specific information can be obtained by focusing on those mutually conserved positions that are retained conserved as the multiple alignment of family members is expanded to include members that are more and more remote. Positions that are conserved among close family members and remain conserved when remote members are added to the alignment, add further support to the putative role of these positions in structure and function determination. On the one hand, examination of conservation among close members may lead to the inclusion of positions that have possibly not yet diverged. On the other hand, examination of conservation among distant family members may be erroneous because of possible arbitrary drift in the aligned sequences (Park et al. 1998; Friedberg et al. 2000b). Therefore, examination of conservation among both close and distant members may pinpoint the critical residues. Such positions are denoted here "mutually persistently conserved" (MPC), and the current study focuses on them. We show that many of these residues are located in positions that play an important role in secondary structure determination, that they are buried in their protein structures, and that many of them form spatial clusters. Moreover, the substitution matrix derived from MPC positions has high relative entropy, indicating that MPC replacements are limited to only certain types of residues.
Results and Discussion
Identifying mutually persistently conserved positions
We have compiled a restrictive database of 118 SSSD protein pairs (see Materials and Methods). The proteins in each pair have the same fold but do not exhibit any significant sequence similarity. It should be noted that although sequence pairwise alignments in SSSD do not have high enough alignment scores to be considered significantly similar, this does not necessarily mean that all pair mates in the SSSD pairs can be considered unrelated. Because the current methods for assessment of statistical significance are constructed to examine whether two proteins may be related by sequence considerations, having found that two sequences are dissimilar by such considerations does not necessarily mean that they are unrelated. It only means that the similarity score is not sufficiently high for them to be considered related. However, as elaborated upon in the Materials and Methods section, we believe that as a population, protein pairs in SSSD are sufficiently dissimilar to be considered unrelated. Furthermore, as no single pair in SSSD has a significant alignment score, then even the high scoring pairs in our database can be considered interesting (if not unrelated).
To identify MPC positions, the persistently conserved positions in each protein family need first to be determined. As noted in the introduction, we wish to identify positions where the conservation is maintained as the alignment is advanced and more remote family members are added. To do that, we need to generate multiple sequence alignments containing close and remote family members. This can be achieved by using PSI-BLAST that identifies remote homologs by an iterative process (Altschul et al. 1997).
Figure 1 ▶ depicts a flowchart of the analysis. Each sequence in the database was run through PSI-BLAST for five iterations, or until convergence. For each sequence, per iteration, the conservation at each position was evaluated by calculating the information content (IC) (see Materials and Methods). Note that the IC of each position is determined by using relative entropy (see Materials and Methods). This means that the conservation of a given amino-acid type in a given position is evaluated relative to its background frequency in the entire database. There exists a certain ambiguity in the use of the term "conservation" when applied to scores assigned to positions in a multiple sequence alignment. Most studies use some variant of the Shannon entropy (IC) formula to assess positional conservation. According to the question at hand, some studies use the information-content formula with scores normalized according to background frequencies, whereas others do not. The use or omission of priors in the computation should depend on the question asked. Here we were interested in determining in the SSSD protein pairs positions that show distinct amino acids and therefore took into account the background frequencies.
Fig. 1.

A schematic flowchart describing the identification of mutually persistently conserved positions. See text for details.
To arrive at conservation scores that are comparable between different sequences, the IC values were expressed in standard deviations from the mean IC of the sequence (ZIC-values). In this study, positions with a conservation score greater than zero (the normalized IC mean) have been considered as conserved. We set the threshold at zero as our other requirements of persistency and mutuality in conservation already narrow down significantly the studied residues, and we did not wish to limit the range of studied residues only to those that are extremely conserved. However, the threshold can be set at any desired value and the analysis repeated on the residues determined by the defined threshold. (Note that although our threshold was put at the average, only 39% of the positions were determined as conserved at the first iteration. This is because the average is computed along the alignment that is frequently longer than the actual length of sequences). As stated above, each sequence was run through PSI-BLAST for several iterations (up to five). Each such iteration provides conservation scores for the positions along the sequence. Positions that had been found to be conserved both at the first and last iteration were determined as persistently conserved. The persistency requirement is especially important given the derivation of the multiple sequence alignment by PSI-BLAST, as PSI-BLAST incorporates very close sequences at the first iteration, and may drift to include nonrelated sequences as the iterations advance (Park et al. 1998; Friedberg et al. 2000b). Thus, the inclusion of residue positions that are identified as conserved both at the first and last iteration of PSI-BLAST is expected to decrease the fraction of positions that were erroneously identified as conserved. Finally, the MPC positions were determined by selecting only those persistently conserved positions that were "mutually" conserved in the two pair mates.
Out of 118 protein pairs in our database, 93 pairs had more than a single PSI-BLAST iteration for both pair mates. Seventy-four percent of the positions identified as conserved at the first iteration were persistently conserved. Among all persistently conserved positions, 45% show mutual conservation, while 55% show persistent conservation only in one pair mate. It is evident from the above statistics that the application of the two requirements of persistency and mutuality of conservation directs us to a strictly defined subset of residues. These positions, together with the mutually conserved positions in protein families that converged after the first iteration, make up the MPC positions, 2603 in total. This result is highly statistically significant (p<.0001; see Materials and Methods). Eight hundred thirty-eight of these positions (32%) are occupied by identical residues in the two pair mates, and 1765 positions (68%) show different residues. These proportions deviate significantly from what is observed for all aligned positions in the database, where only 12% (1962/15,566) of the aligned positions are occupied by identical residues and 88% (13,604/15,566) show different residues. The total number of residues in the 2603 MPC positions added up to 3701 (and not to 2603×2, as some proteins appeared in more than one pair and in quite a few cases exhibited the same MPC positions). We assume that the MPC positions were maintained persistently conserved in a corresponding manner in the two remote protein families because they play important roles in structure and/or function determination, and we turn to find out what these roles might be.
Over-represented amino acid residues in mutually persistently conserved positions
Comparison of the amino-acid frequency distribution in MPC positions with their frequency distribution in all positions in the data revealed a significant difference (p≤.01 by a χ2 test). By applying a χ2 test to the individual amino acids we could point out the amino acids that contributed mostly to the significant deviation, and to identify those residues that were significantly over-represented (or under-represented) in MPC positions. As illustrated in Figure 2 ▶, aspartic acid, isoleucine, glycine, proline, histidine, cysteine, tryptophan, phenylalanine, and tyrosine were found to be significantly over-represented in MPC positions in comparison to their background frequencies. In many cases, those residues were maintained unchanged in the structurally aligned positions of the two pair mates. Previously, we have shown that conserved identical residues in aligned positions have distinct roles, mainly in or near the active sites of the proteins (Friedberg et al. 2000a).
Fig. 2.

Distribution of residue types in mutually persistently conserved (MPC) positions expressed as the log-odds ratio between the frequency of a residue in MPC positions (obs) and its frequency in the entire database of SSSD proteins (exp). All frequency differences were found to be statistically significant by a κ2 test, except for Leucine, Asparagine, and Valine (marked with ‘∧’).
Substitution matrix derived from mutually persistently conserved positions
A log-odds substitution matrix based on the data from MPC positions describes the favorableness of substitutions in these positions, and provides a tool for analyzing the allowable substitutions at positions that are suspected to be important for structural/ functional determination. We derived such a matrix (Fig. 3a ▶), as described in the Materials and Methods section. The pairs of residues that were introduced to the matrix's cells were the pairs of residues in the aligned proteins, at positions denoted as MPC. For comparison, we derived also a matrix from all structurally aligned positions in the database (Fig. 3b ▶). The structurally derived matrix was quite similar to such matrices derived by others (Naor et al. 1996; Prlic et al. 2000). Comparison of the amino-acid pair distribution forming the structurally derived matrix to those forming the matrices of the BLOSUM series (Henikoff and Henikoff 1992) has revealed an interesting pattern of similarity (Fig. 4 ▶). The amino-acid pair distributions were compared by computing the Jensen-Shannon divergence (Lin 1991; see Materials and Methods for details). We found that as the BLOSUM series number rises, the similarity between the distributions decreases (Fig. 4 ▶). In other words, the structurally derived matrix is most similar to the BLOSUM matrix derived from low-similarity sequences. On the contrary, the Jensen-Shannon divergence between the distributions used to derive the MPC matrix and those of the BLOSUM series remained almost constant regardless of BLOSUM serial number, and it was usually higher than the corresponding divergence obtained with the structurally derived distribution.
Fig. 3.


Amino-acid residue substitution matrices derived from (a) mutually persistently conserved positions and (b) all structurally aligned positions. Values are scaled to 1/10 bit.
Fig. 4.

Comparison between sequence-derived and structure-derived substitution matrices. The amino-acid pair frequency distributions that were used for the derivation of the substitution matrices were compared by the Jensen-Shannon divergence. A series of BLOSUM matrices were compared with the mutually persistently conserved-derived matrix (filled squares) and with the structurally derived matrix (open circles).
The relative entropy of the MPC-derived matrix was found to be 1.015 bits and highly statistically significant (p≤.01). For comparison, the relative entropy of the structurally derived matrix is 0.17 bits. Generally, when a substitution matrix is derived from a multiple sequence alignment, the relative entropy of the matrix decreases as the evolutionary distances among the sequences increase. This is because the observed and background distributions draw closer as evolutionary distance increases. Interestingly, although the MPC-derived matrix is constructed from protein pairs with no detectable sequence similarity, its relative entropy value is quite high, correlating with those matrices incorporating close homologs, such as BLOSUM 85. However, the high relative entropy values in matrices incorporating close homologs is because of the fact that they have not yet diverged, and the rate of substitutions among them is quite low. By definition, the majority of substitutions between similar sequences are synonymous. In the MPC-derived matrix, the same high frequency of synonymy prevails, but for a completely different reason: MPC positions will tend to be synonymous by virtue of their irreplaceability, and not because they have not yet diverged in evolution. The MPC-derived matrix therefore combines two components of vastly different types of substitution matrices: on the one hand, it is a matrix derived from the alignments of proteins that are remote in sequence. On the other hand, the actual data from which this matrix is derived is that of structurally aligned positions that are extremely well-conserved. We propose that by determining the MPC positions, we include in the matrix only those positions that, although substituted between distant proteins, still maintain a necessary role in their respective structures. Inspection of the MPC-derived matrix (Fig. 3a ▶) reveals that favorably substituted residues are located along the diagonal and within three distinct groups: aromatic, aliphatic, and charged. The high log-odds values along the diagonal indicate that the maintenance of certain residues as unchanged is of high significance in MPC positions. Other replacements are limited to residues with similar physicochemical characteristics. Within the aromatic residues, we note that substitutions among tyrosine, phenylalanine, and tryptophan are highly favorable. Likewise, substitutions among the aliphatic residues are highly favorable. While such general preferences have been observed in the other matrices, the actual relative contributions of the various pairwise substitutions are different, as demonstrated in Figure 4 ▶. It is conceivable therefore, that the MPC-derived matrix captures valuable information regarding critical structural determinants, putting high restrictions on allowable substitutions in positions that are supposedly critical for the structure and function determination.
Mutually persistently conserved positions in secondary structure elements
One role key positions may have in structure determination is in the stabilization of secondary structures. Extensive work has been done in determining the preferences of certain residue types in α helices and their role in determining helix structure, including helix initiation and termination (Aurora and Rose 1998; Kumar and Bansal 1998). Here, we investigate the presence of MPC residues in specific positions along secondary-structure elements, both in α helices and β strands. The MPC frequencies at each position in the vicinity of the termini of secondary structure elements and their flanking regions were determined and compared to the frequencies expected at random (see Materials and Methods). Figure 5 ▶ shows the comparison between observed and expected frequencies of MPC positions along the positions of the secondary-structure elements, expressed by their log-odds values. As demonstrated in Figure 5a ▶, there is a clear preference for MPC positions to be present at the flanking regions of α helices, both at their N and C termini. These tendencies were found to be highly statistically significant by a χ2 test, especially in positions N"`, N`, and C" (p≤.01). Thus, these residues probably play a role in the helix initiation and termination. Notably, among the MPC positions at N` and N", there is over-representation of amino-acid residues with hydrogen bond acceptors in their side chain, consistent with a possible role in determination of the N-terminus of the helix. Similarly, the MPC positions at position C` show over-representation of amino-acid residues with a hydrogen-bond donor in their side chains, consistent with the stabilization of the C-terminus of the helix, which is relatively negatively charged. Specific positioning of MPC residues in β strands also is observed, although less prominently (Fig. 5b ▶). It was found that MPC residues were preferred at the terminal position of the β strand and in its vicinity (p≤.01). Thus, we have demonstrated that one of the roles MPC residues may have is in the determination and stabilization of secondary structure elements along the protein sequence. A similar observation was reported by Reddy et al. (2001) for residues in CKAAPs.
Fig. 5.

Frequency of mutually persistently conserved (MPC) positions in secondary structure elements. The X-axis shows the positions in and flanking the secondary structure element (nomenclature after Aurora and Rose 1998). The flanking regions are marked with apostrophes, the in-element residues with digits, and the initial and terminal (capping) residues with a "c." The Y-axis is the logarithm of the ratio between the actual frequency of MPC residues in a position and that expected at random, based on the overall frequency of MPC positions in the data. The positions in which MPCs were found to be significantly over- or under-represented are marked with an "*." (a) α helices; (b) β strands.
Solvent accessibility
The solvent accessibility values of MPC positions were compared with those of all paired positions in the database. Figure 6 ▶ shows the fractions of exposed and buried MPC positions in comparison to all aligned database residues. By applying a t-test to the actual solvent accessibility values, we show that the MPC positions significantly differ from the positions in the whole data set (p≤0.02). This suggests that MPC positions tend to be located in the protein's interior, lending further support to their possible role as maintainers of structure/function.
Fig. 6.

Distribution of mutually persistently conserved positions (white bars) by solvent accessibility compared to all aligned residues (black bars). Residues were defined as buried when the solvent accessibility was <30% and exposed otherwise.
Spatial proximity of mutually persistently conserved positions
We describe the spatial proximity of MPC positions by using a graph representation. A graph in this respect denotes a network of nodes connected by edges. Graph-theoretical approaches have been used in structural biology, for example, for protein structure prediction (Samudrala and Moult 1998), for estimating the number of possible topologies for a protein built from a given number of elements (Jaroszewski and Godzik 2000), and for identifying side-chain clusters in protein structures (Kannan and Vishveshwara 1999). Here, we use such a representation to describe the structural proximity of MPC residues, using the structural information of both respective protein structures that make a pair. Any given pair of structurally aligned proteins is represented by a graph, where aligned residue positions are presented as vertices, and edges are drawn between those residue pairs that are aligned and are in spatial proximity in both pair mates. Residues consecutive in sequence are not joined by edges. From this graph, we extract the MPC subgraph: a graph whose nodes are those aligned positions that are MPC, and are in spatial proximity in both respective, aligned chains. By taking into account the number of edges and their sequence distance given the protein fold, a quantitative measure is assigned to each MPC subgraph, representing the spatial proximity of its constituents (see Materials and Methods for a detailed description). The higher this score, the closer in space are the examined residues.
At the end of this procedure, each protein pair is assigned a quantitative value that depicts the structural proximity of its MPC positions. To assess the significance of this spatial proximity score, we performed a Monte-Carlo run under the null hypothesis that the spatial proximity score for MPC positions is no better than that expected at random. Given Np MPC positions in a protein pair, we selected from the aligned proteins Np aligned positions at random, and derived their proximity score, as done for the MPC positions. This process was repeated 500 times for each protein pair. If less than 25 Monte-Carlo runs had a score greater than the MPC score (p<.05), then the null hypothesis was rejected, and the spatial proximity of the MPC positions was determined as statistically significant. Out of the 118 protein pairs, 69 pairs were found to have MPC positions whose spatial proximity was better than that expected at random. This suggests that a fraction of the MPC positions form spatial clusters of interacting residues that may have a functional or structural role. Thus, an additional role of these residues may be in establishing folding nuclei and/or special substructures associated with the functional sites.
A case study
For a close impression of the possible roles of MPC positions, we look at one example of a protein pair in our database and its MPC constituents: Lipase B from Candida antarctica (CALB; PDB entry 1tca [Uppenberg et al. 1994]) and haloalkane dehalogenase from Xanthobacter autotrophicus (XADL; PDB entry 1ede [Verschueren et al. 1993]), two enzymes that show a very high structural similarity, yet no detectable sequence similarity. CALB belongs to the lipase family, a diverse group of enzymes that hydrolyze triglycerides at lipid-water interfaces, and which all have a catalytic triad similar to the one found in serine proteases (Ser-His-Asp/Glu). XADL is a haloalkane dehalogenase, which converts 1-haloalkanes into primary alcohols and a halide ion by hydrolytic cleavage of the carbon-halogen bond. CALB and XADL belong to the SCOP superfamily of α/β hydrolases (Murzin et al. 1995), although they are classified in different families, fungal lipases and haloalkane dehalogenases, respectively. In CATH, CALB and XADL are classified in the same homology group despite their sequence dissimilarity, as a result of the high structural similarity of the two structures (Orengo et al. 1997). XADL is of length 310 amino acids and CALB is of length 317 amino acids, and there are 39 positions determined as MPC along the structural alignment. Inspection of the locations of these residues along the structures of the proteins shows the same patterns that were revealed for the whole database: many of the MPC positions are located at the termini of secondary structures, and a relatively large fraction are located in turns, thus their importance in the structure determination is by maintaining the turns that are critical for the overall fold of the molecules. Most interesting are those residues that form spatial clusters. Here, we discuss in detail one such cluster in the two proteins, which is related to the active site (Table 1).
Table 1.
Spatially close mutually persistently conserved positions in XADL and CALB that establish and stabilize the active site
| XADL | CALB | Parallel function |
| D260 | D167 | catalytic triad |
| D124 | S105 | catalytic triad |
| W125 | Q106 | substrate stabilization |
| G55 | G39 | active-site stabilization |
| G126 | G107 | active-site stabilization |
| G127 | G108 | active-site stabilization |
Looking at the alignment, both catalytic triads are structurally aligned, and XADL:D260/CALB:D187 and XADL: D124/CALB:S105 are identifiable as MPC. XADL:H289/CALB:H224 was not identifiable using MPC positions, as CALB:H224 is not a conserved residue in the first PSI-BLAST iteration, although it is conserved in subsequent iterations. The other residues identified as MPC also play a role in maintaining and stabilizing the active site. These include XADL:W125/CALB:Q106 that are MPC in both proteins. At least XADL:W125 is known to play a critical role in binding the halide atom of the substrate, as confirmed by mutation studies (Kennes et al. 1995). In addition, several glycines appear in this cluster as MPC. Glycine residues serve as "filler residues" when a side chain might interfere with necessary functionality or may disrupt secondary structure. Following the MPC XADL:W125/CALB:Q106, there are two consecutive glycines in both proteins XADL:G126/CALB:G107 and XADL:G127/CALB:G108. These are MPC positions with high conservation scores. Both are within a 7.0 A distance from XADL:D124/CALB:S105 active site nucleophile, and from XADL:W125/CALB:Q106, which is a substrate stabilizer. It is obvious that replacement of these glycines with a side-chain–containing residue will cause disruption of the proteins' function, by being in contact with the active site nucleophile, or with the substrate stabilizing residue. Another conserved glycine is observed in XADL:G55/CALB:G39. This glycine is well conserved in most of the α/β hydrolases. Indeed, its ZIC score is quite high: 3.47 in XADL and 4.65 in CALB. A side chain at this position would create a close contact between its Cβ and the Cβ of the active site position XADL:D124/CALB:S105 and possibly disturb interactions. Therefore, glycine is mostly suitable at this position. Thus, we have seen that spatially close MPC residues have clearly been maintained for a reason in this example for maintenance and stabilization of the structure of the active site.
Concluding remarks
Evolutionary information has been used in various studies to identify residues that may be important for structure and function determination (Mirny and Shakhnovich 1999, 2001; Ptitsyn and Ting 1999; Reddy et al. 2001). In most of these studies, the identification of candidate residues was obtained based on structural and sequence information, using different data sets for the structural and sequence analysis, and different approaches to estimate conservation of a suspected residue. It is mostly important that interpretation of the results should be done in view of the different parameters used in the analysis. In the current study, the data set of protein sequences used is constructed in a very stringent fashion. It includes pairs of similar-structure proteins that exhibit only 12% of identical residues on the average. The positions that are candidates for maintaining important structural/functional characteristics are structurally aligned positions that show residue conservation in their respective protein sequences. It is important to note that conservation is evaluated by calculating the information content of a position, considering the 20 amino acids without clustering and taking into account the background frequencies of the amino acids in the data. Positions with information content above the average of all the sequence positions are determined as conserved. Furthermore, only positions that were found to be conserved in both close and remote family members of the two corresponding protein families are considered in the analysis.
These definitions direct us to a specific subset of residues (MPC) that are shown to be relevant for structure/function determination. Among these residues stand out the aromatic residues, and cysteine, glycine, and proline as appearing above their background frequencies. The bulky aromatic residues play important roles in the packaging of the protein, while the cysteines are maintained conserved for preserving the disulfide bridges, and proline and glycine are located in critical structural positions, most often flanking secondary structures or near active sites. Notably, we do not identify as standing out the aliphatic residues that are frequently found in the protein hydrophobic cores and are important for protein stabilization. Because these residues are interchangeable among themselves and are highly abundant in the data, they cannot be singled out by our procedure that uses a 20-letter alphabet for the amino acids and considers their background frequencies in the information content calculation. Thus, the analysis directs us to different types of residues and structural roles. The residues that we have identified are mostly buried within their protein structures, but only in 70% of the proteins they form spatial clusters more than expected at random. In many of these cases, these clusters are found to be related to the active site of the protein. Thus, residues in MPC positions are important for establishing and maintaining the substructure around the active site. One very distinct feature of the identified MPC positions is their location in the termini of secondary structure elements. Thus, it is important to conserve certain types of residues in these positions to maintain secondary structure elements through evolution.
To obtain the MPC positions, we perform in each family a strict conservation analysis, but we do not require that the same residues be maintained in both proteins. This enables us to capture the interchangeability between MPC positions by analyzing the pairs of residues found in MPC positions of the two structurally aligned proteins. The substitution matrix obtained is very informative (H = 1.015) and defines the restrictions of allowable substitutions in these critical positions. We observe interchangeability only within groups of same-character amino acids, i.e., within the aliphatic, hydroxyl-containing aliphatic, aromatic, and charged amino acids. Interestingly, some interchanges between positively and negatively charged residues are observed, consistent with other structurally derived matrices (Naor et al. 1996; Blake and Cohen 2001). From a predictive perspective, such a matrix provides valuable information regarding the allowable substitutions for maintaining a desired structure. When comparing two multiple alignments of protein families, it may be used to identify those persistently conserved positions that should be aligned as mutually conserved, and serve as the anchor positions for homology modeling.
Materials and methods
Construction of a database of structurally similar, sequence-dissimilar proteins
The FSSP (fold classification based on structure-structure alignment of proteins) (Holm and Sander 1996) and distant aligned protein sequences (DAPS) (Rice and Eisenberg 1998, http://siren.bio.indiana.edu/daps) databases were used as a starting point for the database of 118 SSSD protein pairs. Briefly, FSSP is a database based on an exhaustive all-versus-all structural alignment of proteins in the PDB database (Berman et al. 2000). The classification and alignments are automatically maintained and continuously updated using the DALI program (Holm and Sander 1996). Each FSSP file has a single structural representative against which all structurally similar proteins are aligned in decreasing order of structural similarity. The DAPS database is based on FSSP and contains alignments of all protein pairs sharing <25% identical residues. These pairs of proteins were based on the PDB_SELECT25 list (Hobohm and Sander 1994). We verified that the structural alignments are not different from those based on structural alignments obtained by SSAP (Orengo and Taylor 1996). DALI, the structural alignment method used to construct FSSP, is based on a Monte-Carlo optimization for the alignment of Cα−Cα distance matrices. SSAP aligns proteins as a set of Cβ vectors. We found that ∼80% of the paired positions were coaligned when either alignment was used as a standard.
For generation of the SSSD database used in this study, the DAPS database was filtered using the following criteria (Friedberg et al. 2000a): minimal protein length of 30 residues for both pair members; resolution better than 3.5Å for each pair member; difference in lengths within a protein pair does not exceed 50% of the shorter member; the alignment length is at least 60% of the longer member's length; and the sequences of the pair members should not be well aligned using sequence alignment methods. A good sequence alignment, regardless of compatibility with the FSSP structural alignment, denotes a sequence similarity that we wish to avoid. Each pair was checked for similarity using the BESTFIT program from the GCG package (version 10, Genetics Computing Group), an implementation of the Smith-Waterman algorithm (Smith and Waterman 1981). Statistical significance was evaluated by comparing the actual alignment score to a sample of random scores obtained by alignment of one sequence to shuffled sequences with the same amino-acid composition as the second sequence. Sequence pairs with alignment scores that deviated more than six standard deviations from the average random score were excluded (Z≥6). The average Z score of the sequence pairs in our data was found to be 1.16 with a standard deviation of 1.63. Therefore, we can say that in SSSD, no proteins within a pair are similar. As a population, they are dissimilar enough to be considered unrelated by sequence.
The SSSD database is available at http://bioinfo.md.huji.ac.il/marg/SSSD/.
Assessment of conservation
Generally, per any given PSI-BLAST iteration, the information content for a single position j in a multiple sequence alignment (IC(j)) would be:
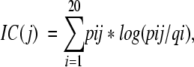 |
where: pij is the frequency of residue i at position j, and qi is the frequency of residue i in the database. We used the information content values provided by PSI-BLAST, which are actually corrected information content values incorporating the following caveats: sequence weighting in the MSA is performed so as to assign a smaller weight to sequences having more relatives in the MSA, thus preventing them from "outvoting" more divergent sequences; and estimation of pij is further complicated by sample size and knowledge of relationships among residues. PSI-BLAST solves this problem using pseudocounts (Altschul et al. 1997).
Upon obtaining the IC(j) for a given position in a given iteration, the normalized value ZIC(j) was calculated by
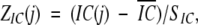 |
where IC is the mean of IC(j) values along the sequence in a particular iteration, and SIC is its standard deviation. A position was considered to be conserved when ZIC(j)>0.
Statistical significance of number of mutually persistently conserved positions found
To evaluate if the observed number of MPC positions deviates significantly from that expected at random, we applied a normal approximation to a binomial test. The null hypothesis is that the fraction of MPC positions observed is the same as that expected at random. The expected number of MPC positions is calculated as follows: as there is a differential conservation in buried locations compared with exposed locations, we partitioned all positions according to their solvent exposure. Fifty-six percent of the positions were found to be buried (<30% solvent exposure) and 44% exposed. The fraction of PCs in buried and exposed positions is 0.3784 and 0.1914, respectively. The fraction of MPC positions expected at random is therefore 0.56 × 0.37842 + 0.44 × 0.19142 = 0.096. By using a normal approximation to a binomial test, we show that the deviation between observed and expected at random is highly statistically significant (Z = 30; p<.0001).
Residue distribution in secondary structure elements
Analysis of residue distribution in secondary structure elements was carried out as follows: helix and strand locations were determined using DSSP (Kabsch and Sander 1983). Helices or strands whose lengths were less than seven residues were discarded. Each MPC position was assigned in a secondary structure position, or a flanking region. We have named the positions as in Aurora and Rose (1998): the order of N4`, N"`,N",N`,Ncap,N1,N2,N3, N4. . .C4,C3,C2,C1,Ccap. . .C4` for flanking and in-element positions is given. The flanking regions are marked with apostrophes, the in-element residues with digits, and the initial and terminal (capping) residues with a "c."
We aligned all helices by the determined positions and calculated the relative occurrence of MPC residues in each position. The same was done for β strands. The occurrence of MPC residues in a position was expressed as log(Nj/Ej), where Nj is the actual number of MPC residues at position j, and Ej is the expected number of MPC residues, based on the fraction of MPC residues in the data.
Solvent accessibility
Solvent accessibility (SA) values in Å2, were taken from the FSSP database. For each residue, these were divided by the accessible surface area of the extended conformation of that residue (Miller et al. 1987) and expressed in percentages. The analysis was carried out both by using these values and by clustering the residues into two solvent-accessibility categories: buried (SA<30%) and exposed (SA≥30%).
Assessing spatial proximity of mutually persistently conserved positions
As described in the Results section, we assess the spatial proximity of MPC positions using a graph representation of the residues in the protein. A quantitative measure of the spatial proximity of residues in an MPC subgraph would be the number of edges in it compared to the number of vertices. However, because we compare the actual measure to those obtained by a Monte-Carlo procedure that uses the same number of vertices, the constant number of vertices is canceled out. In addition, instead of just counting the number of edges, the spatial proximity is better represented by weighting the edges according to the probability of having contacting residues within that sequence distance in the particular fold examined. Generally, an edge drawn between contacting residues distant in sequence receives a higher weight than an edge drawn between contacting residues that are close in sequence. However, because of the different folds of different proteins, the weighting function should not be universal. Therefore, it was constructed according to the contact map of each chain. For example, upon examining a particular fold, it might be shown that residues within a sequence distance of 50 positions have a higher probability of being in contact than residues within a distance of 40 or 60 positions. This phenomenon is a result of regularity in the protein's tertiary structure, and will vary between different fold patterns. Thus, for computing the edge weights, the frequency of contacting residues with the same sequence separation was taken into account.
The calculation in detail is shown below. Given two structurally aligned proteins A and B:
For protein A build a vector A = <a2,. . .,an−2>, ak being the number of contacting residue pairs that are k residues distant in sequence, and n is the chain length (2≤ k ≤n−2). Contacting residues are those with a distance ≤7.0Å between β-carbons. This process is repeated for the second protein in the alignment, generating B = <b2,. . .,bm−2> (m being the length of the second protein sequence).
Determine the probability for two residues separated by a sequence distance of k positions to be in contact:
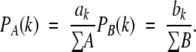 |
The weight of the edge drawn between any two contacting residues separated by a sequence distance of k positions (WA(k) for protein A and WB(k) for protein B) is calculated as follows:
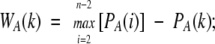 |
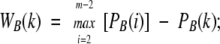 |
where max [PA(i)] and max [PB(i)] are the maximal probabilities for two i-separated residues to be in contact in proteins A and B, respectively. This weighting function provides an equal baseline score of zero for proteins in a pair.
Ai and Aj are two contacting residues in protein A. They are aligned to Bk and Bl respectively, which are also contacting. Therefore, one vertex would be [Ai,Bk] and the other would be [Aj,Bl].
The edge weight between the vertices [Ai,Bk] and [Aj,Bl] would be
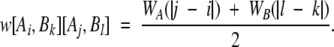 |
Without the weighting function, the quantitative measure for the spatial proximity of residues in a subgraph would have been obtained by the summation of the edges. Similarly, now the weighted number of edges is summed.
Finally, assessment of the spatial proximity of the MPC positions is performed using a Monte-Carlo procedure. For each protein pair we repeat the above analysis with randomly picked aligned positions. The number of those positions is the same as the number of vertices in the MPC graph. The randomization is repeated 500 times. If <25 randomization scores (5% of 500) have a better spatial proximity score than the MPC score, the result is considered significant.
Generation of log-odds matrices
Generation of the matrix derived from mutually persistently conserved positions
All the aligned positions that were determined to be MPC were tallied. For each two residues Ai and Aj (1≤i≤j≤20), we count the number of times that they appear as aligned in MPC positions. This provides the number of substitutions between Ai and Aj. A substitution matrix was derived as described in (Naor et al. 1996). The values that appear in the matrix in Figure 3 ▶ were obtained by
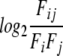 |
where Fij are the observed frequencies of substitutions between amino acids Ai and Aj, and Fi•Fj are the expected frequencies, based on the frequencies of amino acids Ai and Aj in the data. The values are scaled to 1/10 bits.
Generation of the structurally derived matrix
For the structurally derived matrix, all the aligned positions in the SSSD protein pairs were tallied, and the matrix was derived as described above.
Comparing frequency distributions by the Jensen-Shannon divergence
The Jensen-Shannon (JS) divergence of two distributions p1 and p2 is defined as in (Lin 1991): JS = H(π1p1+π2p2) – π1H(p1) – π2H(p2), where H(pi) is the entropy of distribution pi, and πi is the weight given to that distribution. π1, π2>0 and π1+π2 = 1. We used JS divergence with π1 = π2 = 0.5 to compare between the observed amino-acid pair frequency distributions in the BLOSUM matrices and the MPC and structurally derived matrices.
Acknowledgments
This study was supported by the Israeli Science Foundation administered by the Israeli Academy of Sciences and Humanities. We thank Naftali Tishby for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
PC, persistently conserved
MPC, mutually persistently conserved
SSSD, structurally similar sequence dissimilar
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.18602
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora, R. and Rose, G.D. 1998. Helix capping. Protein Sci. 7 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The protein data bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake, J.D. and Cohen, F.E. 2001. Pairwise sequence alignment below the twilight zone. J. Mol. Biol. 307 721–735. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U., Reidhaar-Olson, J.F., Lim, W.A., and Sauer, R.T. 1990. Deciphering the message in protein sequences: Tolerance to amino acid substitutions. Science 247 1306–1310. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E. and Levitt, M. 2000. Expectations from structural genomics. Protein Sci. 9 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirel, M.C., Atilgan, A.R., Jernigan, R.L., Erman, B., and Bahar, I. 1998. Identification of kinetically hot residues in proteins. Protein Sci. 7 2522–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi, Z., Fiser, A., and Simon, I. 1997. Stabilization centers in proteins: Identification, characterization and predictions. J. Mol. Biol. 272 597–612. [DOI] [PubMed] [Google Scholar]
- Friedberg, I., Kaplan, T., and Margalit, H. 2000a. Glimmers in the midnight zone: Characterization of aligned identical residues in sequence-dissimilar proteins sharing a common fold. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8 162–170. [PubMed] [Google Scholar]
- Friedberg, I., Kaplan, T., and Margalit, H. 2000b. Evaluation of PSI-BLAST alignment accuracy in comparison to structural alignments. Protein Sci. 9 2278–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobohm, U. and Sander, C. 1994. Enlarged representative set of protein structures. Protein Sci. 3 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. The FSSP database: Fold classification based on structure-structure alignment of proteins. Nucleic Acids Res. 24 206–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroszewski, L. and Godzik, A. 2000. Search for a new description of protein topology and local structure. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8 211–217. [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kannan, N. and Vishveshwara, S. 1999. Identification of side-chain clusters in protein structures by a graph spectral method. J. Mol. Biol. 292 441–464. [DOI] [PubMed] [Google Scholar]
- Kennes, C., Pries, F., Krooshof, G.H., Bokma, E., Kingma, J., and Janssen, D.B. 1995. Replacement of tryptophan residues in haloalkane dehalogenase reduces halide binding and catalytic activity. Eur. J. Biochem. 228 403–407. [PubMed] [Google Scholar]
- Koehl, P. and Levitt, M., 1999. Structure-based conformational preferences of amino acids. Proc. Natl. Acad. Sci. 96 12524–12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppensteiner, W.A., Lackner, P., Wiederstein, M., and Sippl, M.J. 2000. Characterization of novel proteins based on known protein structures. J. Mol. Biol. 296 1139–1152. [DOI] [PubMed] [Google Scholar]
- Kumar, S. and Bansal, M. 1998. Dissecting α-helices: Position-specific analysis of α-helices in globular proteins. Proteins 31 460–476. [DOI] [PubMed] [Google Scholar]
- Lim, W.A. and Sauer, R.T. 1989. Alternative packing arrangements in the hydrophobic core of lambda repressor. Nature 339 31–36. [DOI] [PubMed] [Google Scholar]
- Lin, J. 1991. Divergence measures based on the Shannon entropy. IEEE Transactions on Information Theory 37 145–151. [Google Scholar]
- Markiewicz, P., Kleina, L.G., Cruz, C., Ehret, S. and Miller, J.H. 1994. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as "spacers" which do not require a specific sequence. J. Mol. Biol. 240 421–433. [DOI] [PubMed] [Google Scholar]
- Milla, M.E., Brown, B.M., and Sauer, R.T. 1994. Protein stability effects of a complete set of alanine substitutions in Arc repressor. Nat. Struct. Biol. 1 518–523. [DOI] [PubMed] [Google Scholar]
- Miller, S., Janin, J., Lesk, A.M., and Chothia, C. 1987. Interior and surface of monomeric proteins. J. Mol. Biol. 196 641–656. [DOI] [PubMed] [Google Scholar]
- Mirny, L. and Shakhnovich, E. 2001. Evolutionary conservation of the folding nucleus. J. Mol. Biol. 308 123–129. [DOI] [PubMed] [Google Scholar]
- Mirny, L.A. and Shakhnovich, E.I. 1999. Universally conserved positions in protein folds: Reading evolutionary signals about stability, folding kinetics and function. J. Mol. Biol. 291 177–196. [DOI] [PubMed] [Google Scholar]
- Mirny, L.A., Abkevich, V.I., and Shakhnovich, E.I., 1998. How evolution makes proteins fold quickly. Proc. Natl. Acad. Sci. 95 4976–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Naor, D., Fischer, D., Jernigan, R.L., Wolfson, H.J., and Nussinov, R. 1996. Amino acid pair interchanges at spatially conserved locations. J. Mol. Biol. 256 924–938. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A. and Taylor, W.R. 1996. SSAP: Sequential structure alignment program for protein structure comparison. Methods Enzymol. 266 617–635. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Jones, D.T., and Thornton, J.M. 1994. Protein superfamilies and domain superfolds. Nature 372 631–634. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Michie, A.D., Jones, S., Jones, D.T., Swindells, M.B., and Thornton, J.M. 1997. CATH—a hierarchic classification of protein domain structures. Structure 5 1093–1108. [DOI] [PubMed] [Google Scholar]
- Park, J., Karplus, K., Barrett, C., Hughey, R., Haussler, D., Hubbard, T., and Chothia, C. 1998. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 284 1201–1210. [DOI] [PubMed] [Google Scholar]
- Prlic, A., Domingues, F.S., and Sippl, M.J. 2000. Structure-derived substitution matrices for alignment of distantly related sequences. Protein Eng. 13 545–550. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. and Ting, K.L. 1999. Non-functional conserved residues in globins and their possible role as a folding nucleus. J. Mol. Biol. 291 671–682. [DOI] [PubMed] [Google Scholar]
- Reddy, B.V., Li, W.W., Shindyalov, I.N., and Bourne, P.E. 2001. Conserved key amino acid positions (CKAAPs) derived from the analysis of common substructures in proteins. Proteins 42 148–163. [DOI] [PubMed] [Google Scholar]
- Rennell, D., Bouvier, S.E., Hardy, L.W., and Poteete, A.R. 1991. Systematic mutation of bacteriophage T4 lysozyme. J. Mol. Biol. 222 67–88. [DOI] [PubMed] [Google Scholar]
- Samudrala, R. and Moult, J. 1998. A graph-theoretic algorithm for comparative modeling of protein structure. J. Mol. Biol. 279 287–302. [DOI] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Suckow, J., Markiewicz, P., Kleina, L.G., Miller, J., Kisters-Woike, B., and Muller-Hill, B. 1996. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J. Mol. Biol. 261 509–523. [DOI] [PubMed] [Google Scholar]
- Uppenberg, J., Hansen, M.T., Patkar, S., and Jones, T.A. 1994. The sequence, crystal structure determination and refinement of two crystal forms of lipase B from Candida antarctica. Structure 2 293–308. [DOI] [PubMed] [Google Scholar]
- Verschueren, K.H., Franken, S.M., Rozeboom, H.J., Kalk, K.H., and Dijkstra, B.W. 1993. Refined x-ray structures of haloalkane dehalogenase at pH 6.2 and pH 8.2 and implications for the reaction mechanism. J. Mol. Biol. 232 856–872. [DOI] [PubMed] [Google Scholar]